
The Role of the Adrenal–Gut–Brain Axis on Comorbid Depressive Disorder Development in Diabetes
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Diabetes, the HPA Axis, and Microbiota
3. Depression, the HPA Axis, and Microbiota
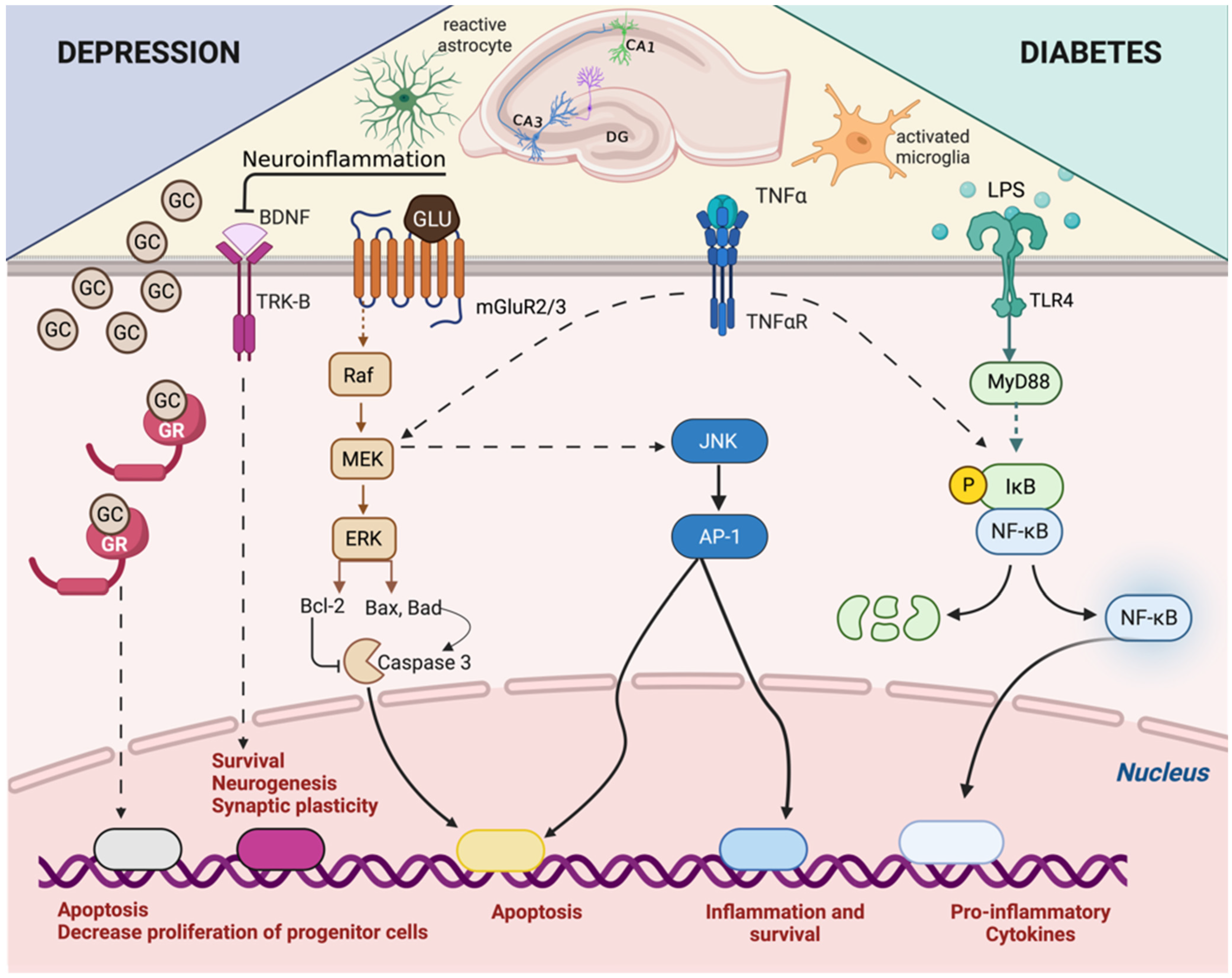
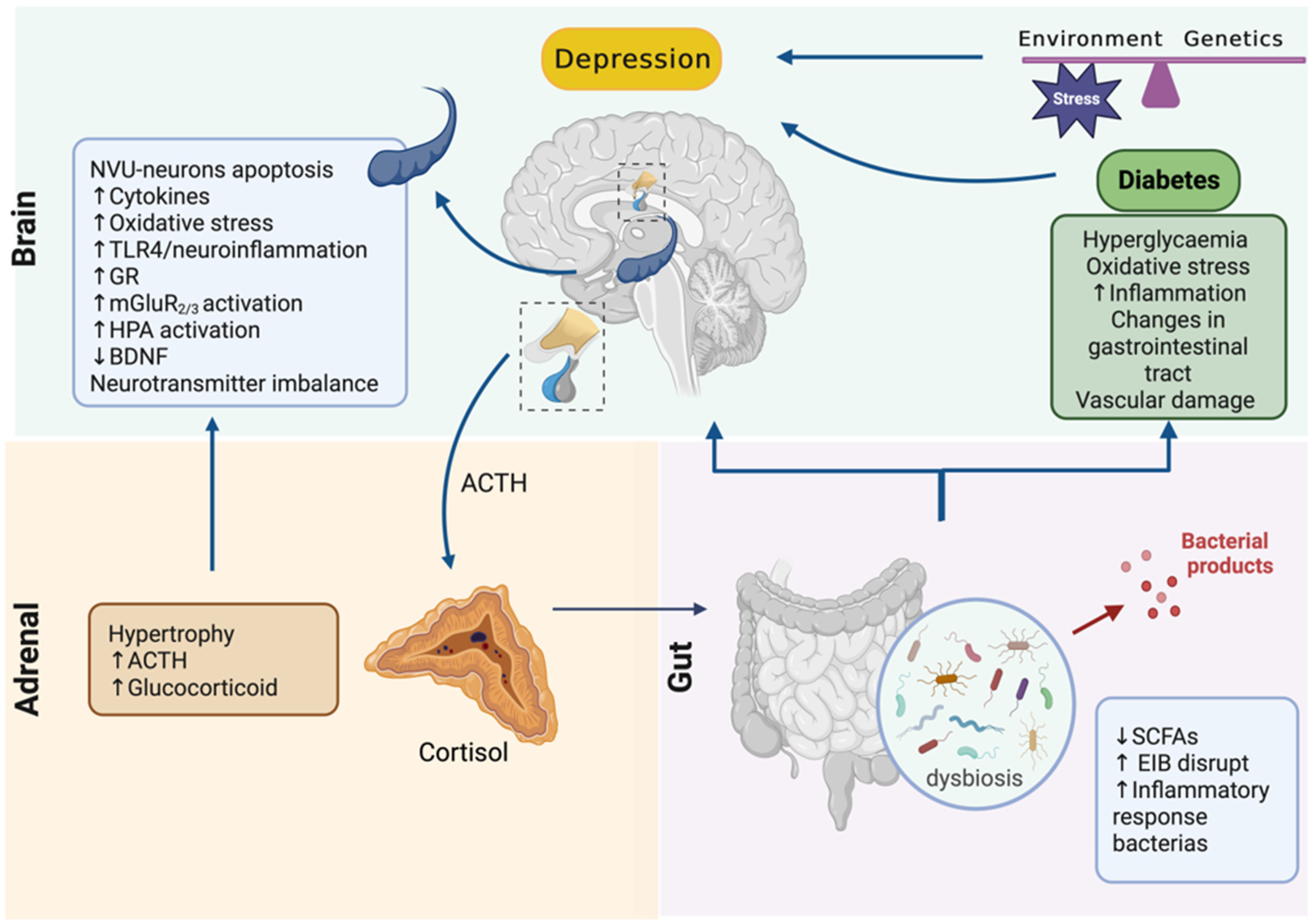
4. Adrenal–Gut Axis Imbalance Is Central to Depressive Disorder Development and Aggravation in Diabetes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Fiedorowicz, J.G.; Palagummi, N.M.; Forman-Hoffman, V.L.; Miller, D.D.; Haynes, W.G. Elevated Prevalence of Obesity, Metabolic Syndrome, and Cardiovascular Risk Factors in Bipolar Disorder. Ann. Clin. Psychiatry 2008, 20, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Fiedorowicz, J.G.; He, J.; Merikangas, K.R. The Association between Mood and Anxiety Disorders with Vascular Diseases and Risk Factors in a Nationally Representative Sample. J. Psychosom. Res. 2011, 70, 145–154. [Google Scholar] [CrossRef]
- Liu, J.; Han, Y.-S.; Liu, L.; Tang, L.; Yang, H.; Meng, P.; Zhao, H.-Q.; Wang, Y.-H. Abnormal Glu/MGluR2/3/PI3K Pathway in the Hippocampal Neurovascular Unit Leads to Diabetes-Related Depression. Neural Regen. Res. 2021, 16, 727–733. [Google Scholar] [CrossRef]
- Borbély, É.; Simon, M.; Fuchs, E.; Wiborg, O.; Czéh, B.; Helyes, Z. Novel Drug Developmental Strategies for Treatment-Resistant Depression. Br. J. Pharmacol. 2022, 179, 1146–1186. [Google Scholar] [CrossRef]
- Bryan, C.; Songer, T.; Brooks, M.M.; Rush, A.J.; Thase, M.E.; Gaynes, B.; Balasubramani, G.K.; Trivedi, M.H.; Fava, M.; Wisniewski, S.R. The Impact of Diabetes on Depression Treatment Outcomes. Gen. Hosp. Psychiatry 2010, 32, 33–41. [Google Scholar] [CrossRef]
- Zhou, X.-Y.; Zhang, F.; Hu, X.-T.; Chen, J.; Tang, R.-X.; Zheng, K.-Y.; Song, Y.-J. Depression Can Be Prevented by Astaxanthin through Inhibition of Hippocampal Inflammation in Diabetic Mice. Brain Res. 2017, 1657, 262–268. [Google Scholar] [CrossRef]
- Ergenc, M.; Ozacmak, H.S.; Turan, I.; Ozacmak, V.H. Melatonin Reverses Depressive and Anxiety Like-Behaviours Induced by Diabetes: Involvement of Oxidative Stress, Age, Rage and S100B Levels in the Hippocampus and Prefrontal Cortex of Rats. Arch. Physiol. Biochem. 2022, 128, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Thangaraj, D.; Radhakrishnan, M. A Novel 5HT3 Antagonist 4i (N-(3-Chloro-2-Methylphenyl)Quinoxalin-2-Carboxamide) Prevents Diabetes-Induced Depressive Phenotypes in Mice: Modulation of Serotonergic System. Behav. Brain Res. 2016, 297, 41–50. [Google Scholar] [CrossRef]
- Moosaie, F.; Mohammadi, S.; Saghazadeh, A.; Dehghani Firouzabadi, F.; Rezaei, N. Brain-Derived Neurotrophic Factor in Diabetes Mellitus: A Systematic Review and Meta-Analysis. PLoS ONE 2023, 18, e0268816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, D.; Law, H.K.-W.; Wu, Y.; Zhu, G.-H.; Huang, W.-Y.; Kang, Y. Integrative Analysis of Gut Microbiota and Fecal Metabolites in Rats after Prednisone Treatment. Microbiol. Spectr. 2021, 9, e0065021. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Yang, L.; Jiang, J.; Ni, Y.; Zhu, J.; Zheng, X.; Wang, Q.; Lu, X.; Fu, Z. Chronic Glucocorticoid Treatment Induced Circadian Clock Disorder Leads to Lipid Metabolism and Gut Microbiota Alterations in Rats. Life Sci 2018, 192, 173–182. [Google Scholar] [CrossRef]
- Ergang, P.; Vagnerová, K.; Hermanová, P.; Vodička, M.; Jágr, M.; Šrůtková, D.; Dvořáček, V.; Hudcovic, T.; Pácha, J. The Gut Microbiota Affects Corticosterone Production in the Murine Small Intestine. Int. J. Mol. Sci. 2021, 22, 4229. [Google Scholar] [CrossRef]
- Speer, K.E.; Semple, S.; Naumovski, N.; D’Cunha, N.M.; McKune, A.J. HPA Axis Function and Diurnal Cortisol in Post-Traumatic Stress Disorder: A Systematic Review. Neurobiol. Stress 2019, 11, 100180. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhou, M.; Wang, J.; Yao, J.; Yu, J.; Liu, W.; Wu, L.; Wang, J.; Gao, R. Involvement of the Microbiota-Gut-Brain Axis in Chronic Restraint Stress: Disturbances of the Kynurenine Metabolic Pathway in Both the Gut and Brain. Gut Microbes 2021, 13, 1869501. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Marchesi, J.R.; Scully, P.; Codling, C.; Ceolho, A.-M.; Quigley, E.M.M.; Cryan, J.F.; Dinan, T.G. Early Life Stress Alters Behavior, Immunity, and Microbiota in Rats: Implications for Irritable Bowel Syndrome and Psychiatric Illnesses. Biol. Psychiatry 2009, 65, 263–267. [Google Scholar] [CrossRef]
- Jahnke, J.R.; Roach, J.; Azcarate-Peril, M.A.; Thompson, A.L. Maternal Precarity and HPA Axis Functioning Shape Infant Gut Microbiota and HPA Axis Development in Humans. PLoS ONE 2021, 16, e0251782. [Google Scholar] [CrossRef]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis during Early Life Regulates the Hippocampal Serotonergic System in a Sex-Dependent Manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal Microbial Colonization Programs the Hypothalamic-Pituitary-Adrenal System for Stress Response in Mice: Commensal Microbiota and Stress Response. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Chan, O.; Chan, S.; Inouye, K.; Vranic, M.; Matthews, S.G. Molecular Regulation of the Hypothalamo-Pituitary-Adrenal Axis in Streptozotocin-Induced Diabetes: Effects of Insulin Treatment. Endocrinology 2001, 142, 4872–4879. [Google Scholar] [CrossRef] [PubMed]
- Allin, K.H.; Nielsen, T.; Pedersen, O. Mechanisms in Endocrinology: Gut Microbiota in Patients with Type 2 Diabetes Mellitus. Eur. J. Endocrinol. 2015, 172, R167–R177. [Google Scholar] [CrossRef]
- Joseph, J.J.; Golden, S.H. Cortisol Dysregulation: The Bidirectional Link between Stress, Depression, and Type 2 Diabetes Mellitus. Ann. N. Y. Acad. Sci. 2017, 1391, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Farzi, A.; Hassan, A.M.; Zenz, G.; Holzer, P. Diabesity and Mood Disorders: Multiple Links through the Microbiota-Gut-Brain Axis. Mol. Asp. Med. 2019, 66, 80–93. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the Immune System. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Sagara, R.; Inoue, T.; Sonoda, N.; Yano, C.; Motoya, M.; Umakoshi, H.; Sakamoto, R.; Ogawa, Y. Association between Cortisol and Left Ventricular Diastolic Dysfunction in Patients with Diabetes Mellitus. J. Diabetes Investig. 2022, 13, 344–350. [Google Scholar] [CrossRef]
- Roy, M.S.; Roy, A.; Brown, S. Increased Urinary-Free Cortisol Outputs in Diabetic Patients. J. Diabetes Its Complicat. 1998, 12, 24–27. [Google Scholar] [CrossRef]
- Chiodini, I.; Adda, G.; Scillitani, A.; Coletti, F.; Morelli, V.; Di Lembo, S.; Epaminonda, P.; Masserini, B.; Beck-Peccoz, P.; Orsi, E.; et al. Cortisol Secretion in Patients With Type 2 Diabetes. Diabetes Care 2007, 30, 83–88. [Google Scholar] [CrossRef]
- Roy, M.S.; Roy, A.; Gallucci, W.T.; Collier, B.; Young, K.; Kamilaris, T.C.; Chrousos, G.P. The Ovine Corticotropin-Releasing Hormone-Stimulation Test in Type I Diabetic Patients and Controls: Suggestion of Mild Chronic Hypercortisolism. Metabolism 1993, 42, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Inouye, K.; Vranic, M.; Matthews, S.G. Hyperactivation of the Hypothalamo-Pituitary-Adrenocortical Axis in Streptozotocin-Diabetes Is Associated with Reduced Stress Responsiveness and Decreased Pituitary and Adrenal Sensitivity. Endocrinology 2002, 143, 1761–1768. [Google Scholar] [CrossRef]
- Kim, S.; Park, E.S.; Chen, P.R.; Kim, E. Dysregulated Hypothalamic-Pituitary-Adrenal Axis Is Associated With Increased Inflammation and Worse Outcomes After Ischemic Stroke in Diabetic Mice. Front. Immunol. 2022, 13, 864858. [Google Scholar] [CrossRef]
- Carvalho, V.F.; Barreto, E.O.; Diaz, B.L.; Serra, M.F.; Azevedo, V.; Cordeiro, R.S.B.; Martins, M.A.; Silva, P.M.R.E. Systemic Anaphylaxis Is Prevented in Alloxan-Diabetic Rats by a Mechanism Dependent on Glucocorticoids. Eur. J. Pharmacol. 2003, 472, 221–227. [Google Scholar] [CrossRef]
- Torres, R.C.; Prevatto, J.P.; Martins, M.A.; Carvalho, V.F. From Type-1 Diabetes HPA Axis to the Disease Complications. J. Diabetes Metab. 2013, S12, 1–8. [Google Scholar] [CrossRef]
- Silva, P.M.R.E.; Carvalho, V.F.; Cordeiro, R.S.B.; Martins, M.A. Down-Regulation of Allergic Responses in Conditions of Experimental Diabetes: A Role for Glucocorticoids? Neuroimmunomodulation 2009, 16, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Chaves, A.D.S.; Magalhães, N.S.; Insuela, D.B.R.; Silva, P.M.R.E.; Martins, M.A.; Carvalho, V.F. Effect of the Renin-Angiotensin System on the Exacerbation of Adrenal Glucocorticoid Steroidogenesis in Diabetic Mice: Role of Angiotensin-II Type 2 Receptor. Front. Endocrinol. 2022, 13, 1040040. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.C.; Magalhães, N.S.; Silva, P.M.R.E.; Martins, M.A.; Carvalho, V.F. Activation of PPAR-γ Reduces HPA Axis Activity in Diabetic Rats by up-Regulating PI3K Expression. Exp. Mol. Pathol. 2016, 101, 290–301. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting Innate Immune Mediators in Type 1 and Type 2 Diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; Van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut Metagenome in European Women with Normal, Impaired and Diabetic Glucose Control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; Van Den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Durazzo, M.; Ferro, A.; Gruden, G. Gastrointestinal Microbiota and Type 1 Diabetes Mellitus: The State of Art. J. Clin. Med. 2019, 8, 1843. [Google Scholar] [CrossRef]
- Demirci, M.; Bahar Tokman, H.; Taner, Z.; Keskin, F.E.; Çağatay, P.; Ozturk Bakar, Y.; Özyazar, M.; Kiraz, N.; Kocazeybek, B.S. Bacteroidetes and Firmicutes Levels in Gut Microbiota and Effects of Hosts TLR2/TLR4 Gene Expression Levels in Adult Type 1 Diabetes Patients in Istanbul, Turkey. J. Diabetes Complicat. 2020, 34, 107449. [Google Scholar] [CrossRef] [PubMed]
- Radwan, S.; Gilfillan, D.; Eklund, B.; Radwan, H.M.; El Menofy, N.G.; Lee, J.; Kapuscinski, M.; Abdo, Z. A Comparative Study of the Gut Microbiome in Egyptian Patients with Type I and Type II Diabetes. PLoS ONE 2020, 15, e0238764. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, X.; Sun, L.; Liu, Y.; Lv, Y.; Gang, X.; Wang, G. Gut Microbiota Profile in Patients with Type 1 Diabetes Based on 16S RRNA Gene Sequencing: A Systematic Review. Dis. Markers 2020, 2020, 3936247. [Google Scholar] [CrossRef]
- Ejtahed, H.-S.; Hoseini-Tavassol, Z.; Khatami, S.; Zangeneh, M.; Behrouzi, A.; Ahmadi Badi, S.; Moshiri, A.; Hasani-Ranjbar, S.; Soroush, A.-R.; Vaziri, F.; et al. Main Gut Bacterial Composition Differs between Patients with Type 1 and Type 2 Diabetes and Non-Diabetic Adults. J. Diabetes Metab. Disord. 2020, 19, 265–271. [Google Scholar] [CrossRef]
- Guilloteau, P.; Martin, L.; Eeckhaut, V.; Ducatelle, R.; Zabielski, R.; Van Immerseel, F. From the Gut to the Peripheral Tissues: The Multiple Effects of Butyrate. Nutr. Res. Rev. 2010, 23, 366–384. [Google Scholar] [CrossRef]
- Ochocińska, A.; Wysocka-Mincewicz, M.; Groszek, A.; Rybak, A.; Konopka, E.; Bierła, J.B.; Trojanowska, I.; Szalecki, M.; Cukrowska, B. Could I-FABP Be an Early Marker of Celiac Disease in Children with Type 1 Diabetes? Retrospective Study from the Tertiary Reference Centre. Nutrients 2022, 14, 414. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.-H.; Xie, Q.-S.; Chen, G.-C.; Huang, C.-L.; Yu, T.; Chen, Q.-K.; Li, J.-Y. Impaired Intestinal Barrier Function in Type 2 Diabetic Patients Measured by Serum LPS, Zonulin, and IFABP. J. Diabetes Its Complicat. 2021, 35, 107766. [Google Scholar] [CrossRef]
- Nighot, M.; Rawat, M.; Al-Sadi, R.; Castillo, E.F.; Nighot, P.; Ma, T.Y. Lipopolysaccharide-Induced Increase in Intestinal Permeability Is Mediated by TAK-1 Activation of IKK and MLCK/MYLK Gene. Am. J. Pathol. 2019, 189, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.M.G.; de Assis Costa, J.; Alfenas, R.d.C.G. Metabolic Endotoxemia and Diabetes Mellitus: A Systematic Review. Metabolism 2017, 68, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Jayashree, B.; Bibin, Y.S.; Prabhu, D.; Shanthirani, C.S.; Gokulakrishnan, K.; Lakshmi, B.S.; Mohan, V.; Balasubramanyam, M. Increased Circulatory Levels of Lipopolysaccharide (LPS) and Zonulin Signify Novel Biomarkers of Proinflammation in Patients with Type 2 Diabetes. Mol. Cell. Biochem. 2014, 388, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal Mucosal Adherence and Translocation of Commensal Bacteria at the Early Onset of Type 2 Diabetes: Molecular Mechanisms and Probiotic Treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- Snelson, M.; De Pasquale, C.; Ekinci, E.I.; Coughlan, M.T. Gut Microbiome, Prebiotics, Intestinal Permeability and Diabetes Complications. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101507. [Google Scholar] [CrossRef]
- Lim, G.Y.; Tam, W.W.; Lu, Y.; Ho, C.S.; Zhang, M.W.; Ho, R.C. Prevalence of Depression in the Community from 30 Countries between 1994 and 2014. Sci. Rep. 2018, 8, 2861. [Google Scholar] [CrossRef]
- Friedrich, M.J. Depression Is the Leading Cause of Disability around the World. JAMA 2017, 317, 1517. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Millson, B.; Power, G.S. Burden of Treatment Resistant Depression (TRD) in Patients with Major Depressive Disorder in Ontario Using Institute for Clinical Evaluative Sciences (ICES) Databases: Economic Burden and Healthcare Resource Utilization. J. Affect. Disord. 2020, 277, 30–38. [Google Scholar] [CrossRef]
- Arnaud, A.; Benner, J.; Suthoff, E.; Werneburg, B.; Reinhart, M.; Sussman, M.; Kessler, R.C. The Impact of Early Remission on Disease Trajectory and Patient Outcomes in Major Depression Disorder (MDD): A Targeted Literature Review and Microsimulation Modeling Approach Based on the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. J. Affect. Disord. 2023, 325, 264–272. [Google Scholar] [CrossRef]
- Zajkowska, Z.; Walsh, A.; Zonca, V.; Gullett, N.; Pedersen, G.A.; Kieling, C.; Swartz, J.R.; Karmacharya, R.; Fisher, H.L.; Kohrt, B.A.; et al. A Systematic Review of the Association between Biological Markers and Environmental Stress Risk Factors for Adolescent Depression. J. Psychiatr. Res. 2021, 138, 163–175. [Google Scholar] [CrossRef]
- Gonda, X.; Petschner, P.; Eszlari, N.; Baksa, D.; Edes, A.; Antal, P.; Juhasz, G.; Bagdy, G. Genetic Variants in Major Depressive Disorder: From Pathophysiology to Therapy. Pharmacol. Ther. 2019, 194, 22–43. [Google Scholar] [CrossRef] [PubMed]
- Bădescu, S.V.; Tătaru, C.; Kobylinska, L.; Georgescu, E.L.; Zahiu, D.M.; Zăgrean, A.M.; Zăgrean, L. The Association between Diabetes Mellitus and Depression. J. Med. Life 2016, 9, 120–125. [Google Scholar] [PubMed]
- OuYang, H.; Chen, B.; Abdulrahman, A.-M.; Li, L.; Wu, N. Associations between Gestational Diabetes and Anxiety or Depression: A Systematic Review. J. Diabetes Res. 2021, 2021, 9959779. [Google Scholar] [CrossRef]
- Barch, D.M.; Tillman, R.; Kelly, D.; Whalen, D.; Gilbert, K.; Luby, J.L. Hippocampal Volume and Depression among Young Children. Psychiatry Res. Neuroimaging 2019, 288, 21–28. [Google Scholar] [CrossRef]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal Volume Reduction in Major Depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Stockmeier, C.A.; Mahajan, G.J.; Konick, L.C.; Overholser, J.C.; Jurjus, G.J.; Meltzer, H.Y.; Uylings, H.B.M.; Friedman, L.; Rajkowska, G. Cellular Changes in the Postmortem Hippocampus in Major Depression. Biol. Psychiatry 2004, 56, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Czéh, B.; Lucassen, P.J. What Causes the Hippocampal Volume Decrease in Depression? Are Neurogenesis, Glial Changes and Apoptosis Implicated? Eur. Arch. Psychiatry Clin. Neurosci. 2007, 257, 250–260. [Google Scholar] [CrossRef]
- Willard, S.L.; Riddle, D.R.; Forbes, M.E.; Shively, C.A. Cell Number and Neuropil Alterations in Subregions of the Anterior Hippocampus in a Female Monkey Model of Depression. Biol. Psychiatry 2013, 74, 890–897. [Google Scholar] [CrossRef]
- Chen, F.; Bertelsen, A.B.; Holm, I.E.; Nyengaard, J.R.; Rosenberg, R.; Dorph-Petersen, K.-A. Hippocampal Volume and Cell Number in Depression, Schizophrenia, and Suicide Subjects. Brain Res. 2020, 1727, 146546. [Google Scholar] [CrossRef]
- Beauquis, J.; Roig, P.; De Nicola, A.F.; Saravia, F. Neuronal Plasticity and Antidepressants in the Diabetic Brain. Ann. N. Y. Acad. Sci. 2009, 1153, 203–208. [Google Scholar] [CrossRef]
- Czéh, B.; Michaelis, T.; Watanabe, T.; Frahm, J.; de Biurrun, G.; van Kampen, M.; Bartolomucci, A.; Fuchs, E. Stress-Induced Changes in Cerebral Metabolites, Hippocampal Volume, and Cell Proliferation Are Prevented by Antidepressant Treatment with Tianeptine. Proc. Natl. Acad. Sci. USA 2001, 98, 12796–12801. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F. A Hypothesis of Monoamine (5-HT)-Glutamate/GABA Long Neural Circuit: Aiming for Fast-Onset Antidepressant Discovery. Pharmacol. Ther. 2020, 208, 107494. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, F.; Zhai, M.; He, M.; Hu, Y.; Feng, L.; Li, Y.; Yang, J.; Wu, C. Hyperactive Neuronal Autophagy Depletes BDNF and Impairs Adult Hippocampal Neurogenesis in a Corticosterone-Induced Mouse Model of Depression. Theranostics 2023, 13, 1059–1075. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Choe, S.; Woo, H.; Jeong, H.; An, H.-K.; Moon, H.; Ryu, H.Y.; Yeo, B.K.; Lee, Y.W.; Choi, H.; et al. Autophagic Death of Neural Stem Cells Mediates Chronic Stress-Induced Decline of Adult Hippocampal Neurogenesis and Cognitive Deficits. Autophagy 2020, 16, 512–530. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Wu, H.-R.; Zhang, S.-S.; Xiao, H.-L.; Yu, J.; Ma, Y.-Y.; Zhang, Y.-D.; Liu, Q. Catalpol Ameliorates Depressive-like Behaviors in CUMS Mice via Oxidative Stress-Mediated NLRP3 Inflammasome and Neuroinflammation. Transl. Psychiatry 2021, 11, 353. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and Depression: A Review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Herbison, C.E.; Allen, K.; Robinson, M.; Newnham, J.; Pennell, C. The Impact of Life Stress on Adult Depression and Anxiety Is Dependent on Gender and Timing of Exposure. Dev. Psychopathol. 2017, 29, 1443–1454. [Google Scholar] [CrossRef]
- Wulsin, A.C.; Wick-Carlson, D.; Packard, B.A.; Morano, R.; Herman, J.P. Adolescent Chronic Stress Causes Hypothalamo-Pituitary-Adrenocortical Hypo-Responsiveness and Depression-like Behavior in Adult Female Rats. Psychoneuroendocrinology 2016, 65, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Young, E.A.; Haskett, R.F.; Murphy-Weinberg, V.; Watson, S.J.; Akil, H. Loss of Glucocorticoid Fast Feedback in Depression. Arch. Gen. Psychiatry 1991, 48, 693–699. [Google Scholar] [CrossRef]
- Anacker, C.; Zunszain, P.A.; Carvalho, L.A.; Pariante, C.M. The Glucocorticoid Receptor: Pivot of Depression and of Antidepressant Treatment? Psychoneuroendocrinology 2011, 36, 415–425. [Google Scholar] [CrossRef]
- Kahl, K.G.; Schweiger, U.; Pars, K.; Kunikowska, A.; Deuschle, M.; Gutberlet, M.; Lichtinghagen, R.; Bleich, S.; Hüper, K.; Hartung, D. Adrenal Gland Volume, Intra-Abdominal and Pericardial Adipose Tissue in Major Depressive Disorder. Psychoneuroendocrinology 2015, 58, 1–8. [Google Scholar] [CrossRef]
- Druzhkova, T.A.; Yakovlev, A.A.; Rider, F.K.; Zinchuk, M.S.; Guekht, A.B.; Gulyaeva, N.V. Elevated Serum Cortisol Levels in Patients with Focal Epilepsy, Depression, and Comorbid Epilepsy and Depression. Int. J. Mol. Sci. 2022, 23, 10414. [Google Scholar] [CrossRef] [PubMed]
- Sorgdrager, F.J.H.; Doornbos, B.; Penninx, B.W.J.H.; de Jonge, P.; Kema, I.P. The Association between the Hypothalamic Pituitary Adrenal Axis and Tryptophan Metabolism in Persons with Recurrent Major Depressive Disorder and Healthy Controls. J. Affect. Disord. 2017, 222, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Liu, L.; Sheng, C.; Cheng, Z.; Cui, L.; Li, M.; Zhao, Y.; Shi, T.; Yau, T.O.; Li, F.; et al. Increased Serum Levels of Cortisol and Inflammatory Cytokines in People With Depression. J. Nerv. Ment. Dis. 2019, 207, 271–276. [Google Scholar] [CrossRef]
- Lee, E.-H.; Park, J.-Y.; Kwon, H.-J.; Han, P.-L. Repeated Exposure with Short-Term Behavioral Stress Resolves Pre-Existing Stress-Induced Depressive-like Behavior in Mice. Nat. Commun. 2021, 12, 6682. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Ju, J.Y.; Kim, J.H.; Kim, T.-Y.; Yang, B.-H.; Lee, Y.-S.; Son, H. Dexamethasone Inhibits Proliferation of Adult Hippocampal Neurogenesis in Vivo and in Vitro. Brain Res. 2004, 1027, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. The Possibility of Neurotoxicity in the Hippocampus in Major Depression: A Primer on Neuron Death. Biol. Psychiatry 2000, 48, 755–765. [Google Scholar] [CrossRef]
- David, D.J.; Samuels, B.A.; Rainer, Q.; Wang, J.-W.; Marsteller, D.; Mendez, I.; Drew, M.; Craig, D.A.; Guiard, B.P.; Guilloux, J.-P.; et al. Neurogenesis-Dependent and -Independent Effects of Fluoxetine in an Animal Model of Anxiety/Depression. Neuron 2009, 62, 479–493. [Google Scholar] [CrossRef]
- Tanapat, P.; Galea, L.A.; Gould, E. Stress Inhibits the Proliferation of Granule Cell Precursors in the Developing Dentate Gyrus. Int. J. Dev. Neurosci. 1998, 16, 235–239. [Google Scholar] [CrossRef]
- Boulle, F.; Velthuis, H.; Koedam, K.; Steinbusch, H.W.; van den Hove, D.L.A.; Kenis, G.; Gabriel, C.; Mocaer, E.; Franc, B.; Rognan, D.; et al. Behavioral and Neurochemical Characterization of TrkB-Dependent Mechanisms of Agomelatine in Glucocorticoid Receptor-Impaired Mice. Eur. Neuropsychopharmacol. 2016, 26, 65–77. [Google Scholar] [CrossRef]
- Ogłodek, E.A. Changes in the Serum Concentration Levels of Serotonin, Tryptophan and Cortisol among Stress-Resilient and Stress-Susceptible Individuals after Experiencing Traumatic Stress. Int. J. Environ. Res. Public Health 2022, 19, 16517. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, A.T.D.; Juruena, M.F.; Coimbra, B.M.; Mello, M.F.; Mello, A.F. Increased Immuno-Inflammatory Mediators in Women with Post-Traumatic Stress Disorder after Sexual Assault: 1-Year Follow-Up. J. Psychiatr. Res. 2022, 155, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, L.P.; Mkrtchyan, G.M.; Sukiasyan, S.H.; Boyajyan, A.S. Classic and Alternative Complement Cascades in Post-Traumatic Stress Disorder. Bull. Exp. Biol. Med. 2009, 148, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Xu, H.; Cao, C.; Liu, P.; Luo, S.; Duan, Q.; Ellenbroek, B.; Zhang, X. Characteristics of Pro- and Anti-Inflammatory Cytokines Alteration in PTSD Patients Exposed to a Deadly Earthquake. J. Affect. Disord. 2019, 248, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Küffer, A.; Straus, L.D.; Prather, A.A.; Inslicht, S.S.; Richards, A.; Shigenaga, J.K.; Madden, E.; Metzler, T.J.; Neylan, T.C.; O’Donovan, A. Altered Overnight Levels of Pro-Inflammatory Cytokines in Men and Women with Posttraumatic Stress Disorder. Psychoneuroendocrinology 2019, 102, 114–120. [Google Scholar] [CrossRef]
- Koirala, R.; Aass, H.C.D.; Søegaard, E.G.I.; Dhakal, H.P.; Ojha, S.P.; Hauff, E.; Thapa, S.B. Association of Pro-Inflammatory Cytokines with Trauma and Post-Traumatic Stress Disorder Visiting a Tertiary Care Hospital in Kathmandu. PLoS ONE 2023, 18, e0281125. [Google Scholar] [CrossRef]
- Wang, Z.; Mandel, H.; Levingston, C.A.; Young, M.R.I. An Exploratory Approach Demonstrating Immune Skewing and a Loss of Coordination among Cytokines in Plasma and Saliva of Veterans with Combat-Related PTSD. Hum. Immunol. 2016, 77, 652–657. [Google Scholar] [CrossRef]
- Strawbridge, R.; Hodsoll, J.; Powell, T.R.; Hotopf, M.; Hatch, S.L.; Breen, G.; Cleare, A.J. Inflammatory Profiles of Severe Treatment-Resistant Depression. J. Affect. Disord. 2019, 246, 42–51. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; De Jongh, R.; Kenis, G.; Vandoolaeghe, E.; Neels, H. Increased Serum IL-6 and IL-1 Receptor Antagonist Concentrations in Major Depression and Treatment Resistant Depression. Cytokine 1997, 9, 853–858. [Google Scholar] [CrossRef]
- Murata, S.; Murphy, M.; Khanna, R.; Hoppensteadt, D.; Fareed, J.; Halaris, A. Elevated Salivary Cortisol Predicts Response to Adjunctive Immune Modulation in Treatment-Resistant Bipolar Depression. J. Affect. Disord. Rep. 2021, 4, 100117. [Google Scholar] [CrossRef]
- Voineskos, D.; Daskalakis, Z.J.; Blumberger, D.M. Management of Treatment-Resistant Depression: Challenges and Strategies. Neuropsychiatr. Dis. Treat. 2020, 16, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Thase, M.E.; Pillinger, T. Treatment Resistance in Psychiatry: State of the Art and New Directions. Mol. Psychiatry 2022, 27, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Sikaroodi, M.; Fagan, A.; Heuman, D.; Gilles, H.; Gavis, E.A.; Fuchs, M.; Gonzalez-Maeso, J.; Nizam, S.; Gillevet, P.M.; et al. Posttraumatic Stress Disorder Is Associated with Altered Gut Microbiota That Modulates Cognitive Performance in Veterans with Cirrhosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2019, 317, G661–G669. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Singh, R.; Ghoshal, U.C. Enterochromaffin Cells–Gut Microbiota Crosstalk: Underpinning the Symptoms, Pathogenesis, and Pharmacotherapy in Disorders of Gut-Brain Interaction. J. Neurogastroenterol. Motil. 2022, 28, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sun, T.; Wu, F.; Li, F.; Liu, Y.; Li, W.; Dai, N.; Tan, L.; Li, T.; Song, Y. Correlation of Gut Microbiota and Neurotransmitters in a Rat Model of Post-Traumatic Stress Disorder. J. Tradit. Chin. Med. Sci. 2020, 7, 375–385. [Google Scholar] [CrossRef]
- Lee, B.; Choi, G.M.; Sur, B. Antidepressant-Like Effects of Hesperidin in Animal Model of Post-Traumatic Stress Disorder. Chin. J. Integr. Med. 2021, 27, 39–46. [Google Scholar] [CrossRef]
- Satti, S.; Palepu, M.S.K.; Singh, A.A.; Jaiswal, Y.; Dash, S.P.; Gajula, S.N.R.; Chaganti, S.; Samanthula, G.; Sonti, R.; Dandekar, M.P. Anxiolytic- and Antidepressant-like Effects of Bacillus Coagulans Unique IS-2 Mediate via Reshaping of Microbiome Gut-Brain Axis in Rats. Neurochem. Int. 2023, 163, 105483. [Google Scholar] [CrossRef]
- Gould, E.; Cameron, H.A.; Daniels, D.C.; Woolley, C.S.; McEwen, B.S. Adrenal Hormones Suppress Cell Division in the Adult Rat Dentate Gyrus. J. Neurosci. 1992, 12, 3642–3650. [Google Scholar] [CrossRef]
- Mayer, J.L.; Klumpers, L.; Maslam, S.; de Kloet, E.R.; Joëls, M.; Lucassen, P.J. Brief Treatment with the Glucocorticoid Receptor Antagonist Mifepristone Normalises the Corticosterone-Induced Reduction of Adult Hippocampal Neurogenesis. J. Neuroendocrinol. 2006, 18, 629–631. [Google Scholar] [CrossRef]
- Oomen, C.A.; Mayer, J.L.; de Kloet, E.R.; Joëls, M.; Lucassen, P.J. Brief Treatment with the Glucocorticoid Receptor Antagonist Mifepristone Normalizes the Reduction in Neurogenesis after Chronic Stress. Eur. J. Neurosci. 2007, 26, 3395–3401. [Google Scholar] [CrossRef]
- Young, A.H.; Gallagher, P.; Watson, S.; Del-Estal, D.; Owen, B.M.; Ferrier, I.N. Improvements in Neurocognitive Function and Mood Following Adjunctive Treatment with Mifepristone (RU-486) in Bipolar Disorder. Neuropsychopharmacology 2004, 29, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- DeBattista, C.; Belanoff, J.; Glass, S.; Khan, A.; Horne, R.L.; Blasey, C.; Carpenter, L.L.; Alva, G. Mifepristone versus Placebo in the Treatment of Psychosis in Patients with Psychotic Major Depression. Biol. Psychiatry 2006, 60, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Flores, B.H.; Kenna, H.; Keller, J.; Solvason, H.B.; Schatzberg, A.F. Clinical and Biological Effects of Mifepristone Treatment for Psychotic Depression. Neuropsychopharmacology 2006, 31, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Ding, B.; Feng, C.; Yin, S.; Zhang, T.; Qi, X.; Lv, H.; Guo, X.; Dong, K.; Zhu, Y.; et al. Prevotella and Klebsiella Proportions in Fecal Microbial Communities Are Potential Characteristic Parameters for Patients with Major Depressive Disorder. J. Affect. Disord. 2017, 207, 300–304. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Wang, X.; Wang, Z.; Zhang, J.; Jiang, R.; Wang, X.; Wang, K.; Liu, Z.; Xia, Z.; et al. Similar Fecal Microbiota Signatures in Patients With Diarrhea-Predominant Irritable Bowel Syndrome and Patients With Depression. Clin. Gastroenterol. Hepatol. 2016, 14, 1602–1611.e5. [Google Scholar] [CrossRef]
- Chen, J.-J.; Zheng, P.; Liu, Y.-Y.; Zhong, X.-G.; Wang, H.-Y.; Guo, Y.-J.; Xie, P. Sex Differences in Gut Microbiota in Patients with Major Depressive Disorder. Neuropsychiatr. Dis. Treat. 2018, 14, 647–655. [Google Scholar] [CrossRef]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered Fecal Microbiota Composition in Patients with Major Depressive Disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef]
- Mason, B.L.; Li, Q.; Minhajuddin, A.; Czysz, A.H.; Coughlin, L.A.; Hussain, S.K.; Koh, A.Y.; Trivedi, M.H. Reduced Anti-Inflammatory Gut Microbiota Are Associated with Depression and Anhedonia. J. Affect. Disord. 2020, 266, 394–401. [Google Scholar] [CrossRef]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the Bacterial Butyrate Synthesis Pathways by Analyzing (Meta)Genomic Data. mBio 2014, 5, e00889. [Google Scholar] [CrossRef]
- Leinonen, L.; Joutsiniemi, S.L. Auditory Evoked Potentials and Magnetic Fields in Patients with Lesions of the Auditory Cortex. Acta Neurol. Scand. 1989, 79, 316–325. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant Effects of Ketamine in Depressed Patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Yang, C.; Qu, Y.; Fujita, Y.; Ren, Q.; Ma, M.; Dong, C.; Hashimoto, K. Possible Role of the Gut Microbiota-Brain Axis in the Antidepressant Effects of (R)-Ketamine in a Social Defeat Stress Model. Transl. Psychiatry 2017, 7, 1294. [Google Scholar] [CrossRef]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut Microbiome Remodeling Induces Depressive-like Behaviors through a Pathway Mediated by the Host’s Metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, F.; Hu, X.; Yang, C.; Xu, H.; Yao, Y.; Liu, J. Clostridium Butyricum Attenuates Chronic Unpredictable Mild Stress-Induced Depressive-Like Behavior in Mice via the Gut-Brain Axis. J. Agric. Food Chem. 2018, 66, 8415–8421. [Google Scholar] [CrossRef]
- Miyaoka, T.; Kanayama, M.; Wake, R.; Hashioka, S.; Hayashida, M.; Nagahama, M.; Okazaki, S.; Yamashita, S.; Miura, S.; Miki, H.; et al. Clostridium Butyricum MIYAIRI 588 as Adjunctive Therapy for Treatment-Resistant Major Depressive Disorder: A Prospective Open-Label Trial. Clin. Neuropharmacol. 2018, 41, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Thummasorn, S.; Apichai, S.; Chupradit, S.; Sirisattayawong, P.; Chaiwong, P.; Sriwichaiin, S.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. T2DM Patients with Depression Have Higher Levels of Hyperglycemia and Cognitive Decline than T2DM Patients. PLoS ONE 2022, 17, e0273327. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.M.; Perlman, G.; Kim, N.; Wu, C.-Y.; Daher, V.; Zhou, A.; Mathers, E.H.; Anita, N.Z.; Lanctôt, K.L.; Herrmann, N.; et al. Depression in Type 2 Diabetes: A Systematic Review and Meta-Analysis of Blood Inflammatory Markers. Psychoneuroendocrinology 2021, 134, 105448. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, D.; Dai, Z.; Li, X. Association Between Systemic Immune-Inflammation Index and Diabetic Depression. Clin. Interv. Aging 2021, 16, 97–105. [Google Scholar] [CrossRef]
- Kale, M.; Nimje, N.; Aglawe, M.M.; Umekar, M.; Taksande, B.; Kotagale, N. Agmatine Modulates Anxiety and Depression-like Behaviour in Diabetic Insulin-Resistant Rats. Brain Res. 2020, 1747, 147045. [Google Scholar] [CrossRef]
- Gragnoli, C. Depression and Type 2 Diabetes: Cortisol Pathway Implication and Investigational Needs. J. Cell. Physiol. 2012, 227, 2318–2322. [Google Scholar] [CrossRef]
- Kokkinopoulou, I.; Diakoumi, A.; Moutsatsou, P. Glucocorticoid Receptor Signaling in Diabetes. Int. J. Mol. Sci. 2021, 22, 11173. [Google Scholar] [CrossRef]
- Marissal-Arvy, N.; Campas, M.-N.; Semont, A.; Ducroix-Crepy, C.; Beauvieux, M.-C.; Brossaud, J.; Corcuff, J.-B.; Helbling, J.-C.; Vancassel, S.; Bouzier-Sore, A.-K.; et al. Insulin Treatment Partially Prevents Cognitive and Hippocampal Alterations as Well as Glucocorticoid Dysregulation in Early-Onset Insulin-Deficient Diabetic Rats. Psychoneuroendocrinology 2018, 93, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Seong, J.K.; Yi, S.S. Sequential Alterations of Glucocorticoid Receptors in the Hippocampus of STZ-Treated Type 1 Diabetic Rats. J. Vet. Sci. 2014, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Hao, S.; Erion, J.R.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid Sensitization of Microglia in a Genetic Mouse Model of Obesity and Diabetes. J. Neuroimmunol. 2014, 269, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.-F.; Wang, W.; Niu, L.; Kou, Z.-Z.; Zhu, C.; Wang, W.; Zhao, X.-H.; Luo, D.-S.; Zhang, T.; Zhang, F.-X.; et al. RU486 (Mifepristone) Ameliorates Cognitive Dysfunction and Reverses the down-Regulation of Astrocytic N-Myc Downstream-Regulated Gene 2 in Streptozotocin-Induced Type-1 Diabetic Rats. Neuroscience 2011, 190, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia Drives Intestinal Barrier Dysfunction and Risk for Enteric Infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef]
- Rom, S.; Heldt, N.A.; Gajghate, S.; Seliga, A.; Reichenbach, N.L.; Persidsky, Y. Author Correction: Hyperglycemia and Advanced Glycation End Products Disrupt BBB and Promote Occludin and Claudin-5 Protein Secretion on Extracellular Microvesicles. Sci. Rep. 2020, 10, 18828. [Google Scholar] [CrossRef]
- Martín, R.; Miquel, S.; Chain, F.; Natividad, J.M.; Jury, J.; Lu, J.; Sokol, H.; Theodorou, V.; Bercik, P.; Verdu, E.F.; et al. Faecalibacterium Prausnitzii Prevents Physiological Damages in a Chronic Low-Grade Inflammation Murine Model. BMC Microbiol. 2015, 15, 67. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Hao, S.; Dey, A.; Yu, X.; Baban, B. Blood-Brain Barrier Breakdown Promotes Macrophage Infiltration and Cognitive Impairment in Leptin Receptor-Deficient Mice. J. Cereb. Blood Flow Metab. 2016, 36, 2108–2121. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33–39. [Google Scholar] [CrossRef]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Oliva, A.M.; De Pablos, R.M.; Villarán, R.F.; Argüelles, S.; Venero, J.L.; Machado, A.; Cano, J. Stress Is Critical for LPS-Induced Activation of Microglia and Damage in the Rat Hippocampus. Neurobiol. Aging 2011, 32, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, C.D.; Lepsch, L.B.; Kawamoto, E.M.; Malta, M.B.; Lima, L.D.S.; Werneck Avellar, M.C.; Sapolsky, R.M.; Scavone, C. Chronic Unpredictable Stress Exacerbates Lipopolysaccharide-Induced Activation of Nuclear Factor-ΚB in the Frontal Cortex and Hippocampus via Glucocorticoid Secretion. J. Neurosci. 2006, 26, 3813–3820. [Google Scholar] [CrossRef]
- Frank, M.G.; Hershman, S.A.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Chronic Exposure to Exogenous Glucocorticoids Primes Microglia to Pro-Inflammatory Stimuli and Induces NLRP3 MRNA in the Hippocampus. Psychoneuroendocrinology 2014, 40, 191–200. [Google Scholar] [CrossRef]
- Shukla, P.K.; Meena, A.S.; Pierre, J.F.; Rao, R. Central Role of Intestinal Epithelial Glucocorticoid Receptor in Alcohol- and Corticosterone-Induced Gut Permeability and Systemic Response. FASEB J. 2022, 36, e22061. [Google Scholar] [CrossRef]
- Guo, X.; Shi, Y.; Du, P.; Wang, J.; Han, Y.; Sun, B.; Feng, J. HMGB1/TLR4 Promotes Apoptosis and Reduces Autophagy of Hippocampal Neurons in Diabetes Combined with OSA. Life Sci. 2019, 239, 117020. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Mi, J.; Jiang, Q.-M.; Xu, J.-M.; Tang, Y.-Y.; Tian, G.; Wang, B. Metformin May Produce Antidepressant Effects through Improvement of Cognitive Function among Depressed Patients with Diabetes Mellitus. Clin. Exp. Pharmacol. Physiol. 2014, 41, 650–656. [Google Scholar] [CrossRef]
- Abdallah, M.S.; Mosalam, E.M.; Zidan, A.-A.A.; Elattar, K.S.; Zaki, S.A.; Ramadan, A.N.; Ebeid, A.M. RETRACTED ARTICLE: The Antidiabetic Metformin as an Adjunct to Antidepressants in Patients with Major Depressive Disorder: A Proof-of-Concept, Randomized, Double-Blind, Placebo-Controlled Trial. Neurotherapeutics 2020, 17, 1897–1906. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin Alters the Gut Microbiome of Individuals with Treatment-Naive Type 2 Diabetes, Contributing to the Therapeutic Effects of the Drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Shin, A.C.; Balasubramanian, P.; Suryadevara, P.; Zyskowski, J.; Herdt, T.H.; MohanKumar, S.M.J.; MohanKumar, P.S. Metformin Effectively Restores the HPA Axis Function in Diet-Induced Obese Rats. Int. J. Obes. 2021, 45, 383–395. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, T.; Guo, A.-M.; Chen, W.-B.; Lin, D.; Liu, Z.-Y.; Fei, E.-K. Metformin Ameliorates Lipopolysaccharide-Induced Depressive-Like Behaviors and Abnormal Glutamatergic Transmission. Biology 2020, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.-C.; Tsai, S.-F.; Sie, S.-R.; Kuo, Y.-M. High Glucose Enhances Lipopolysaccharide-Induced Inflammation in Cultured BV2 Microglial Cell Line. Immun. Inflamm. Dis. 2022, 10, e610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, H.; Zhang, S.; Lu, S.; Sun, J.; Qian, Y. Enhancement of LPS-Induced Microglial Inflammation Response via TLR4 Under High Glucose Conditions. Cell. Physiol. Biochem. 2015, 35, 1571–1581. [Google Scholar] [CrossRef]
- Erickson, M.A.; Shulyatnikova, T.; Banks, W.A.; Hayden, M.R. Ultrastructural Remodeling of the Blood-Brain Barrier and Neurovascular Unit by Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2023, 24, 1640. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female Db/Db Mouse. Brain Sci. 2019, 9, 57. [Google Scholar] [CrossRef]
- Hayden, M.R.; Banks, W.A. Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 5427. [Google Scholar] [CrossRef]
- Lin, K.-I.; Johnson, D.R.; Freund, G.G. LPS-Dependent Suppression of Social Exploration Is Augmented in Type 1 Diabetic Mice. Brain Behav. Immun. 2007, 21, 775–782. [Google Scholar] [CrossRef]
- Fischer, S.; Strawbridge, R.; Vives, A.H.; Cleare, A.J. Cortisol as a Predictor of Psychological Therapy Response in Depressive Disorders: Systematic Review and Meta-Analysis. Br. J. Psychiatry 2017, 210, 105–109. [Google Scholar] [CrossRef]
- Haroon, E.; Daguanno, A.W.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.M.; Wommack, E.C.; Felger, J.C.; Miller, A.H. Antidepressant Treatment Resistance Is Associated with Increased Inflammatory Markers in Patients with Major Depressive Disorder. Psychoneuroendocrinology 2018, 95, 43–49. [Google Scholar] [CrossRef]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A Randomized Trial of an N-Methyl-D-Aspartate Antagonist in Treatment-Resistant Major Depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef]
- Siopi, E.; Chevalier, G.; Katsimpardi, L.; Saha, S.; Bigot, M.; Moigneu, C.; Eberl, G.; Lledo, P.-M. Changes in Gut Microbiota by Chronic Stress Impair the Efficacy of Fluoxetine. Cell Rep. 2020, 30, 3682–3690.e6. [Google Scholar] [CrossRef]
- Fontana, A.; Manchia, M.; Panebianco, C.; Paribello, P.; Arzedi, C.; Cossu, E.; Garzilli, M.; Montis, M.A.; Mura, A.; Pisanu, C.; et al. Exploring the Role of Gut Microbiota in Major Depressive Disorder and in Treatment Resistance to Antidepressants. Biomedicines 2020, 8, 311. [Google Scholar] [CrossRef]
- Juruena, M.F.; Cleare, A.J.; Papadopoulos, A.S.; Poon, L.; Lightman, S.; Pariante, C.M. The Prednisolone Suppression Test in Depression: Dose-Response and Changes with Antidepressant Treatment. Psychoneuroendocrinology 2010, 35, 1486–1491. [Google Scholar] [CrossRef]
- Juruena, M.F.; Pariante, C.M.; Papadopoulos, A.S.; Poon, L.; Lightman, S.; Cleare, A.J. The Role of Mineralocorticoid Receptor Function in Treatment-Resistant Depression. J. Psychopharmacol. 2013, 27, 1169–1179. [Google Scholar] [CrossRef]
- Maes, M.; Kubera, M.; Leunis, J.-C.; Berk, M.; Geffard, M.; Bosmans, E. In Depression, Bacterial Translocation May Drive Inflammatory Responses, Oxidative and Nitrosative Stress (O&NS), and Autoimmune Responses Directed against O&NS-Damaged Neoepitopes. Acta Psychiatr. Scand. 2013, 127, 344–354. [Google Scholar] [CrossRef]
- Kéri, S.; Szabó, C.; Kelemen, O. Expression of Toll-Like Receptors in Peripheral Blood Mononuclear Cells and Response to Cognitive-Behavioral Therapy in Major Depressive Disorder. Brain Behav. Immun. 2014, 40, 235–243. [Google Scholar] [CrossRef]
- Kelly, J.R.; Borre, Y.; O’ Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the Blues: Depression-Associated Gut Microbiota Induces Neurobehavioural Changes in the Rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mázala-de-Oliveira, T.; Silva, B.T.; Campello-Costa, P.; Carvalho, V.F. The Role of the Adrenal–Gut–Brain Axis on Comorbid Depressive Disorder Development in Diabetes. Biomolecules 2023, 13, 1504. https://doi.org/10.3390/biom13101504
Mázala-de-Oliveira T, Silva BT, Campello-Costa P, Carvalho VF. The Role of the Adrenal–Gut–Brain Axis on Comorbid Depressive Disorder Development in Diabetes. Biomolecules. 2023; 13(10):1504. https://doi.org/10.3390/biom13101504
Chicago/Turabian StyleMázala-de-Oliveira, Thalita, Bruna Teixeira Silva, Paula Campello-Costa, and Vinicius Frias Carvalho. 2023. "The Role of the Adrenal–Gut–Brain Axis on Comorbid Depressive Disorder Development in Diabetes" Biomolecules 13, no. 10: 1504. https://doi.org/10.3390/biom13101504