
Enabling mRNA Therapeutics: Current Landscape and Challenges in Manufacturing


Abstract
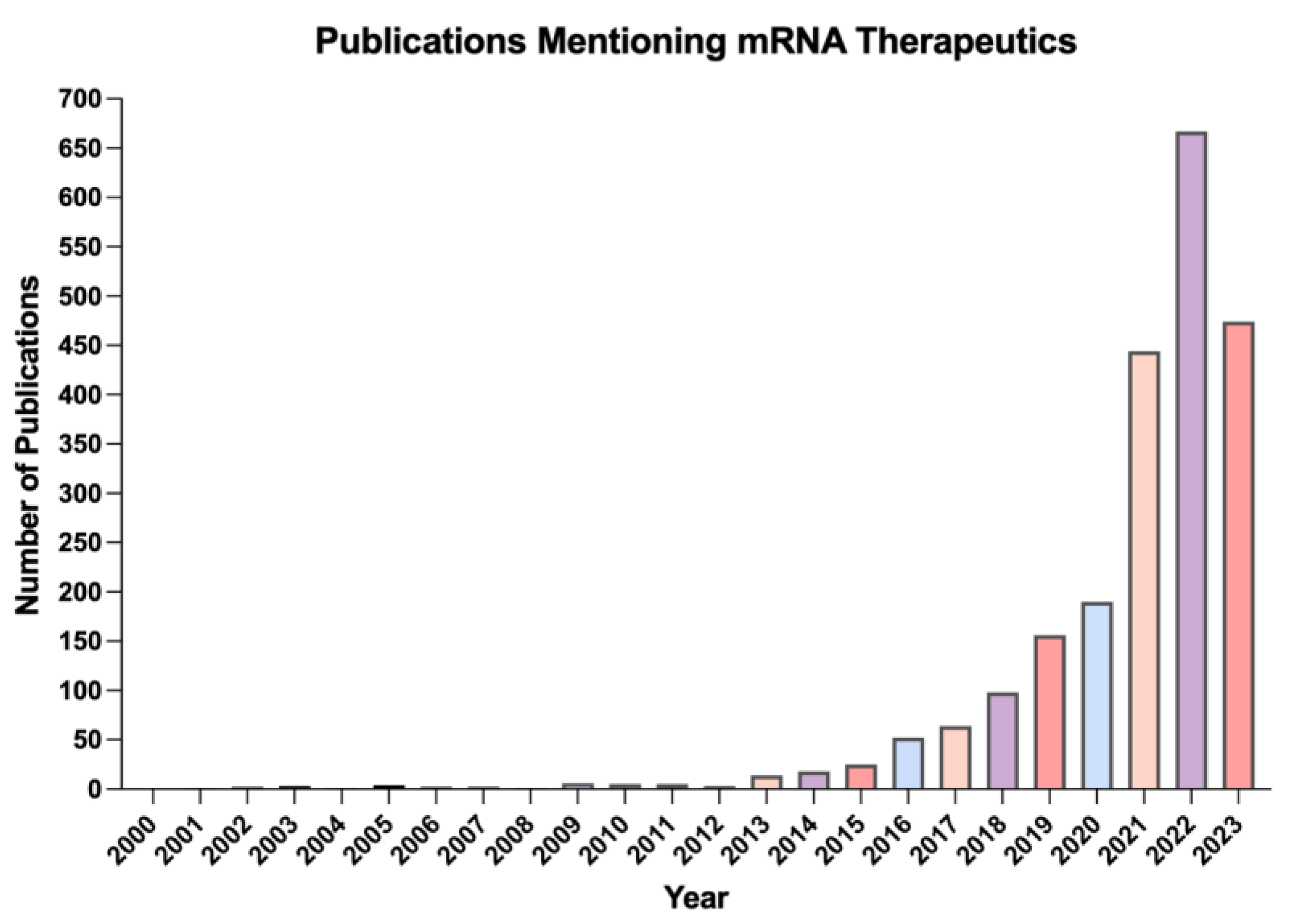
:1. Introduction
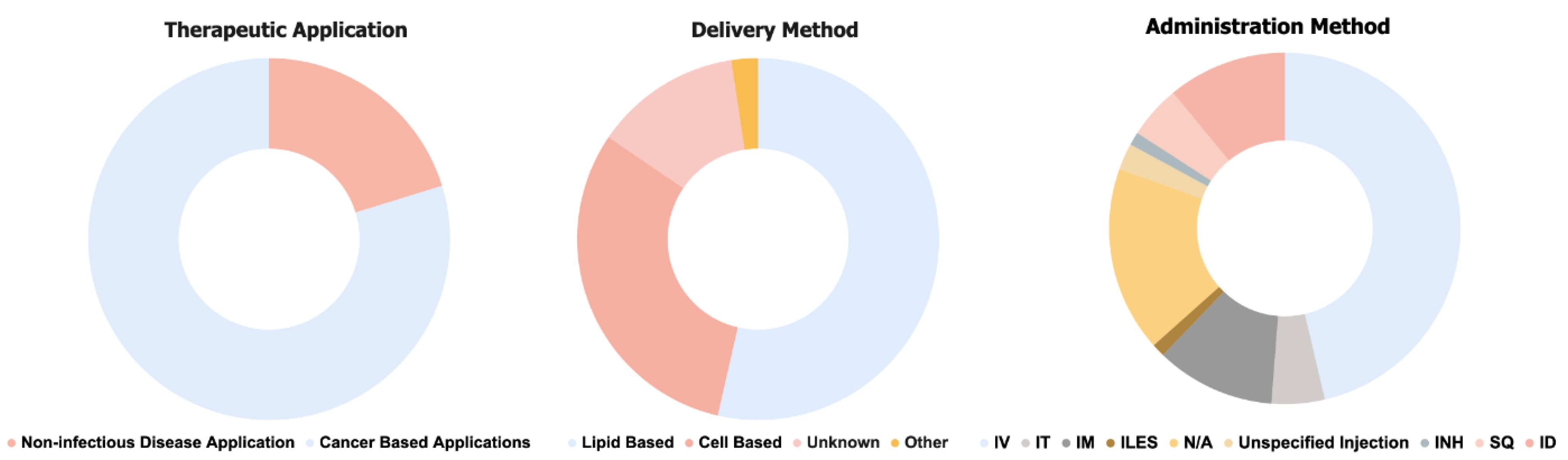
2. Current Therapeutic Applications of mRNA
Ongoing Clinical Trials
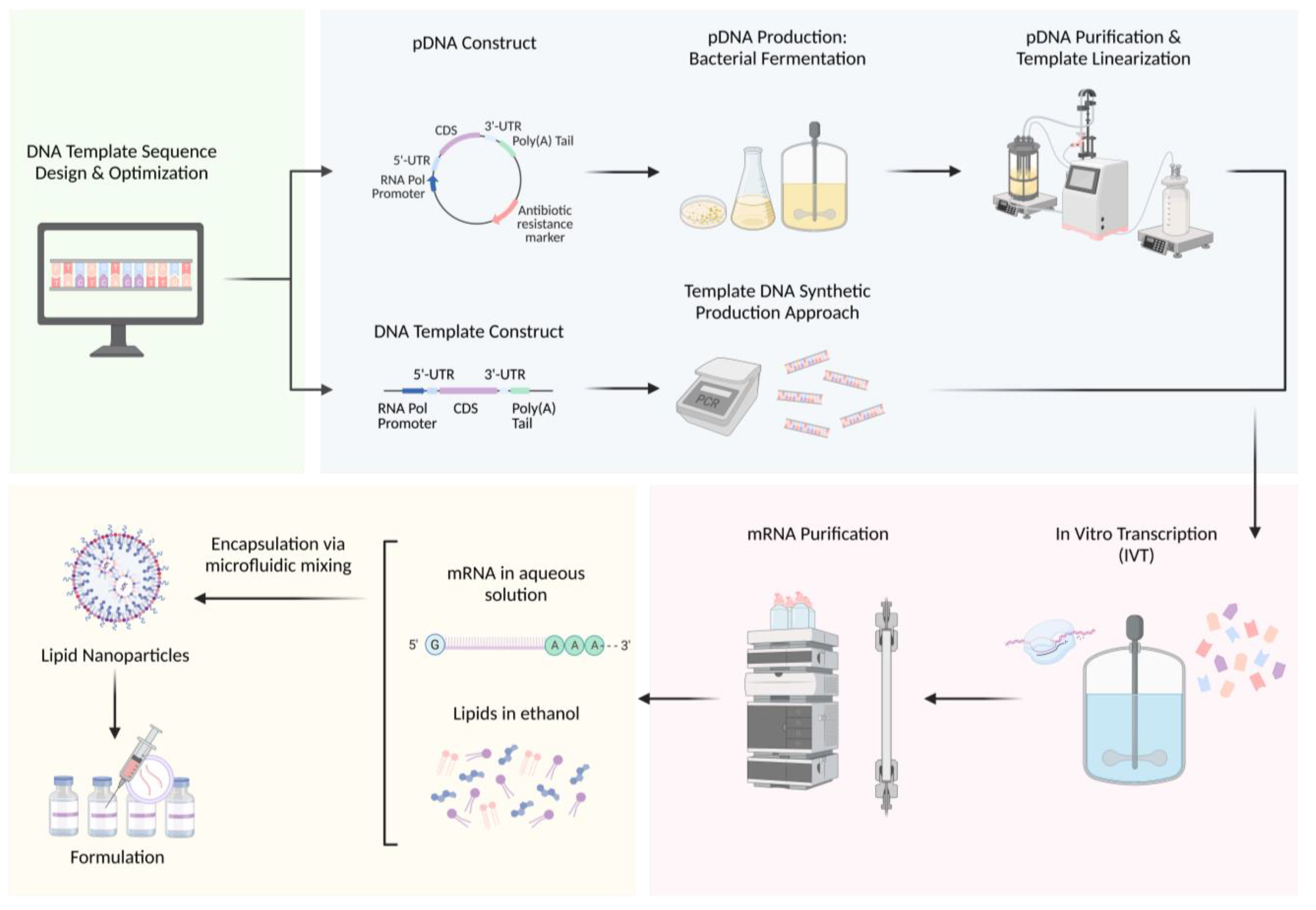
3. Manufacturing Process
3.1. Upstream Process: DNA Template Sequence Design
3.2. Upstream Process: DNA Template Production
3.2.1. Bacterial Fermentation Approach
3.2.2. Synthetic DNA Approach
3.3. Upstream Process: DNA Template Purification
3.4. Upstream Process: mRNA Synthesis
3.4.1. Enzymatic Synthesis
3.4.2. Towards Automated Production of mRNA
3.4.3. Solid-Phase Synthesis
3.5. Downstream Process: mRNA Purification
3.6. Downstream Process: mRNA Delivery
3.6.1. Microfluidic Manufacturing of mRNA-LNPs
3.6.2. mRNA-LNP Formulation
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Rohner, E.; Yang, R.; Foo, K.S.; Goedel, A.; Chien, K.R. Unlocking the promise of mRNA therapeutics. Nat. Biotechnol. 2022, 40, 1586–1600. [Google Scholar] [CrossRef]
- Jain, S.; Venkataraman, A.; Wechsler, M.E.; Peppas, N.A. Messenger RNA-based vaccines: Past, present, and future directions in the context of the COVID-19 pandemic. Adv. Drug Deliv. Rev. 2021, 179, 114000. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Rosa, S.S.; Prazeres, D.M.; Azevedo, A.M.; Marques, M.P. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- Dammes, N.; Peer, D. Paving the Road for RNA Therapeutics. Trends Pharmacol. Sci. 2020, 41, 755–775. [Google Scholar] [CrossRef]
- Geall, A.J.; Ulmer, J.B. Introduction to RNA-based vaccines and therapeutics. Expert Rev. Vaccines 2015, 14, 151–152. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef]
- Moço, P.D.; Xu, X.; Silva, C.A.T.; Kamen, A.A. Production of adeno-associated viral vector serotype 6 by triple transfection of suspension HEK293 cells at higher cell densities. Biotechnol. J. 2023, 18, 2300051. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S. Messenger RNA: The inexpensive biopharmaceutical. J. Multidiscip. Eng. Sci. Technol. 2017, 4, 6937–6941. [Google Scholar]
- Cui, T.; Fakhfakh, K.; Turney, H.; Güler-Gane, G.; Toloczko, A.; Hulley, M.; Turner, R. Comprehensive studies on building a scalable downstream process for mRNAs to enable mRNA therapeutics. Biotechnol. Prog. 2023, 39, e3301. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Tam, Y.Y.C.; Cullis, P.R. Lipid nanoparticles for short interfering RNA delivery. Adv. Genet. 2014, 88, 71–110. [Google Scholar] [PubMed]
- American Society of Cell and Gene Therapy. Gene, Cell, & RNA Therapy Landscape; Q1 2023 Quarterly Data Report; American Society of Cell and Gene Therapy: Waukesha, WI, USA, 2023. [Google Scholar]
- Vervaeke, P.; Borgos, S.; Sanders, N.; Combes, F. Regulatory guidelines and preclinical tools to study the biodistribution of RNA therapeutics. Adv. Drug Deliv. Rev. 2022, 184, 114236. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.; Ip, S.; Bathula, N.V.; Popova, P.; Soriano, S.K.; Ly, H.H.; Eryilmaz, B.; Nguyen Huu, V.A.; Broadhead, R.; Rabel, M. Current status and future perspectives on MRNA drug manufacturing. Mol. Pharm. 2022, 19, 1047–1058. [Google Scholar] [CrossRef]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, production scales and time required for producing RNA vaccines for the global pandemic demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef]
- Michel, T.; Wendel, H.-P.; Krajewski, S. Next-generation therapeutics: mRNA as a novel therapeutic option for single-gene disorders. Mod. Tools Genet. Eng. 2016, 3, 20. [Google Scholar]
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950. [Google Scholar] [CrossRef]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.-S.; Theisen, M.; Hong, S.-J.; Zhou, J.; Rajendran, R. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef]
- Magadum, A.; Kaur, K.; Zangi, L. mRNA-based protein replacement therapy for the heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Zangi, L.; Lui, K.O.; Von Gise, A.; Ma, Q.; Ebina, W.; Ptaszek, L.M.; Später, D.; Xu, H.; Tabebordbar, M.; Gorbatov, R. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013, 31, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Men, K.; Zhang, Y.; Zhang, R.; Yang, L.; Duan, X. Local and systemic delivery of mRNA encoding survivin-T34A by lipoplex for efficient colon cancer gene therapy. Int. J. Nanomed. 2019, 14, 2733–2751. [Google Scholar] [CrossRef] [PubMed]
- Martini, P.G.; Guey, L.T. A new era for rare genetic diseases: Messenger RNA therapy. Hum. Gene Ther. 2019, 30, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Córdoba, K.M.; Jericó, D.; Sampedro, A.; Jiang, L.; Iraburu, M.J.; Martini, P.G.; Berraondo, P.; Avila, M.A.; Fontanellas, A. Messenger RNA as a personalized therapy: The moment of truth for rare metabolic diseases. Int. Rev. Cell Mol. Biol. 2022, 372, 55–96. [Google Scholar] [PubMed]
- Kulkarni, J.A.; Cullis, P.R.; Van Der Meel, R. Lipid nanoparticles enabling gene therapies: From concepts to clinical utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Zhang, M.; Kiss, D.L.; Cooke, J.P. Hospital-based RNA Therapeutics. In Messenger RNA Therapeutics; Springer: Cham, Switzerland, 2022; pp. 73–92. [Google Scholar]
- Burris, H.A.; Patel, M.R.; Cho, D.C.; Clarke, J.M.; Gutierrez, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 2019, 37, 2523. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The limitless future of RNA therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Zhang, Y.; Yin, H. Genome editing with mRNA encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef]
- Foster, J.B.; Barrett, D.M.; Karikó, K. The emerging role of in vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol. Ther. 2019, 27, 747–756. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Intellia Therapeutics. Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of NTLA-2001 in Patients With Hereditary Transthyretin Amyloidosis with Polyneuropathy (ATTRv-PN) and Patients with Transthyretin Amyloidosis-Related Cardiomyopathy (ATTR-CM); Intellia Therapeutics: Cambridge, MA, USA, 2020. [Google Scholar]
- Intellia Therapeutics. NTLA-2002 in Adults With Hereditary Angioedema (HAE); Intellia Therapeutics: Cambridge, MA, USA, 2021. [Google Scholar]
- ModernaTX, Inc. A Long-Term Extension Study to Evaluate the Safety and Clinical Activity of mRNA-3927; ModernaTX, Inc.: Cambridge, MA, USA, 2021. [Google Scholar]
- ModernaTX, Inc. A Study of mRNA-3745 in Adult and Pediatric Participants with Glycogen Storage Disease Type 1a (GSD1a); ModernaTX, Inc.: Cambridge, MA, USA, 2022. [Google Scholar]
- ModernaTX, Inc. Open-Label Study of mRNA-3927 in Participants With Propionic Acidemia; ModernaTX, Inc.: Cambridge, MA, USA, 2021. [Google Scholar]
- ModernaTX, Inc.; Merck Sharp & Dohme LLC. An Efficacy Study of Adjuvant Treatment with the Personalized Cancer Vaccine mRNA-4157 and Pembrolizumab in Participants with High-Risk Melanoma (KEYNOTE-942); ModernaTX, Inc.: Cambridge, MA, USA, 2019. [Google Scholar]
- Memorial Sloan Kettering Cancer Center; Genentech, Inc. Study of Personalized Tumor Vaccines (PCVs) and a PD-L1 Blocker in Patients With Pancreatic Cancer That Can be Treated with Surgery; Memorial Sloan Kettering Cancer Center: New York, NY, USA, 2019. [Google Scholar]
- Gritstone bio, Inc. ; Squibb, B.-M. A Study of a Personalized Cancer Vaccine Targeting Shared Neoantigens; Gritstone bio, Inc.: Emeryville, CA, USA, 2019. [Google Scholar]
- Stemirna Therapeutics; Peking University Cancer Hospital & Institute. Clinical Study of mRNA Vaccine in Patients with Advanced Malignant Solid Tumors; Stemirna Therapeutics: Shanghai, China, 2023. [Google Scholar]
- BioNTech SE. Safety, Pharmacokinetics, Pharmacodynamics, and Preliminary Efficacy Trial of BNT141 in Patients with Unresectable or Metastatic CLDN18.2-Positive Gastric, Pancreatic, Ovarian and Biliary Tract Tumors; BioNTech SE: Mainz, Germany, 2022. [Google Scholar]
- BioNTech SE. Safety and Preliminary Efficacy Trial of BNT142 in Patients with CLDN6-Positive Solid Tumors; BioNTech SE: Mainz, Germany, 2022. [Google Scholar]
- Nitika; Wei, J.; Hui, A.-M. The delivery of mRNA vaccines for therapeutics. Life 2022, 12, 1254. [Google Scholar] [CrossRef] [PubMed]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Patel, M.; Jimeno, A.; Wang, D.; Stemmer, S.; Bauer, T.; Sweis, R.; Geva, R.; Kummar, S.; Reagan, P.; Perets, R. 539 Phase 1 study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L/IL-23/IL-36γ, for intratumoral (ITu) injection+/-durvalumab in advanced solid tumors and lymphoma. Sci. Transl. Med. 2021, 11, eaat9143. [Google Scholar] [CrossRef]
- The Fourth Affiliated Hospital of Zhejiang University School of Medicine. A Dose Escalation and Dose Expansion Clinical Study of STI-7349 in Subjects with Advanced Solid Tumors; The Fourth Affiliated Hospital of Zhejiang University School of Medicine: Yiwu, China, 2023. [Google Scholar]
- Longhurst, H.; Fijen, L.; Lindsay, K.; Butler, J.; Golden, A.; Maag, D.; Xu, Y.; Cohn, D. In vivo CRISPR/Cas9 editing of KLKB1 in patients with Hereditary Angioedema: A First-in-Human Study. Ann. Allergy Asthma Immunol. 2022, 129, S10–S11. [Google Scholar] [CrossRef]
- Cui, T.; Li, B.; Li, W. NTLA-2001: Opening a new era for gene therapy. Life Med. 2022, 1, 49–51. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Zamarin, D.; Marron, T.; Mehmi, I.; Patel, S.P.; Subbiah, V.; El-Khoueiry, A.; Grand, D.; Garcia-Reyes, K.; Goel, S. Abstract CT183: First-in-human study of MEDI1191 (mRNA encoding IL-12) plus durvalumab in patients (pts) with advanced solid tumors. Cancer Res. 2022, 82, CT183. [Google Scholar] [CrossRef]
- Roh, E.H.; Fromen, C.A.; Sullivan, M.O. Inhalable mRNA vaccines for respiratory diseases: A roadmap. Curr. Opin. Biotechnol. 2022, 74, 104–109. [Google Scholar] [CrossRef]
- Mike Smith, O.A.; Brito, L. Stabilized Formulations of Lipid Nanoparticles. U.S. Patent US20200069599A1, 8 November 2017. [Google Scholar]
- Vertex Pharmaceuticals Incorporated. A Phase 1 Study of VX-522 in Participants with Cystic Fibrosis (CF); Vertex Pharmaceuticals Incorporated: Boston, MA, USA, 2023. [Google Scholar]
- Arcturus Therapeutics, Inc.; Novotech CRO. Safety, Tolerability, and Pharmacokinetics of ARCT-032 in Healthy Adult Subjects; Arcturus Therapeutics, Inc.: San Diego, CA, USA, 2023. [Google Scholar]
- Oslo University Hospital. Vaccine Therapy in Curative Resected Prostate Cancer Patients; Oslo University Hospital: Oslo, Norway, 2010. [Google Scholar]
- Oslo University Hospital. Dendritic Cell Immunotherapy Against Cancer Stem Cells in Glioblastoma Patients Receiving Standard Therapy; Oslo University Hospital: Oslo, Norway, 2018. [Google Scholar]
- Berneman, Z.; Kanker, K.O.T.; Kanker, S.; Flanders, R.F. ; Antwerp University Hospital. Efficacy Study of Dendritic Cell Vaccination in Patients with Acute Myeloid Leukemia in Remission; Antwerp University Hospital: Antwerp, Belgium, 2012. [Google Scholar]
- Antwerp University Hospital; Kanker, K.O.T.; Semmy, S.; Olivia Hendrickx Research Fund vzw. Adjuvant Dendritic Cell Immunotherapy for Pediatric Patients With High-Grade Glioma or Diffuse Intrinsic Pontine Glioma; Antwerp University Hospital: Antwerp, Belgium, 2021. [Google Scholar]
- Antwerp University Hospital; Kanker, K.O.T.; Kanker, S. Autologous Dendritic Cell Vaccination in Mesothelioma; Antwerp University Hospital: Antwerp, Belgium, 2017. [Google Scholar]
- Antwerp University Hospital. Adjuvant Dendritic Cell-immunotherapy Plus Temozolomide in Glioblastoma Patients; Antwerp University Hospital: Antwerp, Belgium, 2015. [Google Scholar]
- Affiliated Hospital to Academy of Military Medical Sciences. Clinical Study of DC-AML Cells in the Treatment of Acute Myeloid Leukemia. In Identifier NCT05000801.; 2021. Available online: https://www.clinicaltrials.gov/study/NCT05000801 (accessed on 5 October 2023).
- Sholler, G. PEACH TRIAL- Precision Medicine and Adoptive Cellular Therapy; University of Florida: Gainesville, FL, USA, 2021. [Google Scholar]
- Immunomic Therapeutics, Inc.; University of Florida; National Cancer Institute. Vaccine Therapy for the Treatment of Newly Diagnosed Glioblastoma Multiforme; Immunomic Therapeutics, Inc.: Rockville, MD, USA, 2016. [Google Scholar]
- University of Florida; Immunomic Therapeutics, Inc. RENEW: Feasibility of CMV RNA-Pulsed Dendritic Cells Vaccines for the Treatment of Newly Diagnosed Glioblastoma Patients; University of Florida: Gainesville, FL, USA, 2022. [Google Scholar]
- CoImmune. Dendritic Cell Immunotherapy Plus Standard Treatment of Advanced Renal Cell Carcinoma; CoImmune: Durham, NC, USA, 2020. [Google Scholar]
- Lion TCR Pte. Ltd. Redirected HBV-Specific T Cells in Patients with HBV-Related HCC (SAFE-T-HBV); Lion TCR Pte. Ltd.: Singapore, 2022. [Google Scholar]
- Lion TCR Pte. Ltd. Study of HBV-TCR T Cells (LioCyx-M) as Monotherapy or as Combination with Lenvatinib for HBV-Related HCC; Lion TCR Pte. Ltd.: Singapore, 2022. [Google Scholar]
- Ruijin Hospital; UTC Therapeutics Inc. Anti-Mesothelin CAR-T Cells With Advanced Refractory Solid Tumors; Ruijin Hospital: Shanghai, China, 2021. [Google Scholar]
- CytoMed Therapeutics Pte Ltd.; National University Hospital. Allogeneic NKG2DL-Targeting CAR γδ T Cells (CTM-N2D) in Advanced Cancers; CytoMed Therapeutics Pte Ltd.: Singapore, 2022. [Google Scholar]
- 2Seventy Bio Inc. A Study of bbT369 in Relapsed and/or Refractory B Cell Non-Hodgkin’s Lymphoma (NHL). In Identifier NCT05169489 2022. Available online: https://clinicaltrials.gov/study/NCT05169489 (accessed on 5 October 2023).
- Cartesian Therapeutics. Descartes-11 in Multiple Myeloma; Cartesian Therapeutics: Gaithersburg, MD, USA, 2019. [Google Scholar]
- Cartesian Therapeutics. Descartes-08 CAR-T Cells in Generalized Myasthenia Gravis (MG); Cartesian Therapeutics: Gaithersburg, MD, USA, 2019. [Google Scholar]
- Lyerly, H.; Merck Sharp & Dohme LLC; Duke University. A Study to Evaluate Concurrent VRP-HER2 Vaccination and Pembrolizumab for Patients with Breast Cancer. In Identifier NCT03632941.; 2019. Available online: https://clinicaltrials.gov/study/NCT03632941?tab=history (accessed on 5 October 2023).
- Tang-Du Hospital; Air Force Military Medical University. Exosome-Based Nanoplatform for Ldlr mRNA Delivery in FH; Tang-Du Hospital: Xi’an, China, 2021. [Google Scholar]
- Youn, H.; Chung, J.-K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 2015, 15, 1337–1348. [Google Scholar] [CrossRef]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.-H.; Zheng, L.; Wang, Z. mRNA therapeutics: New vaccination and beyond. Fundam. Res. 2023, 3, 749–759. [Google Scholar] [CrossRef]
- Huang, X.; Kong, N.; Zhang, X.; Cao, Y.; Langer, R.; Tao, W. The landscape of mRNA nanomedicine. Nat. Med. 2022, 28, 2273–2287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Zhou, S.; Dain, L.; Mei, L.; Zhu, G. Circular RNA: An emerging frontier in RNA therapeutic targets, RNA therapeutics, and mRNA vaccines. J. Control. Release 2022, 348, 84–94. [Google Scholar] [CrossRef]
- Marques, R.; Lacerda, R.; Romão, L. Internal Ribosome Entry Site (IRES)-Mediated Translation and Its Potential for Novel mRNA-Based Therapy Development. Biomedicines 2022, 10, 1865. [Google Scholar] [CrossRef]
- de Mey, W.; De Schrijver, P.; Autaers, D.; Pfitzer, L.; Fant, B.; Locy, H.; Esprit, A.; Lybaert, L.; Bogaert, C.; Verdonck, M. A synthetic DNA template for fast manufacturing of versatile single epitope mRNA. Mol. Ther.-Nucleic Acids 2022, 29, 943–954. [Google Scholar] [CrossRef]
- Prather, K.J.; Sagar, S.; Murphy, J.; Chartrain, M. Industrial scale production of plasmid DNA for vaccine and gene therapy: Plasmid design, production, and purification. Enzym. Microb. Technol. 2003, 33, 865–883. [Google Scholar] [CrossRef]
- Deng, Z.; Tian, Y.; Song, J.; An, G.; Yang, P. mRNA vaccines: The dawn of a new era of cancer immunotherapy. Front. Immunol. 2022, 13, 887125. [Google Scholar] [CrossRef]
- Jia, L.; Qian, S.-B. Therapeutic mRNA Engineering from Head to Tail. Acc. Chem. Res. 2021, 54, 4272–4282. [Google Scholar] [CrossRef]
- Xia, X. Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines. Vaccines 2021, 9, 734. [Google Scholar] [CrossRef]
- Trepotec, Z.; Aneja, M.K.; Geiger, J.; Hasenpusch, G.; Plank, C.; Rudolph, C. Maximizing the translational yield of mRNA therapeutics by minimizing 5′-UTRs. Tissue Eng. Part A 2019, 25, 69–79. [Google Scholar] [CrossRef] [PubMed]
- TriLink Biotechnologies. CleanCap Reagent AG Product Insert (Catalog No. N-7113 Version v3). Available online: https://www.trilinkbiotech.com/media/folio3/productattachments/product_insert/n7113_insert_v3.pdf (accessed on 5 October 2023).
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef]
- McClellan, D.A. The codon-degeneracy model of molecular evolution. J. Mol. Evol. 2000, 50, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Mordstein, C.; Savisaar, R.; Young, R.S.; Bazile, J.; Talmane, L.; Luft, J.; Liss, M.; Taylor, M.S.; Hurst, L.D.; Kudla, G. Codon usage and splicing jointly influence mRNA localization. Cell Syst. 2020, 10, 351–362.e358. [Google Scholar] [CrossRef]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166. [Google Scholar]
- Kariko, K.; Kuo, A.; Barnathan, E. Overexpression of urokinase receptor in mammalian cells following administration of the in vitro transcribed encoding mRNA. Gene Ther. 1999, 6, 1092–1100. [Google Scholar] [CrossRef]
- Benteyn, D.; Anguille, S.; Van Lint, S.; Heirman, C.; Van Nuffel, A.M.; Corthals, J.; Ochsenreither, S.; Waelput, W.; Van Beneden, K.; Breckpot, K. Design of an optimized Wilms’ tumor 1 (WT1) mRNA construct for enhanced WT1 expression and improved immunogenicity in vitro and in vivo. Mol. Ther. Nucleic Acids 2013, 2, e134. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Author Correction: Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 673. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef]
- Wan, Y.; Qu, K.; Zhang, Q.C.; Flynn, R.A.; Manor, O.; Ouyang, Z.; Zhang, J.; Spitale, R.C.; Snyder, M.P.; Segal, E. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 2014, 505, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Tureci, O.z.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- von Niessen, A.G.O.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3′ UTRs identified by cellular library screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Pollack, J.L.; Blagev, D.P.; Zaitlen, N.; McManus, M.T.; Erle, D.J. Massively parallel functional annotation of 3′ untranslated regions. Nat. Biotechnol. 2014, 32, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 2018, 156, 172–193. [Google Scholar] [CrossRef]
- Kim, S.C.; Sekhon, S.S.; Shin, W.-R.; Ahn, G.; Cho, B.-K.; Ahn, J.-Y.; Kim, Y.-H. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Mol. Cell. Toxicol. 2021, 18, 1–8. [Google Scholar] [CrossRef]
- Kormann, M.S.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef]
- Jalkanen, A.L.; Coleman, S.J.; Wilusz, J. Determinants and implications of mRNA poly (A) tail size–does this protein make my tail look big? Semin. Cell Dev. Biol. 2014, 34, 24–32. [Google Scholar] [CrossRef]
- Trepotec, Z.; Geiger, J.; Plank, C.; Aneja, M.K.; Rudolph, C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA 2019, 25, 507–518. [Google Scholar] [CrossRef]
- Lara, A.R.; Knabben, I.; Regestein, L.; Sassi, J.; Caspeta, L.; Ramírez, O.T.; Büchs, J. Comparison of oxygen enriched air vs. pressure cultivations to increase oxygen transfer and to scale-up plasmid DNA production fermentations. Eng. Life Sci. 2011, 11, 382–386. [Google Scholar] [CrossRef]
- Listner, K.; Bentley, L.; Okonkowski, J.; Kistler, C.; Wnek, R.; Caparoni, A.; Junker, B.; Robinson, D.; Salmon, P.; Chartrain, M. Development of a highly productive and scalable plasmid DNA production platform. Biotechnol. Prog. 2006, 22, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Lahijani, R.; Hulley, G.; Soriano, G.; Horn, N.A.; Marquet, M. High-yield production of pBR322-derived plasmids intended for human gene therapy by employing a temperature-controllable point mutation. Hum. Gene Ther. 1996, 7, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.R.; McKie, E.A.; Ward, J.M.; Keshavarz-Moore, E. Impact of intrinsic DNA structure on processing of plasmids for gene therapy and DNA vaccines. J. Biotechnol. 2004, 114, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Selas Castiñeiras, T.; Williams, S.G.; Hitchcock, A.G.; Smith, D.C. E. coli strain engineering for the production of advanced biopharmaceutical products. FEMS Microbiol. Lett. 2018, 365, fny162. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.; Eiteman, M.A.; Altman, E. DNA plasmid production in different host strains of Escherichia coli. J. Ind. Microbiol. Biotechnol. 2009, 36, 521–530. [Google Scholar] [CrossRef]
- Lopes, M.B.; Gonçalves, G.A.; Felício-Silva, D.; Prather, K.L.; Monteiro, G.A.; Prazeres, D.M.; Calado, C.R. In situ NIR spectroscopy monitoring of plasmid production processes: Effect of producing strain, medium composition and the cultivation strategy. J. Chem. Technol. Biotechnol. 2015, 90, 255–261. [Google Scholar] [CrossRef]
- Carnes, A.E.; Hodgson, C.P.; Williams, J.A. Inducible Escherichia coli fermentation for increased plasmid DNA production. Biotechnol. Appl. Biochem. 2006, 45, 155–166. [Google Scholar]
- Xu, Z.-N.; Shen, W.-H.; Chen, H.; Cen, P.-L. Effects of medium composition on the production of plasmid DNA vector potentially for human gene therapy. J. Zhejiang Univ. SCIENCE B 2005, 6, 396–400. [Google Scholar] [CrossRef]
- Ohlson, J. Plasmid manufacture is the bottleneck of the genetic medicine revolution. Drug Discov. Today 2020, 25, 1891. [Google Scholar] [CrossRef]
- Rosa, S.S.; Nunes, D.; Antunes, L.; Prazeres, D.M.; Marques, M.P.; Azevedo, A.M. Maximizing mRNA vaccine production with Bayesian optimization. Biotechnol. Bioeng. 2022, 119, 3127–3139. [Google Scholar] [CrossRef]
- Sun, B.; Yu, X.; Yin, Y.; Liu, X.; Wu, Y.; Chen, Y.; Zhang, X.; Jiang, C.; Kong, W. Large-scale purification of pharmaceutical-grade plasmid DNA using tangential flow filtration and multi-step chromatography. J. Biosci. Bioeng. 2013, 116, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Bimboim, H.C.; Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979, 7, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.; Flaschel, E. Method for Producing Extra-Chromosomal Nucleic Acid Molecules. U.S. Patent US7842481B2, 30 November 2010. [Google Scholar]
- Schmeer, M.; Schleef, M. Pharmaceutical grade large-scale plasmid DNA manufacturing process. In DNA Vaccines: Methods and Protocols; Humana: New York, NY, USA, 2014; pp. 219–240. [Google Scholar]
- Holmes, D.S.; Quigley, M. A rapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 1981, 114, 193–197. [Google Scholar] [CrossRef]
- Zhu, K.; Jin, H.; He, Z.; Zhu, Q.; Wang, B. A continuous method for the large-scale extraction of plasmid DNA by modified boiling lysis. Nat. Protoc. 2006, 1, 3088–3093. [Google Scholar] [CrossRef] [PubMed]
- Eon-Duval, A.; Gumbs, K.; Ellett, C. Precipitation of RNA impurities with high salt in a plasmid DNA purification process: Use of experimental design to determine reaction conditions. Biotechnol. Bioeng. 2003, 83, 544–553. [Google Scholar] [CrossRef]
- Eon-Duval, A.; MacDuff, R.H.; Fisher, C.A.; Harris, M.J.; Brook, C. Removal of RNA impurities by tangential flow filtration in an RNase-free plasmid DNA purification process. Anal. Biochem. 2003, 316, 66–73. [Google Scholar] [CrossRef]
- Latulippe, D.R.; Zydney, A.L. Size exclusion chromatography of plasmid DNA isoforms. J. Chromatogr. A 2009, 1216, 6295–6302. [Google Scholar] [CrossRef] [PubMed]
- Eon-Duval, A.; Burke, G. Purification of pharmaceutical-grade plasmid DNA by anion-exchange chromatography in an RNase-free process. J. Chromatogr. B 2004, 804, 327–335. [Google Scholar] [CrossRef]
- Bo, H.; Wang, J.; Chen, Q.; Shen, H.; Wu, F.; Shao, H.; Huang, S. Using a single hydrophobic-interaction chromatography to purify pharmaceutical-grade supercoiled plasmid DNA from other isoforms. Pharm. Biol. 2013, 51, 42–48. [Google Scholar] [CrossRef]
- Černigoj, U.; Štrancar, A. Scale-up of plasmid DNA downstream process based on chromatographic monoliths. In DNA Vaccines: Methods and Protocols; Humana: New York, NY, USA, 2021; pp. 167–192. [Google Scholar]
- Parker, T.; Cherradi, Y.; Mishra, N. Scalable Purification of Plasmid DNA: Strategies and Considerations for Vaccine and Gene Therapy Manufacturing; Application Note MS-WP7159EN Ver; Millipore Sigma: Burlington, MA, USA, 2020; Volume 1. [Google Scholar]
- Hornblower, B.; Robb, G.B.; Tzertzinis, G. Minding Your Caps and Tails—Considerations for Functional mRNA Synthesis. Available online: https://international.neb.com/tools-and-resources/feature-articles/minding-your-caps-and-tails (accessed on 5 October 2023).
- Bancel, S.; Issa, W.J.; Aunins, J.G.; Chakraborty, T. Manufacturing Methods for Production of RNA Transcripts. U.S. Patent US10138507B2, 27 November 2018. [Google Scholar]
- Kwon, S.; Kwon, M.; Im, S.; Lee, K.; Lee, H. mRNA vaccines: The most recent clinical applications of synthetic mRNA. Arch. Pharmacal Res. 2022, 45, 245–262. [Google Scholar] [CrossRef]
- Nance, K.D.; Meier, J.L. Modifications in an emergency: The role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Dousis, A.; Ravichandran, K.; Hobert, E.M.; Moore, M.J.; Rabideau, A.E. An engineered T7 RNA polymerase that produces mRNA free of immunostimulatory byproducts. Nat. Biotechnol. 2023, 41, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef]
- Durbin, A.F.; Wang, C.; Marcotrigiano, J.; Gehrke, L. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio 2016, 7, e00833-16. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Jo, M.H.; Lin, C.; Wu, B.; Orme-Johnson, M.; Walz, T.; Hohng, S.; Hur, S. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc. Natl. Acad. Sci. USA 2012, 109, E3340–E3349. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Asahara, H.; Tzertzinis, G.; Roy, B. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA 2020, 26, 345–360. [Google Scholar] [CrossRef]
- Piao, X.; Yadav, V.; Wang, E.; Chang, W.; Tau, L.; Lindenmuth, B.E.; Wang, S.X. Double-stranded RNA reduction by chaotropic agents during in vitro transcription of messenger RNA. Mol. Ther. Nucleic Acids 2022, 29, 618–624. [Google Scholar] [CrossRef]
- Kern, J.A.; Davis, R.H. Application of Solution Equilibrium Analysis to inVitro RNA Transcription. Biotechnol. Prog. 1997, 13, 747–756. [Google Scholar] [CrossRef]
- Nemec, K.S.; Livk, A.G.; Celjar, A.M.; Skok, J.; Sekirnik, R.; Kostelec, T.; Gagnon, P.; Štrancar, A. Effect of Mg2+ Ion Concentration on IVT Reaction Kinetics Determined by Novel Rapid Analytical HPLC Assay; Sartorius Company: Göttingen, Germany, 2021. [Google Scholar]
- Young, J.S.; Ramirez, W.F.; Davis, R.H. Modeling and optimization of a batch process for in vitro RNA production. Biotechnol. Bioeng. 1997, 56, 210–220. [Google Scholar] [CrossRef]
- Nikolic, M.; Gasiūnienė, M.; Asa, D.; Šeputienė, V. Determination of the Optimal Buffer Conditions and Nucleotide Concentrations to Maximize mRNA Yield Using In Vitro Transcription; ThermoFisher Scientific: Waltham, MA, USA, 2022. [Google Scholar]
- Samnuan, K.; Blakney, A.K.; McKay, P.F.; Shattock, R.J. Design-of-experiments in vitro transcription yield optimization of self-amplifying RNA. bioRxiv 2021. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkic-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef]
- Yang, L.; Tang, L.; Zhang, M.; Liu, C. Recent advances in the molecular design and delivery technology of mRNA for vaccination against infectious diseases. Front. Immunol. 2022, 13, 896958. [Google Scholar] [CrossRef]
- Borden, K.L. The eukaryotic translation initiation factor eIF4E wears a “cap” for many occasions. Translation 2016, 4, e1220899. [Google Scholar] [CrossRef]
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.-Y.; Verrier, B. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef]
- Fang, E.; Liu, X.; Li, M.; Zhang, Z.; Song, L.; Zhu, B.; Wu, X.; Liu, J.; Zhao, D.; Li, Y. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 2022, 7, 94. [Google Scholar] [CrossRef]
- Pradère, U.; Halloy, F.; Hall, J. Chemical synthesis of long RNAs with terminal 5′-phosphate groups. Chem. Eur. J. 2017, 23, 5210–5213. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; Di Piazza, A.T. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedzwiecka, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl (3′-O-methyl) GpppG and 7-methyl (3′-deoxy) GpppG. RNA 2001, 7, 1486–1495. [Google Scholar]
- Kuhn, A.; Diken, M.; Kreiter, S.; Selmi, A.; Kowalska, J.; Jemielity, J.; Darzynkiewicz, E.; Huber, C.; Türeci, Ö.; Sahin, U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010, 17, 961–971. [Google Scholar] [CrossRef]
- Henderson, J.M.; Ujita, A.; Hill, E.; Yousif-Rosales, S.; Smith, C.; Ko, N.; McReynolds, T.; Cabral, C.R.; Escamilla-Powers, J.R.; Houston, M.E. Cap 1 messenger RNA synthesis with co-transcriptional cleancap® analog by in vitro transcription. Curr. Protoc. 2021, 1, e39. [Google Scholar] [CrossRef]
- Pregeljc, D.; Skok, J.; Vodopivec, T.; Mencin, N.; Krušič, A.; Ličen, J.; Nemec, K.Š.; Štrancar, A.; Sekirnik, R. Increasing yield of in vitro transcription reaction with at-line high pressure liquid chromatography monitoring. Biotechnol. Bioeng. 2023, 120, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.A.; Davis, R.H. Application of a fed-batch system to produce RNA by in vitro transcription. Biotechnol. Prog. 1999, 15, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Skok, J.; Megušar, P.; Vodopivec, T.; Pregeljc, D.; Mencin, N.; Korenč, M.; Krušič, A.; Celjar, A.M.; Pavlin, N.; Krušič, J. Gram-Scale mRNA Production Using a 250-mL Single-Use Bioreactor. Chem. Ing. Tech. 2022, 94, 1928–1935. [Google Scholar] [CrossRef]
- Helgers, H.; Hengelbrock, A.; Schmidt, A.; Strube, J. Digital twins for continuous mRNA production. Processes 2021, 9, 1967. [Google Scholar] [CrossRef]
- Vetter, F.L.; Zobel-Roos, S.; Mota, J.P.B.; Nilsson, B.; Schmidt, A.; Strube, J. Toward Autonomous Production of mRNA-Therapeutics in the Light of Advanced Process Control and Traditional Control Strategies for Chromatography. Processes 2022, 10, 1868. [Google Scholar] [CrossRef]
- Ouranidis, A.; Davidopoulou, C.; Tashi, R.-K.; Kachrimanis, K. Pharma 4.0 continuous mRNA drug products manufacturing. Pharmaceutics 2021, 13, 1371. [Google Scholar] [CrossRef]
- Liu, C.; Shi, Q.; Huang, X.; Koo, S.; Kong, N.; Tao, W. mRNA-based cancer therapeutics. Nat. Rev. Cancer 2023, 23, 526–543. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Beaucage, S.; Caruthers, M. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981, 22, 1859–1862. [Google Scholar] [CrossRef]
- RL, L.; Mahadevan, V. Oligonucleotide synthesis on a polymer support. J. Am. Chem. Soc. 1965, 87, 3526–3527. [Google Scholar]
- Li, N.-S.; Frederiksen, J.K.; Piccirilli, J.A. Automated solid-phase synthesis of RNA oligonucleotides containing a nonbridging phosphorodithioate linkage via phosphorothioamidites. J. Org. Chem. 2012, 77, 9889–9892. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, Y.S. Large-scale automated synthesis of therapeutic oligonucleotides: A status update. Adv. Nucleic Acid Ther. 2019, 68, 453–473. [Google Scholar]
- Cedillo, I.; Chreng, D.; Engle, E.; Chen, L.; McPherson, A.K.; Rodriguez, A.A. Synthesis of 5′-GalNAc-conjugated oligonucleotides: A comparison of solid and solution-phase conjugation strategies. Molecules 2017, 22, 1356. [Google Scholar] [CrossRef]
- Kumar, R.K.; Guzaev, A.P.; Rentel, C.; Ravikumar, V.T. Efficient synthesis of antisense phosphorothioate oligonucleotides using a universal solid support. Tetrahedron 2006, 62, 4528–4534. [Google Scholar] [CrossRef]
- Ryczek, M.; Pluta, M.; Błaszczyk, L.; Kiliszek, A. Overview of Methods for Large-Scale RNA Synthesis. Appl. Sci. 2022, 12, 1543. [Google Scholar] [CrossRef]
- Flamme, M.; McKenzie, L.K.; Sarac, I.; Hollenstein, M. Chemical methods for the modification of RNA. Methods 2019, 161, 64–82. [Google Scholar] [CrossRef]
- Yu, C.-H.; Kabza, A.M.; Sczepanski, J.T. Assembly of long l-RNA by native RNA ligation. Chem. Commun. 2021, 57, 10508–10511. [Google Scholar] [CrossRef]
- Kershaw, C.J.; O’Keefe, R.T. Splint ligation of RNA with T4 DNA ligase. In Recombinant and In Vitro RNA Synthesis: Methods and Protocols; Springer Science + Business Media: Berlin/Heidelberg, Germany, 2012; pp. 257–269. [Google Scholar]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the messenger: Advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef]
- Whitley, J.; Zwolinski, C.; Denis, C.; Maughan, M.; Hayles, L.; Clarke, D.; Snare, M.; Liao, H.; Chiou, S.; Marmura, T. Development of mRNA manufacturing for vaccines and therapeutics: mRNA platform requirements and development of a scalable production process to support early phase clinical trials. Transl. Res. 2022, 242, 38–55. [Google Scholar] [CrossRef]
- Ouranidis, A.; Vavilis, T.; Mandala, E.; Davidopoulou, C.; Stamoula, E.; Markopoulou, C.K.; Karagianni, A.; Kachrimanis, K. mRNA therapeutic modalities design, formulation and manufacturing under pharma 4.0 principles. Biomedicines 2021, 10, 50. [Google Scholar] [CrossRef]
- Von Der Mülbe, F.; Reidel, L.; Ketterer, T.; Gontcharova, L.; Bauer, S.; Pascolo, S.; Probst, J.; Schmid, A. Method for Producing RNA. U.S. Patent US1001 7826B2, 2018.
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther.-Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.B.; Wen, Y.; Geall, A.; Porter, F. RNA Purification Methods. U.S. Patent US20210214388A1, 2016. [Google Scholar]
- Phua, K.K.; Leong, K.W.; Nair, S.K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J. Control. Release 2013, 166, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Sultana, N.; Magadum, A.; Hadas, Y.; Kondrat, J.; Singh, N.; Youssef, E.; Calderon, D.; Chepurko, E.; Dubois, N.; Hajjar, R.J. Optimizing cardiac delivery of modified mRNA. Mol. Ther. 2017, 25, 1306–1315. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Kornbrust, D.J.; Foy, J.W.; Solano, E.C.; Schneider, D.J.; Feinstein, E.; Molitoris, B.A.; Erlich, S. Toxicological and pharmacokinetic properties of chemically modified siRNAs targeting p53 RNA following intravenous administration. Nucleic Acid Ther. 2012, 22, 255–264. [Google Scholar] [CrossRef]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid nanoparticles─ from liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Suys, E.J.; Lee, J.S.; Nguyen, D.H.; Park, K.D.; Truong, N.P. Lipid-based nanoparticles in the clinic and clinical trials: From cancer nanomedicine to COVID-19 vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.S.; Lee, A.C.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetsky, M.; Grefhorst, A.; Anderson, N.N.; Racie, T.S.; Bramlage, B.; Akinc, A.; Butler, D.; Charisse, K.; Dorkin, R.; Fan, Y. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA 2008, 105, 11915–11920. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Schlich, M.; Palomba, R.; Costabile, G.; Mizrahy, S.; Pannuzzo, M.; Peer, D.; Decuzzi, P. Cytosolic delivery of nucleic acids: The case of ionizable lipid nanoparticles. Bioeng. Transl. Med. 2021, 6, e10213. [Google Scholar] [CrossRef]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1510, 152–166. [Google Scholar] [CrossRef]
- Aldosari, B.N.; Alfagih, I.M.; Almurshedi, A.S. Lipid nanoparticles as delivery systems for RNA-based vaccines. Pharmaceutics 2021, 13, 206. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing considerations for the development of lipid nanoparticles using microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.J.; Higgins, J.; Woods, A.; Levy, B.; Xia, Y.; Hsiao, C.J.; Acosta, E.; Almarsson, Ö.; Moore, M.J.; Brito, L.A. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Control. Release 2021, 335, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, C.; Zuidema, J.; Crommelin, D.J.A.; Storm, G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection.: II. Influence of liposomal size, lipid composition and lipid dose. Biochim. Biophys. Acta (BBA)-Biomembr. 1997, 1328, 261–272. [Google Scholar] [CrossRef]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef]
- Chen, J.; Ye, Z.; Huang, C.; Qiu, M.; Song, D.; Li, Y.; Xu, Q. Lipid nanoparticle-mediated lymph node–targeting delivery of mRNA cancer vaccine elicits robust CD8+ T cell response. Proc. Natl. Acad. Sci. USA 2022, 119, e2207841119. [Google Scholar] [CrossRef]
- Parhiz, H.; Brenner, J.S.; Patel, P.N.; Papp, T.E.; Shahnawaz, H.; Li, Q.; Shi, R.; Zamora, M.E.; Yadegari, A.; Marcos-Contreras, O.A. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE). J. Control. Release 2022, 344, 50–61. [Google Scholar] [CrossRef]
- Anjaneyulu Dirisala, J.L.; Gonzalez-Carter, D.; Wang, Z. Editorial: Delivery systems in biologics-based therapeutics. Front. Bioeng. Biotechnol. 2023, 11, 1274210. [Google Scholar] [CrossRef]
- Żak, M.M.; Zangi, L. Lipid nanoparticles for organ-specific mRNA therapeutic delivery. Pharmaceutics 2021, 13, 1675. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Andretto, V.; Repellin, M.; Pujol, M.; Almouazen, E.; Sidi-Boumedine, J.; Granjon, T.; Zhang, H.; Remaut, K.; Jordheim, L.P.; Briançon, S. Hybrid core-shell particles for mRNA systemic delivery. J. Control. Release 2023, 353, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Dirisala, A.; Uchida, S.; Toh, K.; Li, J.; Osawa, S.; Tockary, T.A.; Liu, X.; Abbasi, S.; Hayashi, K.; Mochida, Y. Transient stealth coating of liver sinusoidal wall by anchoring two-armed PEG for retargeting nanomedicines. Sci. Adv. 2020, 6, eabb8133. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, B.; Poon, W.; Zhang, Y.-N.; Lin, Z.P.; Kingston, B.R.; Tavares, A.J.; Zhang, Y.; Chen, J.; Valic, M.S.; Syed, A.M. The dose threshold for nanoparticle tumour delivery. Nat. Mater. 2020, 19, 1362–1371. [Google Scholar] [CrossRef]
- Daniel, S.; Kis, Z.; Kontoravdi, C.; Shah, N. Quality by Design for enabling RNA platform production processes. Trends Biotechnol. 2022, 40, 1213–1228. [Google Scholar] [CrossRef]
- Wang, X.; Liu, S.; Sun, Y.; Yu, X.; Lee, S.M.; Cheng, Q.; Wei, T.; Gong, J.; Robinson, J.; Zhang, D. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat. Protoc. 2023, 18, 265–291. [Google Scholar] [CrossRef]
- Gkionis, L.; Campbell, R.A.; Aojula, H.; Harris, L.K.; Tirella, A. Manufacturing drug co-loaded liposomal formulations targeting breast cancer: Influence of preparative method on liposomes characteristics and in vitro toxicity. Int. J. Pharm. 2020, 590, 119926. [Google Scholar] [CrossRef]
- Ripoll, M.; Martin, E.; Enot, M.; Robbe, O.; Rapisarda, C.; Nicolai, M.-C.; Deliot, A.; Tabeling, P.; Authelin, J.-R.; Nakach, M. Optimal self-assembly of lipid nanoparticles (LNP) in a ring micromixer. Sci. Rep. 2022, 12, 9483. [Google Scholar] [CrossRef]
- Shepherd, S.J.; Warzecha, C.C.; Yadavali, S.; El-Mayta, R.; Alameh, M.-G.; Wang, L.; Weissman, D.; Wilson, J.M.; Issadore, D.; Mitchell, M.J. Scalable mRNA and siRNA lipid nanoparticle production using a parallelized microfluidic device. Nano Lett. 2021, 21, 5671–5680. [Google Scholar] [CrossRef]
- Kimura, N.; Maeki, M.; Sato, Y.; Note, Y.; Ishida, A.; Tani, H.; Harashima, H.; Tokeshi, M. Development of the iLiNP device: Fine tuning the lipid nanoparticle size within 10 nm for drug delivery. ACS Omega 2018, 3, 5044–5051. [Google Scholar] [CrossRef] [PubMed]
- Zhigaltsev, I.V.; Belliveau, N.; Hafez, I.; Leung, A.K.; Huft, J.; Hansen, C.; Cullis, P.R. Bottom-up design and synthesis of limit size lipid nanoparticle systems with aqueous and triglyceride cores using millisecond microfluidic mixing. Langmuir 2012, 28, 3633–3640. [Google Scholar] [CrossRef] [PubMed]
- Maeki, M.; Saito, T.; Sato, Y.; Yasui, T.; Kaji, N.; Ishida, A.; Tani, H.; Baba, Y.; Harashima, H.; Tokeshi, M. A strategy for synthesis of lipid nanoparticles using microfluidic devices with a mixer structure. RSC Adv. 2015, 5, 46181–46185. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K. Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Kimura, N.; Maeki, M.; Sato, Y.; Ishida, A.; Tani, H.; Harashima, H.; Tokeshi, M. Development of a microfluidic-based post-treatment process for size-controlled lipid nanoparticles and application to siRNA delivery. ACS Appl. Mater. Interfaces 2020, 12, 34011–34020. [Google Scholar] [CrossRef]
- Wei, W.; Sun, J.; Guo, X.-Y.; Chen, X.; Wang, R.; Qiu, C.; Zhang, H.-T.; Pang, W.-H.; Wang, J.-C.; Zhang, Q. Microfluidic-Based Holonomic Constraints of siRNA in the Kernel of Lipid/Polymer Hybrid Nanoassemblies for Improving Stable and Safe In Vivo Delivery. ACS Appl. Mater. Interfaces 2020, 12, 14839–14854. [Google Scholar] [CrossRef]
- Henderson, M.I.; Eygeris, Y.; Jozic, A.; Herrera, M.; Sahay, G. Leveraging biological buffers for efficient messenger RNA delivery via lipid nanoparticles. Mol. Pharm. 2022, 19, 4275–4285. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, X.; Yan, J.; Du, S.; Xue, Y.; Li, W.; Xiang, G.; Dong, Y. Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact. Mater. 2020, 5, 358–363. [Google Scholar] [CrossRef]
- Ball, R.L.; Bajaj, P.; Whitehead, K.A. Achieving long-term stability of lipid nanoparticles: Examining the effect of pH, temperature, and lyophilization. Int. J. Nanomed. 2017, 305–315. [Google Scholar] [CrossRef]
- Zhang, N.-N.; Li, X.-F.; Deng, Y.-Q.; Zhao, H.; Huang, Y.-J.; Yang, G.; Huang, W.-J.; Gao, P.; Zhou, C.; Zhang, R.-R. A thermostable mRNA vaccine against COVID-19. Cell 2020, 182, 1271–1283.e16. [Google Scholar] [CrossRef]
- Zhang, H.; Leal, J.; Soto, M.R.; Smyth, H.D.; Ghosh, D. Aerosolizable lipid nanoparticles for pulmonary delivery of mRNA through design of experiments. Pharmaceutics 2020, 12, 1042. [Google Scholar] [CrossRef] [PubMed]
- Lamoot, A.; Lammens, J.; De Lombaerde, E.; Zhong, Z.; Gontsarik, M.; Chen, Y.; De Beer, T.R.; De Geest, B.G. Successful batch and continuous lyophilization of mRNA LNP formulations depend on cryoprotectants and ionizable lipids. Biomater. Sci. 2023, 11, 4327–4334. [Google Scholar] [CrossRef] [PubMed]
- Meulewaeter, S.; Nuytten, G.; Cheng, M.H.; De Smedt, S.C.; Cullis, P.R.; De Beer, T.; Lentacker, I.; Verbeke, R. Continuous freeze-drying of messenger RNA lipid nanoparticles enables storage at higher temperatures. J. Control. Release 2023, 357, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Lam, K.; Bajusz, C.; Laczkó, D.; Karikó, K.; Schreiner, P.; Martin, A.; Lutwyche, P.; Heyes, J.; Pardi, N. Lyophilization provides long-term stability for a lipid nanoparticle-formulated, nucleoside-modified mRNA vaccine. Mol. Ther. 2022, 30, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Pardi, M.L.; Wu, J.; Kawasaki, S.; Saito, H. Synthetic RNA-based post-transcriptional expression control methods and genetic circuits. Adv. Drug Deliv. Rev. 2022, 184, 114196. [Google Scholar] [CrossRef]
- Weissman, D.; Karikó, K. mRNA: Fulfilling the promise of gene therapy. Mol. Ther. 2015, 23, 1416–1417. [Google Scholar] [CrossRef]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Kandimalla, R.; Wallen, M.; Tyagi, N.; Wilcher, S.; Yan, J.; Schultz, D.J.; Spencer, W. Exosome-mediated delivery of RNA and DNA for gene therapy. Cancer Lett. 2021, 505, 58–72. [Google Scholar] [CrossRef]
- Ma, C.-C.; Wang, Z.-L.; Xu, T.; He, Z.-Y.; Wei, Y.-Q. The approved gene therapy drugs worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef]
- Berraondo, P.; Martini, P.G.; Avila, M.A.; Fontanellas, A. Messenger RNA therapy for rare genetic metabolic diseases. Gut 2019, 68, 1323–1330. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trial ID | Status | Indication | Treatment Name | Dose Regimen | Administration Method |
|---|---|---|---|---|---|
| NCT04573140 | Recruiting | Adult glioblastoma | Autologous total tumor mRNA and pp65 full-length (fl) lysosomal-associated membrane protein (LAMP) mRNA-loaded DOTAP liposome | Every 2 weeks (3 cycles), monthly (15 cycles) | IV |
| NCT05097911 | Recruiting | Advanced Hepatocellular Carcinoma | MTL-CEBPA | Day 1 and Day 8 of a 21-Day Dosing Schedule | IV |
| NCT05579275 | Recruiting | Advanced malignant solid tumors | JCXH-212 Injection | Every 3 weeks (up to 8 cycles) | Unspecified injection |
| NCT05949775 | Not yet recruiting | Advanced Malignant Solid Tumours | Neoantigen mRNA Personalized Cancer vaccine | Every 3 weeks (9 cycles) | SQ |
| NCT05978102 | Not yet recruiting | Advanced Solid Tumor | STI-7349/IL2v mRNA | Every 3 weeks | IV |
| NCT05533697 | Recruiting | Advanced Solid Tumours | mRNA- 4359 | N/A | IM |
| NCT02872025 | Recruiting | Carcinoma, Intraductal, Noninfiltrating | mRNA 2752 | 2–4 Doses | ILES |
| NCT05659264 | Recruiting | Chronic heart failure | mRNA-0184 | 2 groups: single dose OR 4 doses every 16 weeks | IV |
| NCT05141721 | Recruiting | Colorectal neoplasms | GRT-R902 (samRNA), GRT-C901(viral vector) | 4 doses over first year | IM |
| NCT05712538 | Recruiting | Cystic Fibrosis | ARCT-032 | Single dose | INH |
| NCT05668741 | Recruiting | Cystic Fibrosis | VX-522 | Single dose | INH |
| NCT05938387 | Recruiting | Glioblastoma | CV09050101 mRNA vaccine | 7 doses at different intervals | IM |
| NCT05095727 | Recruiting | Glycogen storage disease | mRNA-3745 | Single dose. Additional dosages after >21 days | IV |
| NCT05497453 | Recruiting | Hepatocellular Carcinoma | OTX-2002 | At least 2 doses | IV |
| NCT04710641 | Recruiting | Hepatocellular Carcinoma | MTL-CEBPA (saRNA) | Every 3 weeks | IV |
| NCT05120830 | Recruiting | Hereditary Angioedema | NTLA-2002 | Single dose | IV |
| NCT05933577 | Recruiting | High-Risk Melanoma | V940 | Every 3 weeks (up to 9 doses) | IM |
| NCT05295433 | Recruiting | Isolated methylmalonic acidemia (MMA) | mRNA-3705 | Every 2–4 weeks | IV |
| NCT04899310 | Recruiting | Isolated methylmalonic acidemia (MMA) | mRNA-3705 | Every 2–4 weeks | IV |
| NCT03289962 | Active, not recruiting | Locally or Advanced Metastatic Cancer | RO7198457 | Every 2 weeks | IV |
| NCT05969041 | Recruiting | Malignant Epithelial Tumours | MT-302 (A) | Weekly–biweekly doses for first 3 doses. Every 4 weeks subsequently | IV |
| NCT05539157 | Active, not recruiting | Malignant solid tumours, etc. | JCXH-211 | Every 4 weeks (up to 3 doses) | IT |
| NCT05714748 | Recruiting | Malignant Tumours | EBV mRNA vaccine | Weekly (4 doses), followed by a 1-month interval (1 dose) | IM |
| NCT03897881 | Active | Melanoma | mRNA-4157 | Every 3 weeks (up to 9 doses) | IM |
| NCT05264974 | Not yet recruiting | Melanoma | Autologous total tumor mRNA loaded DOTAP liposome vaccine | Every 2 weeks | IV |
| NCT04526899 | Recruiting | Melanoma | BNT111 | N/A | IV |
| NCT03871348 | Active, not recruiting | Metastatic Neoplasm | SAR441000 | N/A | IT |
| NCT05142189 | Recruiting | Non-Small Cell Lung Cancer | BNT116 | N/A | IV |
| NCT04442347 | Active, not recruiting | Ornithine Transcarbamylase Deficiency | ARCT-810 | Single dose | IV |
| NCT05526066 | Recruiting | Ornithine Transcarbamylase Deficiency | ARCT-810 | Every 2 weeks (up to 6 doses) | IV |
| NCT04161755 | Active, not recruiting | Pancreatic cancer | RO7198457 | Every week (8 Cycles) | IV |
| NCT03953235 | Active, not recruiting | Personalized cancer vaccine for many cancer types | GRT-R904 (samRNA), GRT-C903 (adenoviral vector) | N/A | N/A |
| NCT05130437 | Recruiting | Propionic Acidemia | mRNA-3927 | Every 3 weeks | IV |
| NCT04159103 | Recruiting | Propionic Acidemia | mRNA-3927 | Every 3 weeks (up to 10 doses) | IV |
| NCT04382898 | Recruiting | Prostate Cancer | BNT112 | N/A | IV |
| NCT05660408 | Not yet recruiting | Pulmonary osteosarcoma | RNA-LP vaccine | Every 2 weeks (2 cycles), monthly (12 cycles) | N/A |
| NCT03739931 | Recruiting | Relapsed solid tumor malignancies/lymphoma | mRNA-2752 | Every 2 weeks | IT |
| NCT04503278 | Recruiting | Solid Tumor | BNT211- CLDN6 CAR-T/CLDN6 CAR-T(A), CLDN6 RNA-LPX | N/A | IV |
| NCT05262530 | Recruiting | Solid Tumor | BNT142 | N/A | IV |
| NCT04710043 | Recruiting | Solid Tumor | BNT152/BNT153 | N/A | IV |
| NCT04455620 | Recruiting | Solid Tumor | BNT151 | N/A | IV |
| NCT03313778 | Active, not recruiting | Solid tumours | mRNA-4157 | 9 cycles (once every 3 weeks) | IM |
| NCT03946800 | Active, not recruiting | Solid tumours | MEDI1191 | Every 3 weeks | IT |
| NCT04683939 | Recruiting | Solid tumours, etc. | BNT141 | N/A | IV |
| NCT04601051 | Recruiting | Transthyretin-Related (ATTR) Familial Amyloid Polyneuropathy | NTLA-2001 | Single dose | IV |
| NCT04534205 | Recruiting | Unresectable Head and Neck Squamous Cell Carcinoma | BNT113 | N/A | IV |
| Microfluidic Architecture | Channel Width | Flow Rate Operation | References |
|---|---|---|---|
| Ring Micromixer (Precision Nanosystems) | 150–280 µm channel widths | Q total = 0.4 mL/min–20 mL/min | [216] |
| Staggered Herringbone Mixers | 150 µm channel width | Q total = 0.024 mL/min–2.4 mL/min | [217] |
| Y junction entry with baffle mixer (iLiNP) | 200 µm channel width | Q total = 0.05–0.5 mL/min | [218] |
| Staggered herringbone mixer with Y junction entry | 200 µm channel width | Q total = 1 mL/min–5 mL/min | [219] |
| Staggered Herringbone mixer | 200 µm channel width | Q total = 0.4 mL/min–1 mL/min | [220] |
| Staggered Herringbone mixer with Y junction entry | 200 µm channel width | Q total = 0.02 to 4 mL/min | [221] |
| Baffle Mixer with Y junction entry | 200 µm channel width | Q total = 0.05 mL/min | [222] |
| Hydrodynamic flow focusing | 150 µm channel width | Q total = 0.20006–0.8001 mL/min | [215] |
| Spiral Mixing | 300 µm channel width | Q total = 0.00666 mL/min | [223] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youssef, M.; Hitti, C.; Puppin Chaves Fulber, J.; Kamen, A.A. Enabling mRNA Therapeutics: Current Landscape and Challenges in Manufacturing. Biomolecules 2023, 13, 1497. https://doi.org/10.3390/biom13101497
Youssef M, Hitti C, Puppin Chaves Fulber J, Kamen AA. Enabling mRNA Therapeutics: Current Landscape and Challenges in Manufacturing. Biomolecules. 2023; 13(10):1497. https://doi.org/10.3390/biom13101497
Chicago/Turabian StyleYoussef, Maryam, Cynthia Hitti, Julia Puppin Chaves Fulber, and Amine A. Kamen. 2023. "Enabling mRNA Therapeutics: Current Landscape and Challenges in Manufacturing" Biomolecules 13, no. 10: 1497. https://doi.org/10.3390/biom13101497