
Biomarker Analysis of Formalin-Fixed Paraffin-Embedded Clinical Tissues Using Proteomics

Abstract
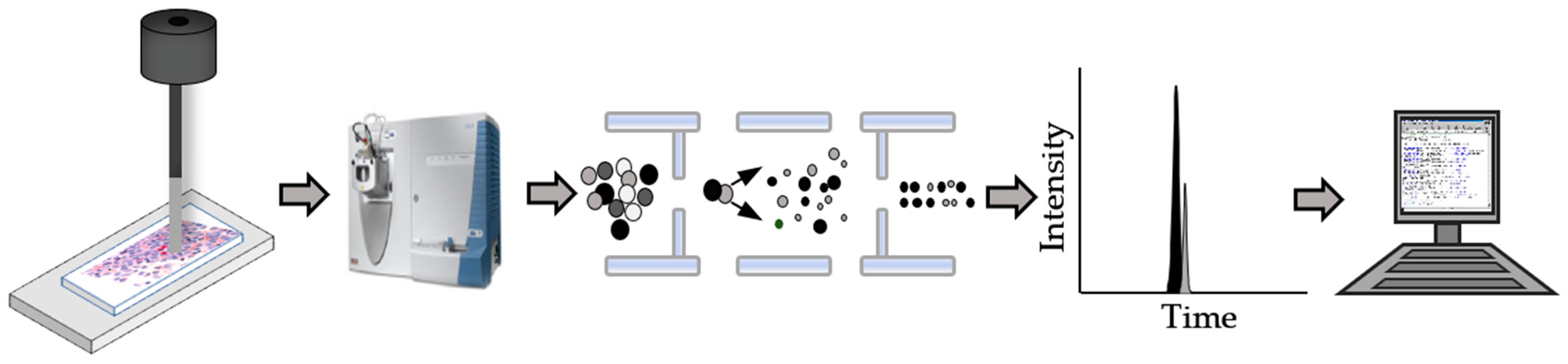
:1. Introduction
2. Blood versus Tissue
3. The Development of FFPE Tissue Proteomics
4. Protein Extraction Methods from FFPE Tissue
5. The Movement to Global Analysis
6. Targeted, Quantitative Biomarker Analysis
7. Challenges
8. Discussion
9. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, Y.; Jacobs, J.M.; Camp, D.G.; Fang, R.; Moore, R.J.; Smith, R.D.; Xiao, W.; Davis, R.W.; Tompkins, R.G. Ultra-high-efficiency strong cation exchange LC/RPLC/MS/MS for high dynamic range characterization of the human plasma proteome. Anal. Chem. 2002, 76, 1134–1144. [Google Scholar] [CrossRef]
- Müller, J.B.; Geyer, P.E.; Colaço, A.R.; Treit, P.V.; Strauss, M.T.; Oroshi, M.; Doll, S.; Virreira Winter, S.; Bader, J.M.; Kohler, N.; et al. The proteome landscape of the kingdoms of life. Nature 2020, 582, 592–596. [Google Scholar] [CrossRef]
- Tirumalai, R.S.; Chan, K.C.; Prieto, D.A.; Issaq, H.J.; Conrads, T.P.; Veenstra, T.D. Characterization of the low molecular weight human serum proteome. Mol. Cell. Proteom. 2003, 2, 1096–1103. [Google Scholar] [CrossRef] [Green Version]
- Uyar, D.S.; Huang, Y.W.; Chesnik, M.A.; Doan, N.B.; Mirza, S.P. Comprehensive serum proteomic analysis in early endometrial cancer. J. Proteom. 2021, 234, 104099–104107. [Google Scholar] [CrossRef]
- Mendes, M.L.; Dittmar, G. Targeted proteomics on its way to discovery. Proteomics 2022, 22, e2100330. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.Y.; Osman, J.; Low, T.Y.; Jamal, R. Plasma/serum proteomics: Depletion strategies for reducing high-abundance proteins for biomarker discovery. Bioanalysis 2019, 11, 1799–1812. [Google Scholar] [CrossRef]
- Krasny, L.; Huang, P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics 2021, 17, 29–42. [Google Scholar] [CrossRef]
- Makawita, S.; Diamandis, E.P. The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-approaches: Current strategies for candidate verification. Clin. Chem. 2010, 56, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Kulyyassov, A.; Fresnais, M.; Longuespee, R. Targeted liquid chromatography-tandem mass spectrometry analysis of proteins: Basic principles, applications, and perspectives. Proteomics 2021, 21, e2100153. [Google Scholar] [CrossRef]
- Johann Jr, D.J.; Wei, B.H.; Prieto, D.A.; Chan, K.C.; Ye, X.; Valera, V.A.; Simpson, R.M.; Rudnick, P.A.; Xiao, Z.; Issaq, H.J.; et al. Combined blood/tisse analysis for cancer biomarker discovery: Application to renal cell carcinoma. Anal. Chem. 2010, 82, 1584–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, A.H.; Fiala, C.A.; Diamandis, E.P.; Kulasingam, V. Pitfalls in cancer biomarker discovery and validation with emphasis on circulating tumor DNA. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2568–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Füzéry, A.K.; Levin, J.; Chan, M.M.; Chan, D.W. Translation of proteomic biomarkers into FDA approved cancer diagnostics: Issues and challenges. Clin. Proteom. 2013, 10, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, H.; Abe, Y.; Nojima, Y.; Aoki, N.; Shoji, H.; Isoyama, J.; Honda, K.; Boku, N.; Mizuguchi, K.; Tomonaga, T.; et al. Temporal dynamics from phosphoproteomics using endoscopic biopsy specimens provides new therapeutic targets in stage IV gastric cancer. Sci. Rep. 2022, 12, 4419. [Google Scholar] [CrossRef] [PubMed]
- Elkjaer, M.L.; Rottger, R.; Baumbach, J.; Illes, Z. A systematic review of tissue and single cell transcriptome/proteome studies of the brain in multiple sclerosis. Front. Immunol. 2022, 13, 761225. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.; Zhang, Z.; Xiang, Z.; Cai, Y.; Zhang, X.; Wu, J. Advances in the application of proteomics in lung cancer. Front. Oncol. 2022, 12, 993781. [Google Scholar] [CrossRef]
- O’Rourke, M.B.; Padula, M.P. Analysis of formalin-fixed, paraffin-embedded (FFPE) tissue via proteomic techniques and misconceptions of antigen retrieval. Biotechniques 2016, 60, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Fox, C.H.; Johnson, F.B.; Whiting, J.; Roller, P.P. Formaldehyde fixation. J. Histochem. Cytochem. 1985, 33, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.-I.; Thumar, J.; Lundgren, D.H.; Rezaul, K.; Mayya, V.; Wu, L.; Eng, J.; Wright, M.E.; Han, D.K. Direct cancer tissue proteomics: A method to identify candidate cancer biomarkers from formalin-fixed paraffin-embedded archival tissues. Oncogene 2007, 26, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Hood, B.L.; Darfler, M.M.; Guiel, T.G.; Furusato, B.; Lucas, D.A.; Ringeisen, B.R.; Sesterhenn, I.A.; Conrads, T.P.; Veenstra, T.D.; Krizman, D.B. Proteomic analysis of formalin-fixed prostate tissue. Mol. Cell. Proteom. 2005, 4, 1741–1753. [Google Scholar] [CrossRef]
- Zeneyedpour, L.; Stingl, C.; Dekker, L.J.M.; Mustafa, D.A.M.; Kros, J.M.; Luider, T.M. Phosphorylation ratio determination in fresh-frozen and formalin-fixed paraffin-embedded tissue with target mass spectrometry. J. Proteome Res. 2020, 19, 4179–4190. [Google Scholar] [CrossRef] [PubMed]
- Giusti, L.; Lucacchini, A. Proteomics studies of formalin-fixed paraffin-embedded tissues. Expert Rev. Proteom. 2013, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- DeSouza, L.V.; Krakovska, O.; Darfler, M.M.; Krizman, D.B.; Romaschin, A.D.; Colgan, T.J.; Siu, K.W.M. mTRAQ-based quantification of potential endometrial carcinoma biomarkers from archived formalin-fixed paraffin-embedded tissues. Proteomics 2010, 10, 3108–3116. [Google Scholar] [CrossRef] [PubMed]
- Sprung, R.W., Jr.; Martinez, M.A.; Carpenter, K.L.; Ham, A.J.; Washington, M.K.; Arteaga, C.L.; Sanders, M.E.; Liebler, D.C. Precision of multiple reaction monitoring mass spectrometry analysis of formalin-fixed paraffin-embedded tissue. J. Proteome Res. 2012, 11, 3498–3505. [Google Scholar] [CrossRef] [PubMed]
- Kuras, M.; Woldmar, N.; Kim, Y.; Hefner, M.; Malm, J.; Moldvay, J.; Döme, B.; Fillinger, J.; Pizzatti, L.; Gil, J.; et al. Proteomic workflows for high-quality quantitative proteome and post-translational modification analysis of clinically relevant samples from formalin-fixed paraffin-embedded archives. J. Proteome Res. 2021, 20, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.R.; Cote, R.J.; Taylor, C.R. Antigen retrieval techniques: Current perspectives. J. Histochem. Cytochem. 2001, 49, 931–937. [Google Scholar] [CrossRef] [Green Version]
- Mantsiou, A.; Makridakis, M.; Fasoulakis, K.; Katafigiotis, I.; Constantinides, C.A.; Zoidakis, J.; Roubelakis, M.G.; Vlahou, A.; Lygirou, V. Proteomics analysis of formalin fixed paraffin embedded tissues in the investigation of prostate cancer. J. Proteome Res. 2020, 19, 2631–2642. [Google Scholar] [CrossRef]
- Azimzadeh, O.; Barjaktarovic, Z.; Aubele, M.; Calzada-Wack, J.; Sarioglu, H.; Atkinson, M.J.; Tapio, S. Formalin-fixed paraffin-embedd (FFPE) proteome analysis using gel-free and gel-based proteomics. J. Proteome Res. 2010, 9, 4710–4720. [Google Scholar] [CrossRef]
- Sun, R.; Hunter, C.; Chen, C.; Ge, W.; Morrice, N.; Liang, S.; Zhu, T.; Yuan, C.; Ruan, G.; Zhang, Q.; et al. Accelerated protein biomarker discovery from FFPE tissue samples using single-shot, short gradient microflow SWATH-MS. J. Proteome Res. 2020, 19, 2732–2741. [Google Scholar] [CrossRef]
- Fu, Z.; Yan, K.; Rosenberg, A.; Jin, Z.; Crain, B.; Athas, G.; Heide, R.S.; Howard, T.; Everett, A.D.; Herrington, D.; et al. Improved protein extraction and protein identification from archival formalin-fixed paraffin-embedded human aortas. Proteom. Clin. Appl. 2013, 7, 217–224. [Google Scholar] [CrossRef]
- Addis, M.F.; Tanca, A.; Pagnozzi, D.; Crobu, S.; Fanciulli, G.; Cossu-Rocca, P.; Uzzau, S. Generation of high-quality protein extracts from formalin-fixed, paraffin-embedded tissues. Proteomics 2009, 9, 3815–3823. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Sasaki, H.; Terasaki, T. Establishet ad validation of highly accurate formalin-fixed paraffin-embedded quantitative proteomics by heat-compatible pressure cycling technology using phase-transfer surfactant and SWATH-MS. Sci. Rep. 2020, 10, 11271. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.B.; O’Leary, T.J.; Mason, J.T. Improving the proteomic analysis of archival tissue by using pressure-assisted protein extraction: A mechanistic approach. J. Proteom. Bioinform. 2014, 7, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refaee, M.; Tezuka, T.; Akasaka, K.; Williamson, M.P. Pressure-dependent changes in the solution structure of hen egg-white lysozyme. J. Mol. Biol. 2003, 327, 857–865. [Google Scholar] [CrossRef]
- Frye, K.J.; Royer, C.A. Probing the contribution of internal cavities to the volume change of protein unfolding under pressure. Protein Sci. 1998, 7, 2217–2222. [Google Scholar] [CrossRef] [Green Version]
- Kobashigawa, Y.; Sakurai, M.; Nitta, K. Effect of hydrostatic pressure on unfolding of alphalactalbumin: Volumetric equivalence of the molten globule and unfolded state. Protein Sci. 1999, 8, 2765–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, C.B.; Cunningham, R.E.; O’Leary, T.J.; Mason, J.T. ‘Tissue surrogates’ as a model for archival formalin-fixed paraffin-embedded tissues. Lab. Investig. 2007, 87, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.B.; Cunningham, R.E.; Waybright, T.J.; Blonder, J.; Veenstra, T.D.; O’Leary, T.J.; Mason, J.T. Elevated hydrostatic pressure promotes protein recovery from formalin-fixed, paraffin-embedded tissue surrogates. Lab. Investig. 2008, 88, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.B.; Chesnick, I.E.; Moore, C.D.; O’Leary, T.J.; Mason, J.T. Elevated pressure improves the extraction and identification of proteins recovered from formalin-fixed paraffin-embedded tissue surrogates. PLoS ONE 2010, 5, e14253. [Google Scholar] [CrossRef] [Green Version]
- Sprung, R.W., Jr.; Brock, J.W.C.; Tanksley, J.P.; Li, M.; Washington, M.K.; Slebos, R.J.C.; Liebler, D.C. Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimensional liquid chromatography-tandem mass spectrometry shotgun proteomic analysis. Mol. Cell. Proteom. 2009, 8, 1988–1998. [Google Scholar] [CrossRef]
- Hughes, C.S.; McConechy, M.K.; Cochrane, D.R.; Nazeran, T.; Karnezis, A.N.; Huntsman, D.G.; Morin, G.B. Quantitative profiling of single formalin fixed tumor sections: Proteomics for translational research. Sci. Rep. 2016, 6, 34949–34963. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lyu, M.; Liang, S.; Ge, W.; Wang, Y.; Ding, X.; Zhang, C.; Zhou, Y.; Chen, S.; Chen, L.; et al. A prostate cancer tissue specific spectral library for targeted proteomic analysis. Proteomics 2022, 22, e2100147. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Sun, M.; Bhargava, R.; Stewart, N.A.; Flint, M.S.; Bigbee, W.L.; Krivak, T.C.; Strange, M.A.; Cooper, K.L.; Zorn, K.K. Proteomic analysis of matched formalin-fixed, paraffin-embedded specimens in patients with advanced serous ovarian carcinoma. Proteomes 2013, 1, 240–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buczak, K.; Ori, A.; Kirkpatrick, J.M.; Holzer, K.; Dauch, D.; Roessler, S.; Endris, V.; Lasitschka, F.; Parca, L.; Schmidt, A.; et al. Spatial tissue proteomics quantifies inter- and intratumor heterogeneity in hepatocellular carcinoma (HCC). Mol. Cell. Proteom. 2018, 17, 810–825. [Google Scholar] [CrossRef] [Green Version]
- Friedich, C.; Schallenberg, S.; Kirchner, M.; Ziehm, M.; Niquet, S.; Haji, M.; Beier, C.; Neudecker, J.; Klauschen, F.; Mertins, P. Comprehensive micro-scaled proteome and phosphoproteome characterization of archived retrospective cancer repositories. Nat. Commun. 2021, 12, 3576–3591. [Google Scholar] [CrossRef]
- Hinneburg, H.; Korac, P.; Schirmeister, F.; Gasparov, S.; Seeberger, P.H.; Zoldos, V.; Kolarich, D. Unlocking cancer glycomes from histopathological formalin-fixed paraffin-embedded (FFPE) tissue microdissections. Mol. Cell. Proteom. 2017, 16, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Lygirou, V.; Fasoulakis, K.; Stroggilos, R.; Makridakis, M.; Latosinska, A.; Frantzi, M.; Katafigiotis, I.; Alamanis, C.; Stravodimos, K.G.; Constantinides, C.A.; et al. Proteomic analysis of prostate cancer FFPE samples reveals markers of disease progression and aggressiveness. Cancers 2022, 14, 3756. [Google Scholar] [CrossRef]
- Guo, T.; Wang, W.; Rudnick, P.A.; Song, T.; Li, J.; Zhuang, Z.; Weil, R.J.; DeVoe, D.L.; Lee, C.S.; Balgley, B.M. Proteome analysis of microdissected formalin-fixed and paraffin-embedded tissue specimens. J. Histochem. Cytochem. 2007, 55, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Jiang, X.; Feng, S.; Tian, R.; Ye, M.; Zou, H. Development of efficient protein extraction methods for shotgun proteome analysis of formalin-fixed tissues. J. Proteome Res. 2007, 6, 1038–1047. [Google Scholar] [CrossRef]
- Zhu, Y.; Weiss, T.; Zhang, Q.; Sun, R.; Wang, B.; Yi, X.; Wu, Z.; Gao, H.; Cai, X.; Ruan, G.; et al. High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification. Mol. Oncol. 2019, 13, 2305–2328. [Google Scholar] [CrossRef]
- Jang, H.N.; Moon, S.J.; Jung, K.C.; Kim, S.W.; Kim, H.; Han, D.; Kim, J.H. Mass spectrometry-based proteomics discovery of prognostic biomarkers in adrenal cortical carcinoma. Cancers 2021, 13, 3890. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Fang, F.; Liu, P.; Zeng, G.; Liu, H.; Zhao, Y.; Xie, X.; Tseng, G.; Randhawa, P.; Xiao, K. Quantitative proteomics for monitoring renal transplant injury. Proteom. Clin. Appl. 2020, 14, e1900036. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Calvo, F.; Bertrand, V.; Longuespée, R.; Delga, A.; Mazzucchelli, G.; Smargiasso, N.; Baiwir, D.; Delvenne, P.; Malaise, M.; De Pauw-Gillet, M.C.; et al. Comparison of two FFPE preparation methods using label-free shotgun proteomics: Applicatin to tissue of diverticulitis patients. J. Proteom. 2015, 112, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Davalieva, K.; Rusevski, A.; Velkov, M.; Noveski, P.; Kubelka-Sabit, K.; Filipovski, V.; Plaseski, T.; Dimovski, A.; Plaseska-Karanfilska, D. Comparative proteomics analysis of human FFPE testicular tissues reveals new candidate biomarkers for distinction among azoospermia types and subtypes. J. Proteom. 2022, 267, 104686. [Google Scholar] [CrossRef]
- Patel, V.; Hood, B.L.; Molinolo, A.A.; Lee, N.H.; Conrads, T.P.; Braisted, J.C.; Krizman, D.B.; Veenstra, T.D.; Gutkind, J.S. Proteomic analysis of laser-captured paraffin-embedded tissues: A molecular portrait of head and neck cancer progression. Clin. Cancer Res. 2008, 14, 1002–1014. [Google Scholar] [CrossRef] [Green Version]
- Bateman, N.W.; Sun, M.; Bhargava, R.; Hood, B.L.; Darfler, M.M.; Kovatich, A.J.; Hooke, J.A.; Krizman, D.B.; Conrads, T.P. Differential proteomic analysis of late-stage and recurrent breast cancer from formalin-fixed paraffin-embedded tissues. J. Proteome Res. 2001, 10, 1323–1332. [Google Scholar] [CrossRef]
- Drummond, E.S.; Nayak, S.; Ueberheide, B.; Wisniewski, T. Proteomic analysis of neurons microdissected from formalin-fixed, paraffin-embedded Alzheimer’s disease brain tissue. Sci. Rep. 2015, 5, 15456. [Google Scholar] [CrossRef] [Green Version]
- Griesser, E.; Wyatt, H.; Ten Have, S.; Stierstorfer, B.; Lenter, M.; Lamond, A.I. Quantitative profiling of the human substantia nigra proteome from laser-capture microdissected FFPE tissue. Mol. Cell. Proteom. 2020, 19, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Moggridge, S.; Sorenson, P.H.; Morin, G.B.; Hughes, C.S. Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. J. Proteome Res. 2018, 4, 1730–1740. [Google Scholar] [CrossRef]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.A.W.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Asleh, K.; Negri, G.L.; Miko, S.E.S.; Colborne, S.; Hughes, C.S.; Wang, X.Q.; Goa, D.; Gilks, C.B.; Chia, S.K.L.; Nielsen, T.O.; et al. Proteomic analysis of archival breast cancer clinical specimens identifies biological subtypes with distinct survival outcomes. Nat. Commun. 2022, 13, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Kittaneh, M.; Montero, A.J.; Gluck, S. Molecular profiling for breast cancer: A comprehensive review. Biomark. Cancer 2013, 5, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Bao, C.; Huang, L.; Wei, J.-F. Current therapeutic strategies for metastatic triple-negative breast cancer: From pharmacists’ perspective. J. Clin. Med. 2022, 11, 6021. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.R.; Key, M.E.; Kalra, K.L. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: An enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J. Histochem. Cytochem. 1991, 39, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Liao, H.-Y.; Zhang, H.-H. Roles of MET in human cancer. Clin. Chem. Acta 2022, 525, 69–83. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Liao, W.-L.; Thyparambil, S.; Henderson, L.; Xu, P.; Zhao, L.; Rambo, B.; Hart, J.; Xiao, S.-Y.; Bengali, K.; et al. Absolute quantitation of Met using mass spectrometry for clinical application: Assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PLoS ONE 2014, 9, e100586. [Google Scholar] [CrossRef]
- Do, M.; Kim, H.; Yeo, I.; Lee, J.; Park, I.A.; Ryu, H.S.; Kim, Y. Clinical application of multiple reaction monitoring-mass spectrometry to human epidermal growth factor receptor 2 measurements as a potential diagnostic tool for breast cancer therapy. Clin. Chem. 2020, 66, 1339–1348. [Google Scholar] [CrossRef]
- Steiner, C.; Lescuyer, P.; Tille, J.-C.; Cutler, P.; Ducret, A. Development of a highly multiplexed SRM assay for biomarker discovery in formalin-fixed paraffin-embedded tissues. Methods Mol. Biol. 2019, 1959, 185–203. [Google Scholar] [CrossRef]
- Maes, E.; Broeckx, V.; Mertens, I.; Sagaert, X.; Prenen, H.; Landuyt, B.; Schoofs, L. Analysis of the formalin-fixed paraffin-embedded tissue proteome: Pitfalls, challenges, and future prospectives. Amino Acids 2013, 45, 205–218. [Google Scholar] [CrossRef]
- Tanca, A.; Pagnozzi, D.; Burrai, G.P.; Polinas, M.; Uzzau, S.; Antuofermo, E.; Addis, M.F. Comparability of differential proteomics data generated from paired archival fresh-frozen and formalin-fixed samples by GeLC-MS/MS and spectral counting. J. Proteom. 2012, 77, 561–576. [Google Scholar] [CrossRef]
- Craven, R.A.; Cairns, D.A.; Zougman, A.; Harnden, P.; Selby, P.J.; Banks, R.E. Proteomic analysis of formalin-fixed paraffin-embedded renal tissue samples by label-free MS: Assessment of overall technical variability and the impact of block age. Proteom. Clin. Appl. 2013, 7, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Piehowski, P.D.; Petyuk, V.A.; Sontag, R.L.; Gritsenko, M.A.; Weitz, K.K.; Fillmore, T.L.; Moon, J.; Makhlouf, H.; Chuaqui, R.F.; Boja, E.S.; et al. Residual tissue repositories as a resource for population-based cancer proteomic studies. Clin. Proteom. 2018, 15, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coscia, F.; Doll, S.; Bech, J.M.; Schweizer, L.; Mund, A.; Lengyel, E.; Lindebjerg, J.; Madsen, G.I.; Moreira, J.M.; Mann, M. A streamlined mass spectrometry-based proteomics workflow for large-scale FFPE tissue analysis. J. Pathol. 2020, 251, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossouw, S.C.; Bendou, H.; Blignaut, R.J.; Bell, L.; Rigby, J.; Christoffels, A. Evaluation of protein purification techniques and effects of storage duration on LC-MS/MS analysis of archived FFPE human CRC tissues. Pathol. Oncol. Res. 2021, 27, 622855. [Google Scholar] [CrossRef]
- Metz, B.; Kersten, G.F.; Baart, G.J.; de Jong, A.; Meiring, H.; ten Hove, J.; van Steenbergen, M.J.; Hennink, W.E.; Crommelin, D.J.; Jiskoot, W. Identification of formaldehyde-induced modifications in proteins: Reactions with insulin. Bioconjugate Chem. 2006, 17, 815–822. [Google Scholar] [CrossRef]
- Bauden, M.; Kristl, T.; Andersson, R.; Marko-Varga, G.; Ansari, D. Characterization of histone-related chemical modifications in formalin-fixed paraffin-embedded and fresh-frozen human pancreatic cancer xenografts using LC-MS/MS. Lab. Investig. 2017, 97, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Neumeister, V.M.; Anagnostou, V.; Siddiqui, S.; England, A.M.; Zarrella, E.R.; Vassilakopoulou, M.; Parisi, F.; Kluger, Y.; Hicks, D.G.; Rimm, D.L. Quantitative assessment of effect of preanalytic cold ischemic time on protein expression in breast cancer tissue. J. Natl. Cancer Inst. 2012, 104, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.Y.; Lee, S.J.; Kris, Y.; Braunschweig, T.; Traicoff, J.L.; Hewitt, S.M. A well-based reverse-phase protein array applicable to extracts from formalin-fixed paraffing-embedded tissue. Proteom. Clin. Appl. 2008, 2, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Gaffney, E.F.; Riegman, P.H.; Grizzle, W.E.; Watson, P.H. Factors that drive the increasing use of FFPE tissue in basic and translational cancer research. Biotech. Histochem. 2018, 93, 373–386. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Number of Peptides Identified | ||
|---|---|---|
| Protein | Frozen Tissue | FFPE Tissue |
| Carbamoyl-phosphate synthase | 91 | 72 |
| 78 kDa Glucose-regulated protein | 21 | 33 |
| ATP synthase β chain | 34 | 32 |
| 60 kDa Heat shock protein | 33 | 32 |
| 10-Formyltetrahydrofolate dehydrogenase | 41 | 32 |
| Catalase | 12 | 30 |
| HMG-CoA synthase | 23 | 29 |
| Acetyl-CoA acyltransferase | 27 | 27 |
| Glutathione-S-transferase | 21 | 25 |
| Pyruvate carboxylase | 21 | 24 |
| Tissue Type | Purpose | Extraction Conditions | Analysis Method | Ref |
|---|---|---|---|---|
| Prostate cancer and BPH | Method development | Liquid-Tissue ID and Liquid-Tissue MS (proprietary) | RP-HPLC ion trap | [20] |
| Prostate cancer | Method development | Samples suspended in 30% ACN, 100 mM NH4HCO3 buffer and boiled for 10 min. | RP-HPLC ion trap | [19] |
| Prostate cancer | Biomarker discovery | Homogenized in 4% SDS, 100 mM DTE, 100 mM TrisHCl pH 7.6. Sonicated 3 times for 10 s and 1 h of heating at 90 °C. | RP-HPLC ion trap | [47] |
| Glioblastoma | Proteome characterization | 8 M urea and 20 mM Tris-HCl at pH 8.0 | cIEF/RP-HPLC ion trap | [48] |
| Mouse Liver | Method development | 1. 40 mM Tris pH 8.2/6 M guanidine-HCl/65 mM DTT, centrifugation 25,000× g 1 h. 2. 40 mM Tris pH 8.2/2% SDS, 100 °C 20 min, 60 °C 2 h. 3. 40 mM Tris pH 8.2/6 M guanidine-HCl/65 mM DTT. 4. 40 mM Tris pH 8.2/6 M guanidine-HCl/ 65 mM DTT, 100 °C 30 min. | RP-HPLC ion trap | [49] |
| Prostate cancer and diffuse large B-cell lymphoma | Tumor stratification | 6 M Urea, 2 M thiourea, 5 mM Na2EDTA in 100 mM NH4HCO3, pH 8.8. Lysed using pressure cycling at 45,000 psi. | RP-HPLC-SWATH | [50] |
| Adrenal cortical carcinoma | Biomarker discovery | 4% SDS, 1 mM TCEP, and 0.3 M Tris pH 8.0. After sonication, the samples were incubated at 95 °C. | RP-HPLC-Orbitrap | [51] |
| Kidney | Renal allograft injury biomarkers | 20 mm Tris, 2% SDS, pH 8.0. Sheared using 18- and 23-gauge needles followed by ultrasonication and heating at 98 °C. | RP-HPLC-Orbitrap | [52] |
| Colorectal cancer | Methods comparison | FASP Kit (proprietary; Expedeon) vs. 10 mM NH4HCO3, pH 6.0 and heated at 95 °C for 1 h. | UPLC-qTOF | [53] |
| Testicular tissue | Azoospermia subtypes biomarkers | 4% SDS, 5 mM MgCl2x6H2O, 10 mM CHAPS, 100 mM NH4HCO3, 0.5 M DTT. Followed by sonication and incubation at 95 °C. | RP-HPLC qTOF with ion mobility | [54] |
| HNSCC | Diagnostic and prognostic biomarkers of HNSCC | Liquid Tissue (proprietary) | RP-HPLC Ion Trap | [55] |
| Breast cancer | Biomarkers | Liquid Tissue (proprietary) | RP-HPLC Ion Trap | [56] |
| Temporal cortex neurons | AD-associated proteins | 20 mM DTT at 57 °C for 1 h followed by 50 mM at room temperature. Method performed with and without RapiGest surfactant | RP-HPLC-Q-Exactive | [57] |
| Substantia nigra | Proteome characterization | 2% SDS in 300 mM Tris-HCl pH 8.0, or 1% SDC in 300 mM Tris-HCl pH 8.5 or 0.2% Rapigest (proprietary) in 50 mM NH4HCO3. Heated at 99 °C and sonicated. | RP-HPLC-Q-Exactive | [58] |
| Cluster | Number of Samples | RFS/OS Rank | Characteristics |
|---|---|---|---|
| 1 | 34 | 2 | Luminal B (n = 18) Her2-enriched (n = 13) |
| 2 | 50 | 4 | Basal-like (n = 41) Her2-enriched (few) |
| 3 | 47 | 1 | Basal-like (n = 31) Her2-enriched (n = 14) |
| 4 | 43 | 3 | Her2-enriched (n = 26) Luminal A (n = 8) Luminal B (n = 8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obi, E.N.; Tellock, D.A.; Thomas, G.J.; Veenstra, T.D. Biomarker Analysis of Formalin-Fixed Paraffin-Embedded Clinical Tissues Using Proteomics. Biomolecules 2023, 13, 96. https://doi.org/10.3390/biom13010096
Obi EN, Tellock DA, Thomas GJ, Veenstra TD. Biomarker Analysis of Formalin-Fixed Paraffin-Embedded Clinical Tissues Using Proteomics. Biomolecules. 2023; 13(1):96. https://doi.org/10.3390/biom13010096
Chicago/Turabian StyleObi, Ekenedirichukwu N., Daniel A. Tellock, Gabriel J. Thomas, and Timothy D. Veenstra. 2023. "Biomarker Analysis of Formalin-Fixed Paraffin-Embedded Clinical Tissues Using Proteomics" Biomolecules 13, no. 1: 96. https://doi.org/10.3390/biom13010096