
Metabolic Maturation Exaggerates Abnormal Calcium Handling in a Lamp2 Knockout Human Pluripotent Stem Cell-Derived Cardiomyocyte Model of Danon Disease
 and
and 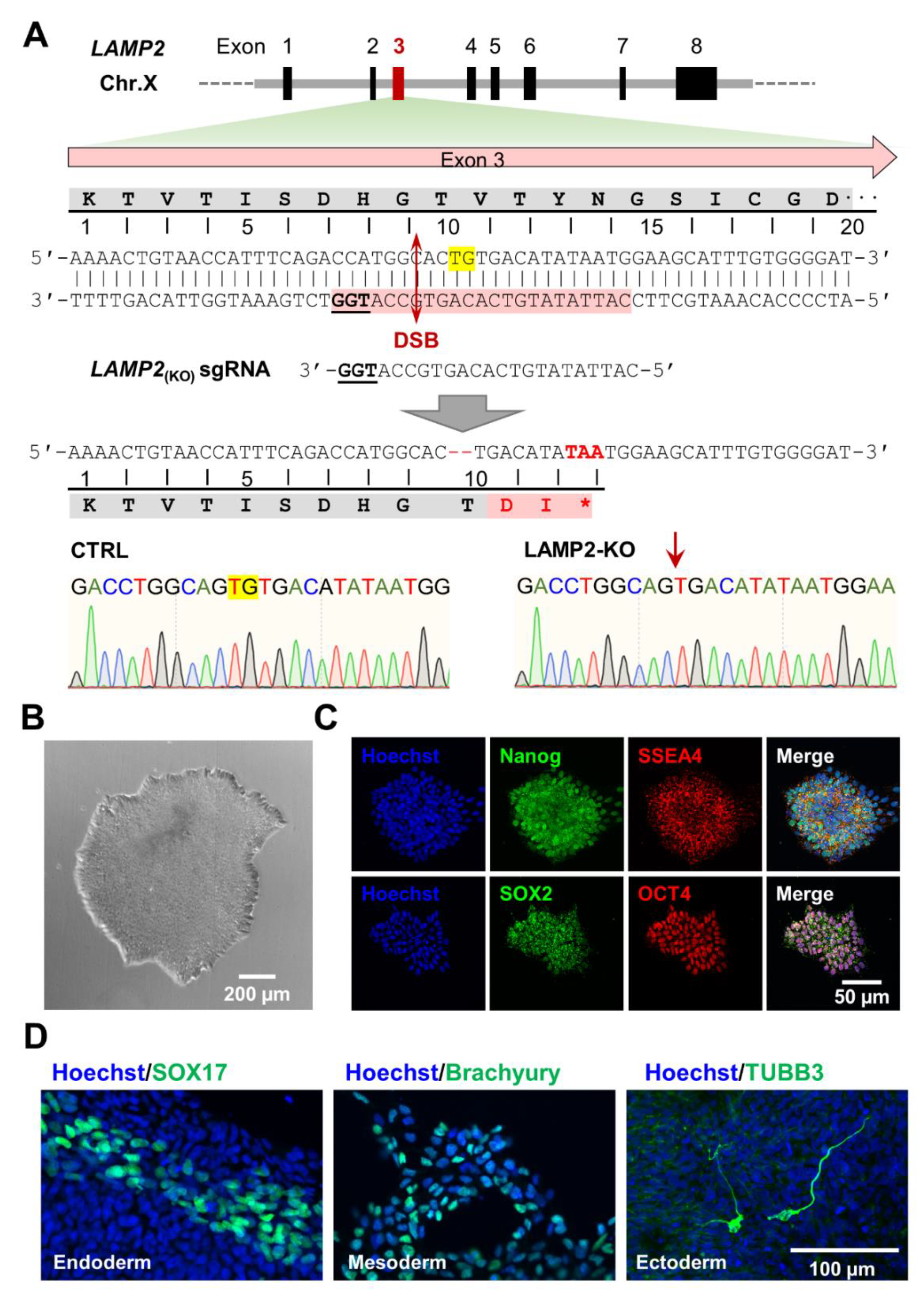{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Maintenance and Differentiation of Human iPSCs
2.2. Metabolic Maturation of iPSC-CMs In Vitro
2.3. Knockout of LAMP2 Using CRISPR/Cas9
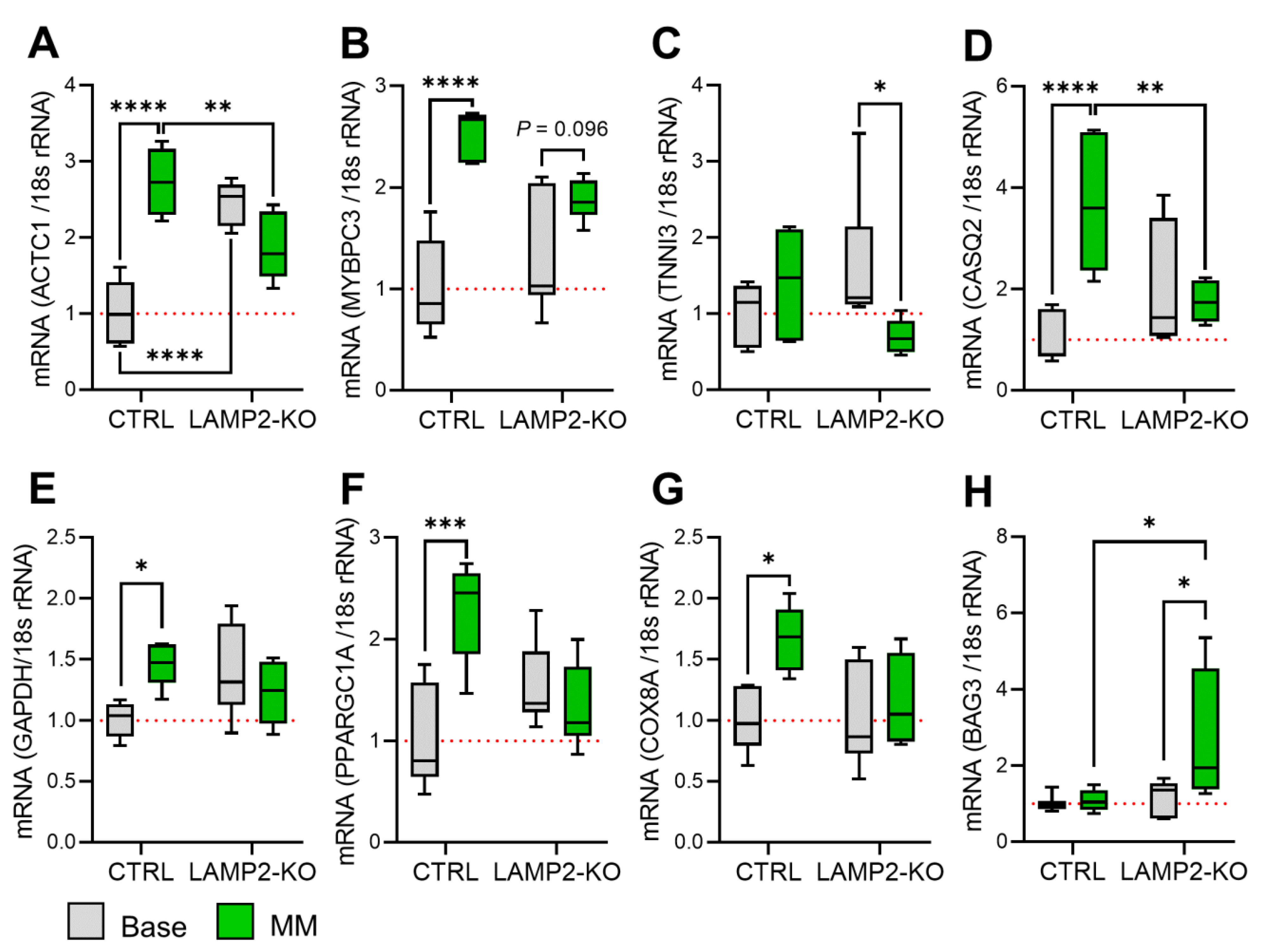
2.4. RNA Extraction of mRNA Expression Quantification by qPCR
2.5. Protein Extraction and Western Blot Analysis
2.6. Immunofluorescence Labeling of Fixed Samples
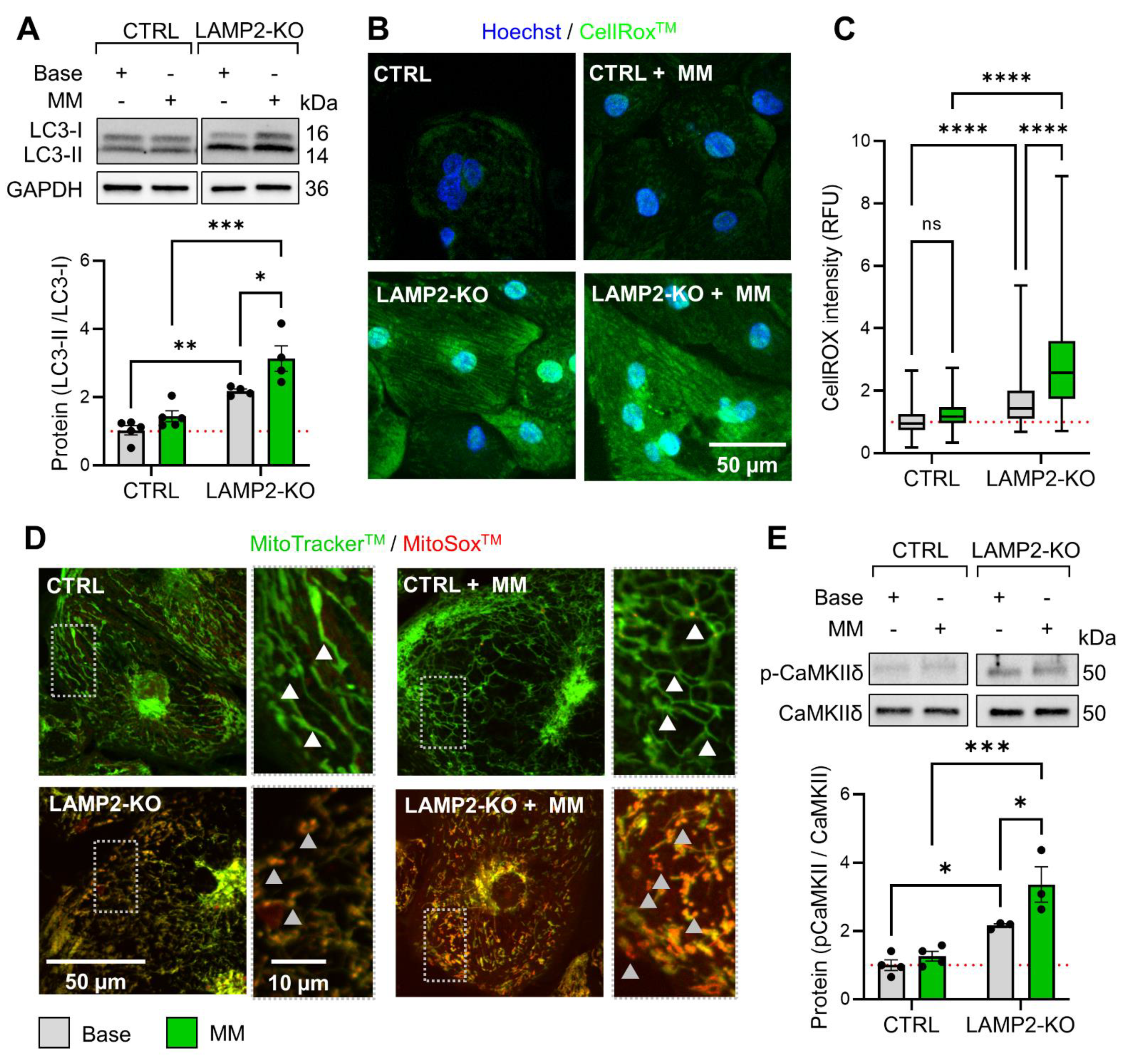
2.7. Measurement of Cytosol and Mitochondria Reactive Oxygen Species (ROS)
2.8. Confocal Imaging and Image Analysis
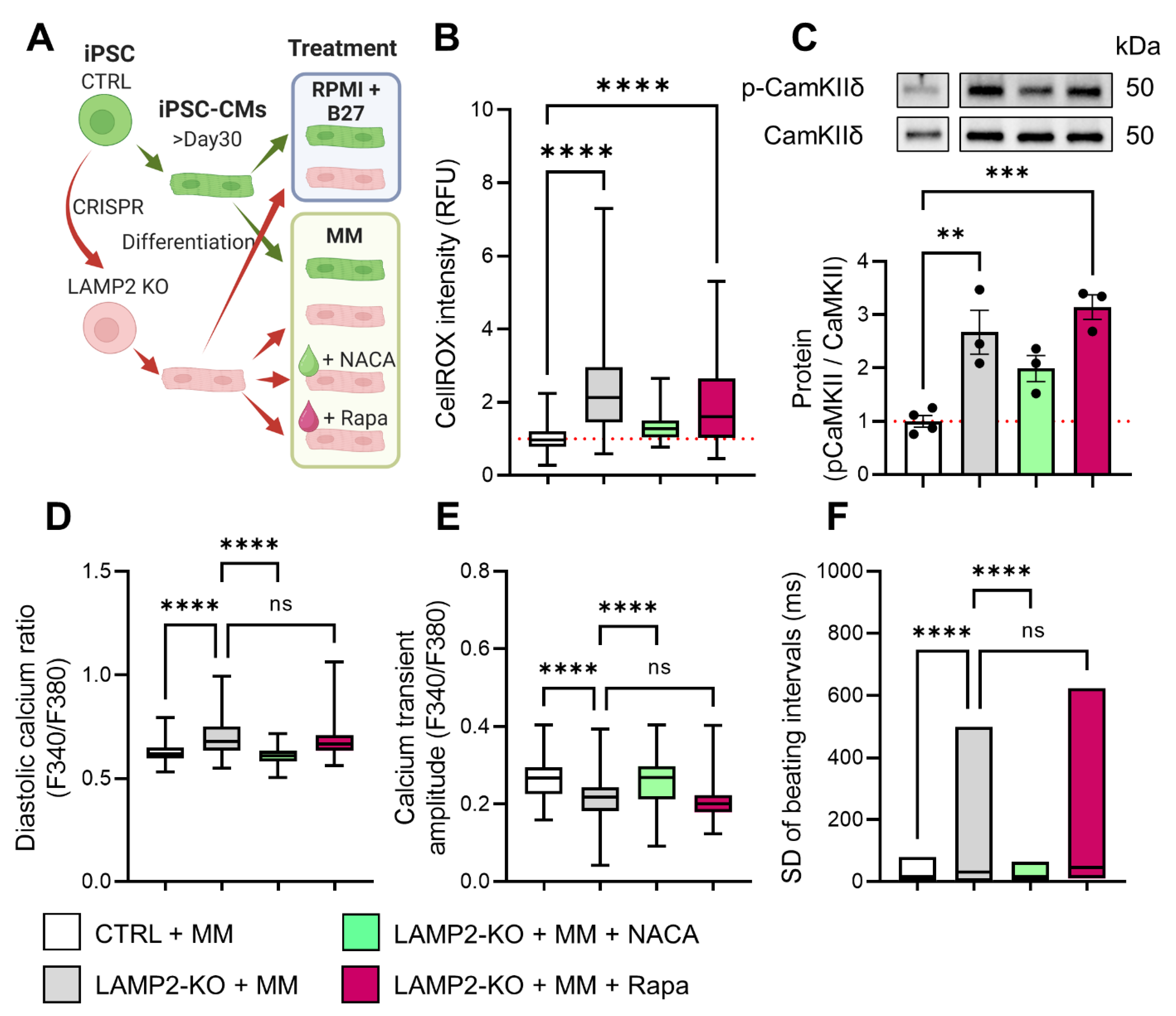
2.9. Calcium Imaging and Data Analysis
2.10. Plots and Statistical Analysis
3. Results
3.1. Generation of Hemizygous LAMP2y/- Knockout Human iPSCs
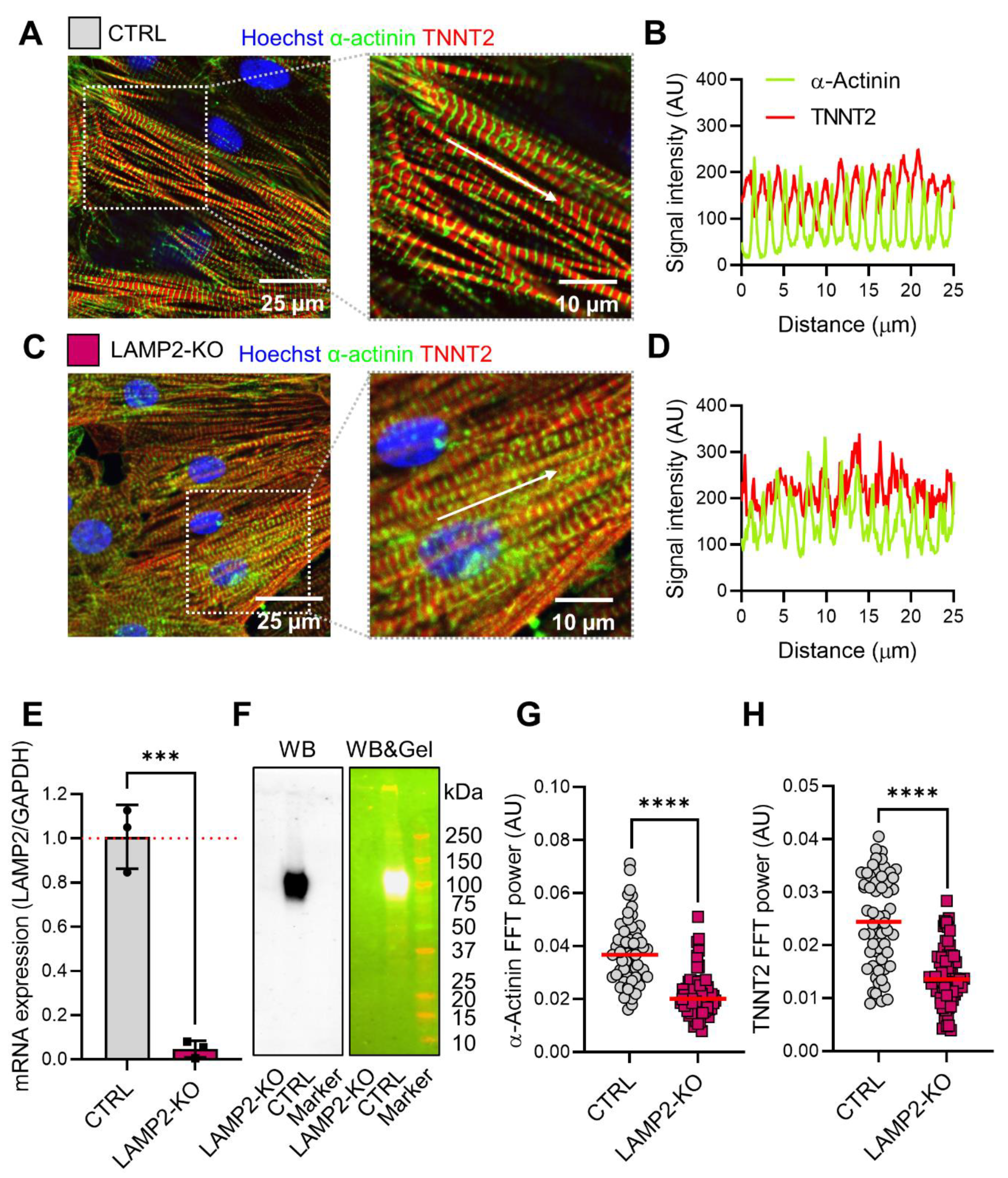
3.2. LAMP2 Knockout Led to Morphological Remodeling of iPSC-Derived Cardiomyocytes
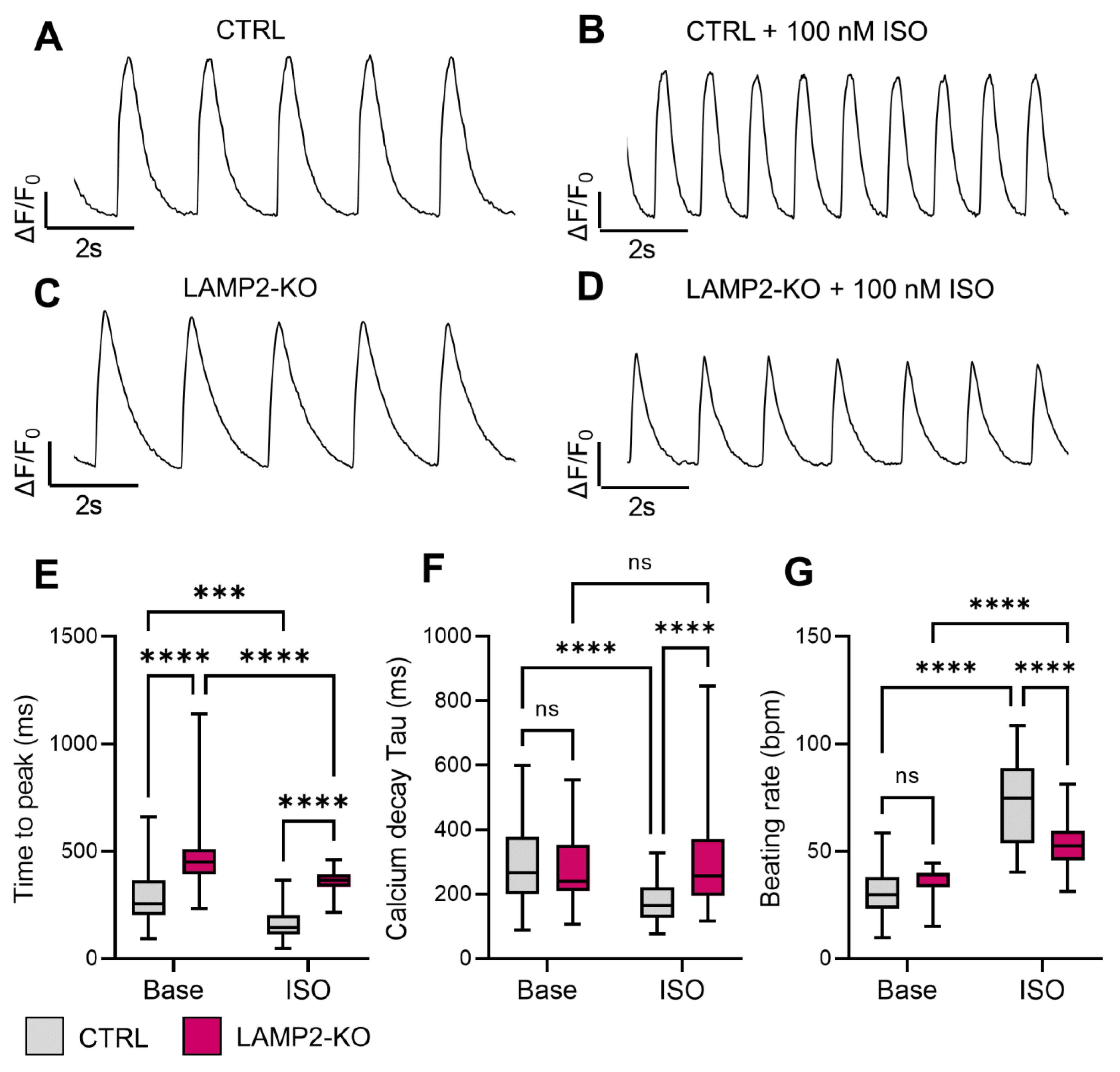
3.3. Blunted Responses of Calcium Handling Regulation to β-Adrenergic Stimulation in LAMP2 KO iPSC-CMs
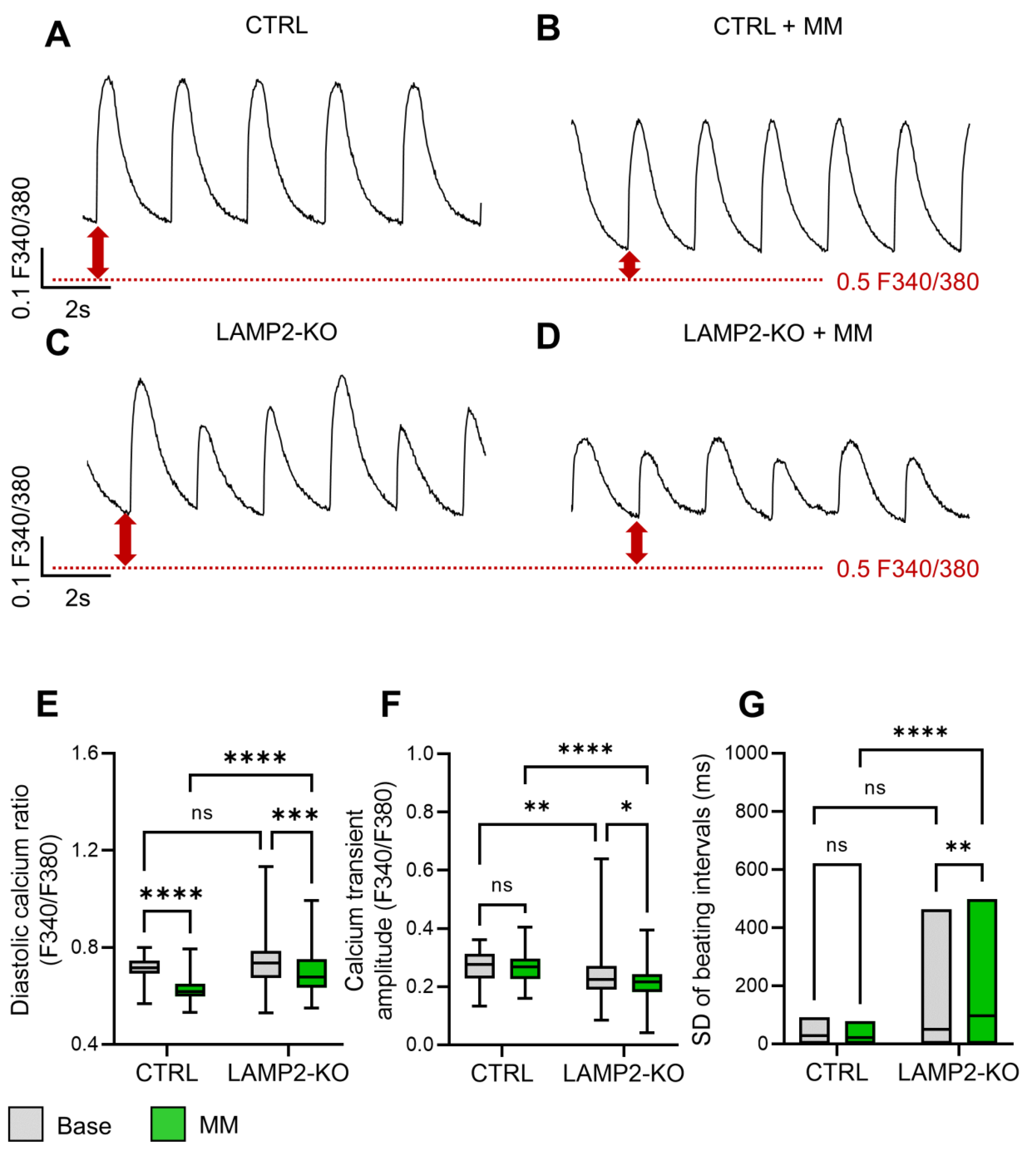
3.4. Metabolic Maturation Medium Exaggerates the Calcium Handling Abnormalities in LAMP2 KO iPSC-CMs
3.5. Metabolic Maturation Promotes ROS Overload and CaMKII Activation in LAMP2 KO CMs
3.6. ROS Scavenger Restored Calcium Homeostasis in LAMP2 KO Cells
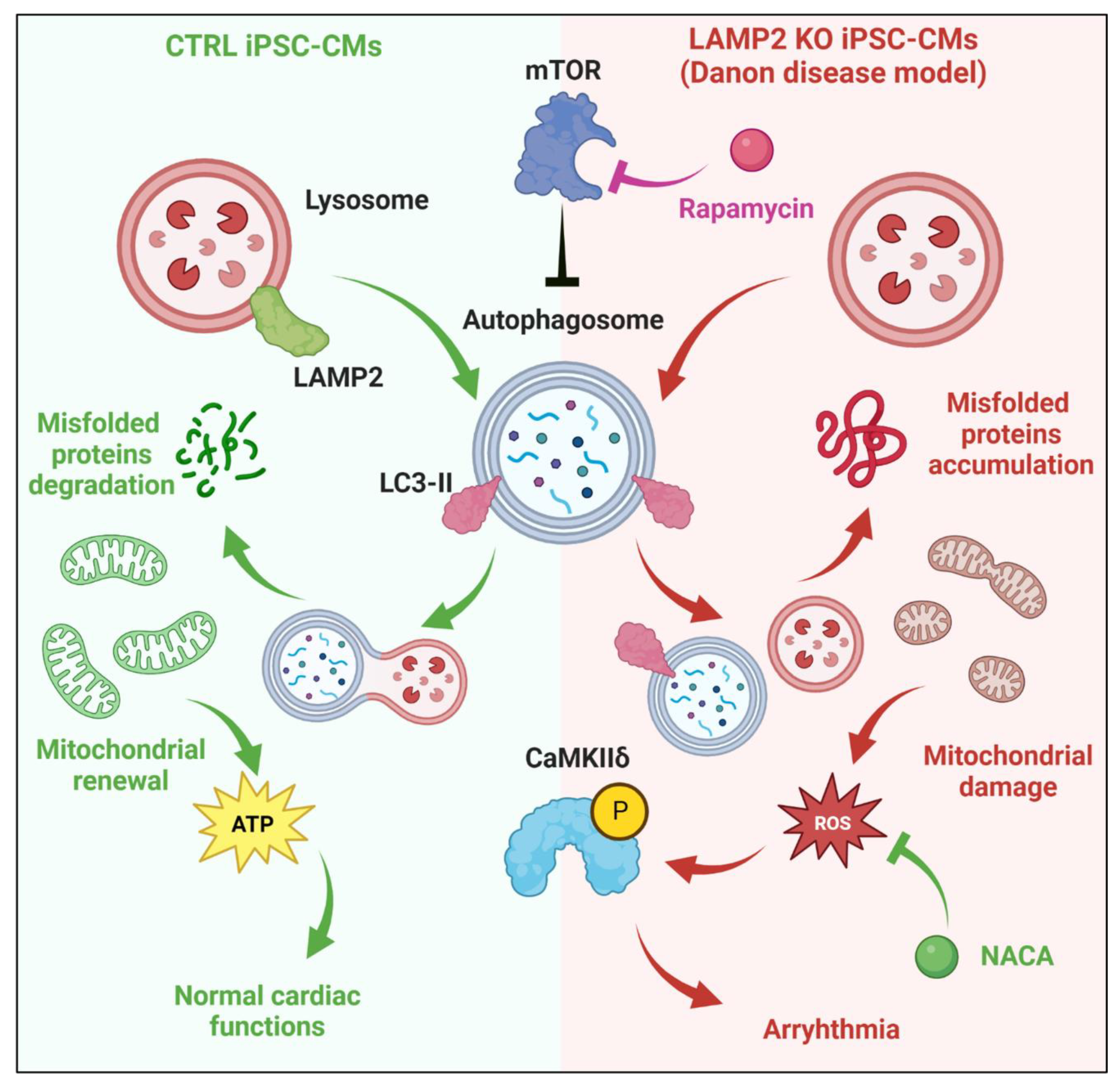
4. Discussion
4.1. Functional Maturation of iPSC-CMs and Modeling of DD
4.2. Cellular Mechanism of Arrhythmia in DD Cardiomyocytes
4.3. Outlook for the Future Treatment of DD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cenacchi, G.; Papa, V.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Review: Danon disease: Review of natural history and recent advances. Neuropathol. Appl. Neurobiol. 2020, 46, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Danon, M.J.; Oh, S.J.; DiMauro, S.; Manaligod, J.R.; Eastwood, A.; Naidu, S.; Schliselfeld, L.H. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981, 31, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Miao, J.; Peng, Y.; Wang, Y.; Dong, J.; Zhao, X. Clinical features of Danon disease and insights gained from LAMP-2 deficiency models. Trends Cardiovasc. Med. 2021, S1050–S1738, 00127-4. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; Von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Beertsen, W.; Eskelinen, E.-L. LAMP-2: A control step for phagosome and autophagosome maturation. Autophagy 2008, 4, 510–512. [Google Scholar] [CrossRef] [Green Version]
- Rowland, T.J.; Sweet, M.E.; Mestroni, L.; Taylor, M.R.G. Danon disease—Dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J. Cell Sci. 2016, 129, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Roberts, W.C.; Arad, M.; Haas, T.S.; Spirito, P.; Wright, G.B.; Almquist, A.K.; Baffa, J.M.; Saul, J.P.; Ho, C.Y.; et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 2009, 301, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; McMahon, C.J.; Smith, L.R.; Bersola, J.; Adesina, A.M.; Breinholt, J.P.; Kearney, D.L.; Dreyer, W.J.; Denfield, S.W.; Price, J.F.; et al. Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation 2005, 112, 1612–1617. [Google Scholar] [CrossRef] [Green Version]
- Brambatti, M.; Caspi, O.; Maolo, A.; Koshi, E.; Greenberg, B.; Taylor, M.R.; Adler, E.D. Danon disease: Gender differences in presentation and outcomes. Int. J. Cardiol. 2019, 286, 92–98. [Google Scholar] [CrossRef]
- Endo, Y.; Furuta, A.; Nishino, I. Danon disease: A phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 2015, 129, 391–398. [Google Scholar] [CrossRef]
- D’Souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R. Danon disease: Clinical features, evaluation, and management. Circ. Heart Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Dvornikov, A.V.; Wang, M.; Yang, J.; Zhu, P.; Le, T.; Lin, X.; Cao, H.; Xu, X. Phenotyping an adult zebrafish lamp2 cardiomyopathy model identifies mTOR inhibition as a candidate therapy. J. Mol. Cell Cardiol. 2019, 133, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Cai, W.; Shi, Y.; Zhou, X.; Guo, G.; Guo, C.; Huang, X.; Han, Z.; Zhang, S.; et al. A Critical Evaluation of Liver Pathology in Humans with Danon Disease and Experimental Correlates in a Rat Model of LAMP-2 Deficiency. Clin. Rev. Allergy Immunol. 2017, 53, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Tanaka, Y.; Lüllmann-Rauch, R.; von Figura, K. Disease model: LAMP-2 enlightens Danon disease. Trends Mol. Med. 2001, 7, 37–39. [Google Scholar] [CrossRef]
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell. Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef]
- Hashem, S.I.; Perry, C.N.; Bauer, M.; Han, S.; Clegg, S.D.; Ouyang, K.; Deacon, D.C.; Spinharney, M.; Panopoulos, A.D.; Izpisua Belmonte, J.C.; et al. Oxidative Stress Mediates Cardiomyocyte Apoptosis in a Human Model of Danon Disease and Heart Failure. Stem Cells 2015, 33, 2343–2350. [Google Scholar] [CrossRef] [Green Version]
- Del Favero, G.; Bonifacio, A.; Rowland, T.J.; Gao, S.; Song, K.; Sergo, V.; Adler, E.D.; Mestroni, L.; Sbaizero, O.; Taylor, M.R.G. Danon Disease-Associated LAMP-2 Deficiency Drives Metabolic Signature Indicative of Mitochondrial Aging and Fibrosis in Cardiac Tissue and hiPSC-Derived Cardiomyocytes. J. Clin. Med. 2020, 9, 2457. [Google Scholar] [CrossRef]
- Vučković, S.; Dinani, R.; Nollet, E.E.; Kuster, D.W.D.; Buikema, J.W.; Houtkooper, R.H.; Nabben, M.; van der Velden, J.; Goversen, B. Characterization of cardiac metabolism in iPSC-derived cardiomyocytes: Lessons from maturation and disease modeling. Stem Cell Res. Ther. 2022, 13, 332. [Google Scholar] [CrossRef]
- Lundy, S.D.; Zhu, W.Z.; Regnier, M.; Laflamme, M.A. Structural and Functional Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef] [PubMed]
- Miki, K.; Deguchi, K.; Nakanishi-Koakutsu, M.; Lucena-Cacace, A.; Kondo, S.; Fujiwara, Y.; Hatani, T.; Sasaki, M.; Naka, Y.; Okubo, C.; et al. ERRγ enhances cardiac maturation with T-tubule formation in human iPSC-derived cardiomyocytes. Nat. Commun. 2021, 12, 3596. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barndt, R.J.; Ma, N.; Tang, Y.; Haugh, M.P.; Alamri, L.S.; Chan, S.Y.; Wu, H. Modeling of dilated cardiomyopathy by establishment of isogenic human iPSC lines carrying phospholamban C25T (R9C) mutation (UPITTi002-A-1) using CRISPR/Cas9 editing. Stem Cell Res. 2021, 56, 102544. [Google Scholar] [CrossRef]
- Wei, S.; Guo, A.; Chen, B.; Kutschke, W.; Xie, Y.-P.; Zimmerman, K.; Weiss, R.M.; Anderson, M.E.; Cheng, H.; Song, L.-S. T-Tubule Remodeling During Transition From Hypertrophy to Heart Failure. Circ. Res. 2010, 107, 520–531. [Google Scholar] [CrossRef] [Green Version]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef]
- Wu, H.; Yang, H.; Rhee, J.-W.; Zhang, J.Z.; Lam, C.K.; Sallam, K.; Chang, A.C.Y.; Ma, N.; Lee, J.; Zhang, H.; et al. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Hear. J. 2019, 40, 3685–3695. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Lee, J.; Vincent, L.G.; Wang, Q.; Gu, M.; Lan, F.; Churko, J.M.; Sallam, K.I.; Matsa, E.; Sharma, A.; et al. Epigenetic Regulation of Phosphodiesterases 2A and 3A Underlies Compromised β-Adrenergic Signaling in an iPSC Model of Dilated Cardiomyopathy. Cell Stem Cell 2015, 17, 89–100. [Google Scholar] [CrossRef]
- Konrad, T.; Sonnenschein, S.; Schmidt, F.P.; Mollnau, H.; Bock, K.; Ocete, B.Q.; Münzel, T.; Theis, C.; Rostock, T. Cardiac arrhythmias in patients with Danon disease. Europace 2016, 19, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Miani, D.; Taylor, M.; Mestroni, L.; D’Aurizio, F.; Finato, N.; Fanin, M.; Brigido, S.; Proclemer, A. Sudden Death Associated With Danon Disease in Women. Am. J. Cardiol. 2012, 109, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on IK1. Pharmacol. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.; Couch, L.; Terracciano, C.M. Excitation-contraction coupling of human induced pluripotent stem cell-derived cardiomyocytes. Front. Cell Dev. Biol. 2015, 3, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedada, F.B.; Wheelwright, M.; Metzger, J.M. Maturation status of sarcomere structure and function in human iPSC-derived cardiac myocytes. Biochim. Biophys. Acta. 2016, 1863, 1829–1838. [Google Scholar] [CrossRef] [Green Version]
- Machiraju, P.; Greenway, S.C. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J. Stem Cells 2019, 11, 33–43. [Google Scholar] [CrossRef]
- Shin, Y.J.; Cho, D.Y.; Chung, T.Y.; Han, S.B.; Hyon, J.Y.; Wee, W.R. Rapamycin reduces reactive oxygen species in cultured human corneal endothelial cells. Curr. Eye Res. 2011, 36, 1116–1122. [Google Scholar] [CrossRef]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox. Signal 2011, 15, 1679–1690. [Google Scholar] [CrossRef]
- Mrschtik, M.; Ryan, K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015, 282, 1858–1870. [Google Scholar] [CrossRef]
- Konecki, D.S.; Foetisch, K.; Zimmer, K.P.; Schlotter, M.; Konecki, U.L. An Alternatively Spliced Form of the Human Lysosome-Associated Membrane Protein-2 Gene Is Expressed in a Tissue-Specific Manner. Biochem. Biophys. Res. Commun. 1995, 215, 757–767. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Cuervo, A.M.; Taylor, M.R.; Nishino, I.; Blum, J.S.; Dice, J.F.; Sandoval, I.V.; Lippincott-Schwartz, J.; August, J.T.; Saftig, P. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic 2005, 6, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, Y.; Hase, K.; Wada, K.; Kabuta, T. An RNautophagy/DNautophagy receptor, LAMP2C, possesses an arginine-rich motif that mediates RNA/DNA-binding. Biochem. Biophys. Res. Commun. 2015, 460, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hase, K.; Fujiwara, Y.; Kikuchi, H.; Aizawa, S.; Hakuno, F.; Takahashi, S.-I.; Wada, K.; Kabuta, T. RNautophagy/DNautophagy possesses selectivity for RNA/DNA substrates. Nucleic Acids Res. 2015, 43, 6439–6449. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, L.; Liu, X.; Dai, Q. A novel LAMP2 p.G93R mutation associated with mild Danon disease presenting with familial hypertrophic cardiomyopathy. Mol. Genet. Genomic Med. 2019, 7, e00941. [Google Scholar] [CrossRef] [Green Version]
- Csányi, B.; Popoiu, A.; Hategan, L.; Hegedűs, Z.; Nagy, V.; Rácz, K.; Hőgye, M.; Sághy, L.; Iványi, B.; Csanády, M.; et al. Identification of Two Novel LAMP2 Gene Mutations in Danon Disease. Can. J. Cardiol. 2016, 32, 1355.e1323–1355.e1330. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; van der Meer, P.; Serra, M.; et al. Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes by Inhibition of HIF1α and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Rog-Zielinska, E.A.; Craig, M.A.; Manning, J.R.; Richardson, R.V.; Gowans, G.J.; Dunbar, D.R.; Gharbi, K.; Kenyon, C.J.; Holmes, M.C.; Hardie, D.G.; et al. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: A role for PGC-1α. Cell Death Differ. 2015, 22, 1106–1116. [Google Scholar] [CrossRef]
- Yoshida, S.; Miyagawa, S.; Fukushima, S.; Kawamura, T.; Kashiyama, N.; Ohashi, F.; Toyofuku, T.; Toda, K.; Sawa, Y. Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes by Soluble Factors from Human Mesenchymal Stem Cells. Mol. Ther. 2018, 26, 2681–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, T.; Gomez, I.G.; Ren, S.; Hudkins, K.; Roach, A.; Alpers, C.E.; Shankland, S.J.; D’Agati, V.D.; Duffield, J.S. Deficient Autophagy Results in Mitochondrial Dysfunction and FSGS. J. Am. Soc. Nephrol. 2015, 26, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balderas-Villalobos, J.; Molina-Muñoz, T.; Mailloux-Salinas, P.; Bravo, G.; Carvajal, K.; Gómez-Viquez, N.L. Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome. Am. J. Physiol. Circ. Physiol. 2013, 305, H1344–H1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafstad, A.D.; Nabeebaccus, A.A.; Shah, A.M. Novel aspects of ROS signalling in heart failure. Basic Res. Cardiol. 2013, 108, 359. [Google Scholar] [CrossRef]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.R.G.; Adler, E.D. Danon Disease. In GeneReviews(®) [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993–2022. [Google Scholar] [PubMed]
- Manso, A.M.; Hashem, S.I.; Nelson, B.C.; Gault, E.; Soto-Hermida, A.; Villarruel, E.; Brambatti, M.; Bogomolovas, J.; Bushway, P.J.; Chen, C.; et al. Systemic AAV9.LAMP2B injection reverses metabolic and physiologic multiorgan dysfunction in a murine model of Danon disease. Sci. Transl. Med. 2020, 12, eaax1744. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Ng, K.M.; Mok, P.Y.; Butler, A.W.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Lai, W.H.; Au, K.W.; Lau, Y.M.; Wong, L.Y.; et al. Amelioration of X-Linked Related Autophagy Failure in Danon Disease With DNA Methylation Inhibitor. Circulation 2016, 134, 1373–1389. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barndt, R.J.; Liu, Q.; Tang, Y.; Haugh, M.P.; Cui, J.; Chan, S.Y.; Wu, H. Metabolic Maturation Exaggerates Abnormal Calcium Handling in a Lamp2 Knockout Human Pluripotent Stem Cell-Derived Cardiomyocyte Model of Danon Disease. Biomolecules 2023, 13, 69. https://doi.org/10.3390/biom13010069
Barndt RJ, Liu Q, Tang Y, Haugh MP, Cui J, Chan SY, Wu H. Metabolic Maturation Exaggerates Abnormal Calcium Handling in a Lamp2 Knockout Human Pluripotent Stem Cell-Derived Cardiomyocyte Model of Danon Disease. Biomolecules. 2023; 13(1):69. https://doi.org/10.3390/biom13010069
Chicago/Turabian StyleBarndt, Robert J., Qing Liu, Ying Tang, Michael P. Haugh, Jeffery Cui, Stephen Y. Chan, and Haodi Wu. 2023. "Metabolic Maturation Exaggerates Abnormal Calcium Handling in a Lamp2 Knockout Human Pluripotent Stem Cell-Derived Cardiomyocyte Model of Danon Disease" Biomolecules 13, no. 1: 69. https://doi.org/10.3390/biom13010069