
Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection

Abstract
:1. Introduction
2. HIF-1
3. Mitochondria
4. Regulation of HIF-1 on Mitochondrial Structure and Function
4.1. Effects of HIF-1 on Mitochondrial Morphology and Structure
4.1.1. HIF-1 and Mitochondrial Population
4.1.2. HIF-1 and Mitochondrial Distribution
4.1.3. HIF-1 and Mitochondrial Structure
4.2. Effects of HIF-1 on Mitochondrial Function
4.2.1. HIF-1 and Mitochondrial Respiratory Chain
4.2.2. HIF-1 and TCA Cycle
4.2.3. HIF-1 and Oxidative Phosphorylation
4.2.4. HIF-1 and Mitochondrial Membrane Potential
5. The Effects of Mitochondria on HIF-1 Activity
5.1. Mitochondrial Metabolic Enzymes and HIF-1 Activity
5.2. Mitochondrial Respiratory Chain and HIF-1 Stability
5.3. Uncoupling Proteins and HIF-1 Activity
5.4. Mitochondrial ATP-Sensitive Potassium Channels and HIF-1 Activity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef] [PubMed]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Kondo, D.; Kim, J.; Lyoo, I.K.; Yurgelun-Todd, D.; Hwang, J.; Renshaw, P.F. Elevating the level of hypoxia inducible factor may be a new potential target for the treatment of depression. Med. Hypotheses 2021, 146, 110398. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Signorile, A.; Roca, E.; Papa, S. cAMP response element-binding protein (CREB) is imported into mitochondria and promotes protein synthesis. FEBS J. 2009, 276, 4325–4333. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Billings, F.T., 4th; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ. Res. 2020, 126, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via denovo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchner-Pfannschmidt, U.; Tug, S.; Kirsch, M.; Fandrey, J. Oxygen-sensing under the influence of nitric oxide. Cell Signal 2010, 22, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Daly, L.A.; Mennerich, D.; Kietzmann, T.; Sée, V. The role of hypoxia-inducible factor post-translational modifications in regulating its localisation, stability, and activity. Int. J. Mol. Sci. 2020, 22, 268. [Google Scholar] [CrossRef] [PubMed]
- Filippopoulou, C.; Simos, G.; Chachami, G. The role of sumoylation in the response to hypoxia: An overview. Cells 2020, 9, 2359. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Yang, C.; Zhong, Z.F.; Wang, S.P.; Vong, C.T.; Yu, B.; Wang, Y.T. HIF-1: Structure, biology and natural modulators. Chin. J. Nat. Med. 2021, 19, 521–527. [Google Scholar] [CrossRef]
- Protasoni, M.; Zeviani, M. Mitochondrial structure and in normal and disease conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Keerthiga, R.; Pei, D.S.; Fu, A. Mitochondrial dysfunction, UPRmt signaling, and targeted therapy in metastasis tumor. Cell Biosci. 2021, 11, 186. [Google Scholar] [CrossRef]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Cur. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Rugarli, E.I.; Langer, T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012, 31, 1336–1349. [Google Scholar] [CrossRef] [Green Version]
- Slot, I.G.; Schols, A.M.; Vosse, B.A.; Kelders, M.C.; Gosker, H.R. Hypoxia differentially regulates muscle oxidative fiber type and metabolism in a HIF-1α-dependent manner. Cell Signal 2014, 26, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Mao, J.; Han, X.; Zhang, W.; Li, Y.; Liu, Y.; Li, Q. Downregulated hypoxia-inducible factor 1α improves myoblast differentiation under hypoxic condition in mouse genioglossus. Mol. Cell Biochem. 2021, 476, 1351–1364. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Zhi, X.; Feng, W.; Rong, Y.; Liu, R. Anatomy of autophagy: From the beginning to the end. Cell Mol. Life Sci. 2018, 75, 815–831. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is a HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.J.; Wang, Z.Y.; Xu, L.; Chen, X.H.; Li, X.X.; Liao, W.T.; Ma, H.K.; Jiang, M.D.; Xu, T.T.; Xu, J.; et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020, 36, 101671. [Google Scholar] [CrossRef]
- Madhu, V.; Boneski, P.K.; Silagi, E.; Qiu, Y.; Kurland, I.; Guntur, A.R.; Shapiro, I.M.; Risbud, M.V. Hypoxic regulation of mitochondrial metabolism and mitophagy in nucleus pulposus cells is dependent on HIF-1α-BNIP3 axis. J. Bone Miner Res. 2020, 35, 1504–1524. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Zhang, J.F.; Yang, Z.J.; Jiang, L.P.; Wei, Y.F.; Lai, Q.N.; Wang, J.B.; Xin, H.B.; Han, X.J. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol. Rep. 2014, 32, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; Zhou, L.; Yin, W.; Bai, J.; Liu, R. MiR-125a induces apoptosis, metabolism disorder and migration impairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int. J. Oncol. 2018, 53, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Zhao, H.; Han, Y.; Li, L.; Xiong, S.; Zeng, L.; Xiao, Y.; Wei, L.; Xiong, X.; Gao, P.; et al. HIF-1α ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif. 2020, 53, e12909. [Google Scholar] [CrossRef]
- Chiche, J.; Rouleau, M.; Gounon, P.; Brahimi-Horn, M.C.; Pouyssegur, J.; Mazure, N.M. Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J. Cell Physiol. 2010, 222, 648–657. [Google Scholar] [CrossRef]
- Chen, X.; Yao, J.M.; Fang, X.; Zhang, C.; Yang, Y.S.; Hu, C.P.; Chen, Q.; Zhong, G.W. Hypoxia promotes pulmonary vascular remodeling via HIF-1α to regulate mitochondrial dynamics. J. Geriatr. Cardiol. 2019, 16, 855–871. [Google Scholar] [CrossRef]
- Miller, K.E.; Sheetz, M.P. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 2004, 117 Pt 13, 2791–2804. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lim, S.; Hoffman, D.; Aspenstrom, P.; Federoff, H.J.; Rempe, D.A. HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J. Cell Biol. 2009, 185, 1065–1081. [Google Scholar] [CrossRef]
- Thomas, L.W.; Staples, O.; Turmaine, M.; Ashcroft, M. CHCHD4 regulates intracellular oxygenation and perinuclear distribution of mitochondria. Front. Oncol. 2017, 7, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentillon, C.; Li, D.; Duan, M.; Yu, W.M.; Preininger, M.K.; Jha, R.; Rampoldi, A.; Saraf, A.; Gibson, G.C.; Qu, C.K.; et al. Targeting HIF-1alpha in combination with PPARalpha activation and postnatal factors promotes the metabolic maturation of human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol. 2019, 132, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial cristae: Where beauty meets functionality. Trends Biochem. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, T.; Spodnik, J.H. Structural changes of mitochondria during free radical-induced apoptosis. Folia Morphol. 2000, 59, 61–75. [Google Scholar]
- Chen, K.C.; Chen, C.R.; Chen, C.Y.; Tzou, K.Y.; Peng, C.C.; Peng, R.Y. Bicalutamide elicits renal damage by causing mitochondrial dysfunction via ROS damage and upregulation of HIF-1. Int. J. Mol. Sci. 2020, 21, 3400. [Google Scholar] [CrossRef] [PubMed]
- Regueira, T.; Lepper, P.M.; Brandt, S.; Ochs, M.; Vuda, M.; Takala, J.; SMJakob Djafarzadeh, S. Hypoxia inducible factor-1alpha induction by tumor necrosis factor-alpha, but not by toll-like receptor agonists, modulates cellular respiration in cultured human hepatocytes. Liver Int. 2009, 29, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Douiev, L.; Miller, C.; Ruppo, S.; Benyamini, H.; Abu-Libdeh, B.; Saada, A. Upregulation of COX4-2 via HIF-1α in Mitochondrial COX4-1 Deficiency. Cells 2021, 10, 452. [Google Scholar] [CrossRef]
- Kirito, K.; Hu, Y.; Komatsu, N. HIF-1 prevents the overproduction of mitochondrial ROS after cytokine stimulation through induction of PDK-1. Cell Cycle 2009, 8, 2844–2849. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- MacVicar, T.; Ohba, Y.; Nolte, H.; Mayer, F.C.; Tatsuta, T.; Sprenger, H.G.; Lindner, B.; Zhao, Y.; Li, J.; Bruns, C.; et al. Lipid signaling drives proteolytic rewiring of mitochondria by YME1L. Nature 2019, 575, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.; et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic heterogeneity of cancer cells: An interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, Y.; Tan, S.; Liu, L.; Hu, S.; Huo, H.; Li, M.; Cui, Q.; Yu, M. Nutrient deprivation-related OXPHOS/glycolysis interconversion via HIF-1α/C-MYC pathway in U251 cells. Tumour Biol. 2016, 37, 6661–6671. [Google Scholar] [CrossRef]
- Watanabe, H.; Bohensky, J.; Freeman, T.; Srinivas, V.; Shapiro, I.M. Hypoxic induction of UCP3 in the growth plate: UCP3 suppresses chondrocyte autophagy. J. Cell Physiol. 2008, 216, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Sasabe, E.; Tatemoto, Y.; Li, D.; Yamamoto, T.; Osaki, T. Mechanism of HIF-1alpha-dependent suppression of hypoxia-induced apoptosis in squamous cell carcinoma cells. Cancer Sci. 2005, 96, 394–402. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinale links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolylhydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, D.C.; Wittig, I.; Brüne, B. TMEM126B deficiency reduces mitochondrial SDH oxidation by LPS, attenuating HIF-1α stabilization and IL-1β expression. Redox Biol. 2019, 20, 204–216. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Zhuang, Y.; Song, X.R.; Niu, Q.; Sun, Q.S.; Li, X.N.; Li, N.; Liu, B.L.; Huang, F.; Qiu, Z.X. Tanshinone IIA prevents LPS-induced inflammatory responses in mice via inactivation of succinate dehydrogenase in macrophages. Acta Pharmacol. 2021, 42, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.K.; Liou, Y.H.; Knudsen, N.H.; Starost, K.A.; Xu, C.; Hyde, A.L.; Liu, S.; Jacobi, D.; Liao, N.S.; Lee, C.H. Bmal1 integrates mitochondrial metabolism and macrophage activation. Elife 2020, 9, e54090. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Ling, J.; Ye, F.; Cheng, N.; Wu, F.; Tang, Z.; Cheng, X.; Liu, H. Paeoniflorin alleviates CFA-induced inflammatory pain by inhibiting TRPV1 and succinate/SUCNR1-HIF-1α/NLPR3 pathway. Int. Immunopharmacol. 2021, 101 Pt B, 108364. [Google Scholar] [CrossRef]
- Ooi, A. Advances in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) research. Semin. Cancer Biol. 2020, 61, 158–166. [Google Scholar] [CrossRef]
- Tseng, C.W.; Kuo, W.H.; Chan, S.H.; Chan, H.L.; Chang, K.J.; Wang, L.H. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the α-Ketoglutarate Signaling Pathway. Cancer Res. 2018, 78, 2799–2812. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Eleni, C.; Spyrou, G.; Brüne, B. The mitochondrial thioredoxin system regulates nitricoxide-induced HIF-1alpha protein. Free Radic. Biol. Med. 2008, 44, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Agani, F.H.; Pichiule, P.; Chavez, J.C.; LaManna, J.C. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J. Biol. Chem. 2000, 275, 35863–35867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; García-Lecea, M.; Cadenas, S.; Hernández, C.; Moncada, S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376 Pt 2, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of hif-1 prolylhydroxylases allows cell metabolism to control basal hif-1. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Mechta-Grigoriou, F. Redox regulation of the hypoxia-inducible factor. Biol. Chem. 2006, 387, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Malo, D.; Hekimi, S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/- mouse mutants. J. Immunol. 2010, 184, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S. Mitochondrial regulation of oxygen sensing. Adv. Exp. Med. Biol. 2010, 661, 339–354. [Google Scholar] [CrossRef]
- Koshikawa, N.; Hayashi, J.; Nakagawara, A.; Takenaga, K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1alpha gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J. Biol. Chem. 2009, 284, 33185–33194. [Google Scholar] [CrossRef] [Green Version]
- Emerling, B.M.; Platanias, L.C.; Black, E.; Nebreda, A.R.; Davis, R.J.; Chandel, N.S. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell Biol. 2005, 25, 4853–4862. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Agani, F.H. Oligomycin inhibits HIF-1alpha expression in hypoxic tumor cells. Am. J. Physiol. Cell Physiol. 2005, 288, C1023–C1029. [Google Scholar] [CrossRef]
- Tan, T.; Marín-García, J.; Damle, S.; Weiss, H.R. Hypoxia inducible factor-1 improves inotropic responses of cardiac myocytes in aging heart without affecting mitochondrial activity. Exp. Physiol. 2010, 95, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, S.W.; Maity, A.; Oprysko, P.R.; Kachur, A.V.; Ayene, I.S.; Biaglow, J.E.; Koch, C.J. Detection of reactive oxygen species via endogenous oxidative pentose phosphate cycle activity in response to oxygen concentration: Implications for the mechanism of HIF-1alpha stabilization under moderate hypoxia. J. Biol. Chem. 2007, 282, 36790–36796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF-1alpha. Science 2003, 302, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Kim, M.H. Targeting the hypoxia inducible factor pathway with mitochondrial uncouplers. Mol. Cell Biochem. 2007, 296, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Yuan, Q.; Qin, N.; Shi, C.; Luo, J.; Fang, Y.; Xu, L.; Sun, Q.; Zen, K.; Jiang, L.; et al. UCP2-induced hypoxia promotes lipid accumulation and tubulointerstitial fibrosis during ischemic kidney injury. Cell Death Dis. 2020, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Boutoual, R.; Meseguer, S.; Villarroya, M.; Martín-Hernández, E.; Errami, M.; Martín, M.A.; Casado, M.; Armengod, M.E. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARγ-UCP2-AMPK axis. Sci. Rep. 2018, 8, 1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, T.U.; Yazihan, N.; Dalgic, A.; Kaya, E.E.; Salman, B.; Kocak, M.; Akcil, E. Role of ATP-dependent K channels in the effects of erythropoietin in renal ischaemia injury. Indian J. Med. Res. 2015, 141, 807–815. [Google Scholar] [CrossRef]
- Li, J.; Zhou, W.; Chen, W.; Wang, H.; Zhang, Y.; Yu, T. Mechanism of the hypoxia inducible factor 1/hypoxic response element pathway in rat myocardial ischemia/diazoxide post-conditioning. Mol. Med. Rep. 2020, 21, 1527–1536. [Google Scholar] [CrossRef]
- An, W. Medical Cell Biology; Peking University Medical Press: Beijing, China, 2019. [Google Scholar]



{kind=link}
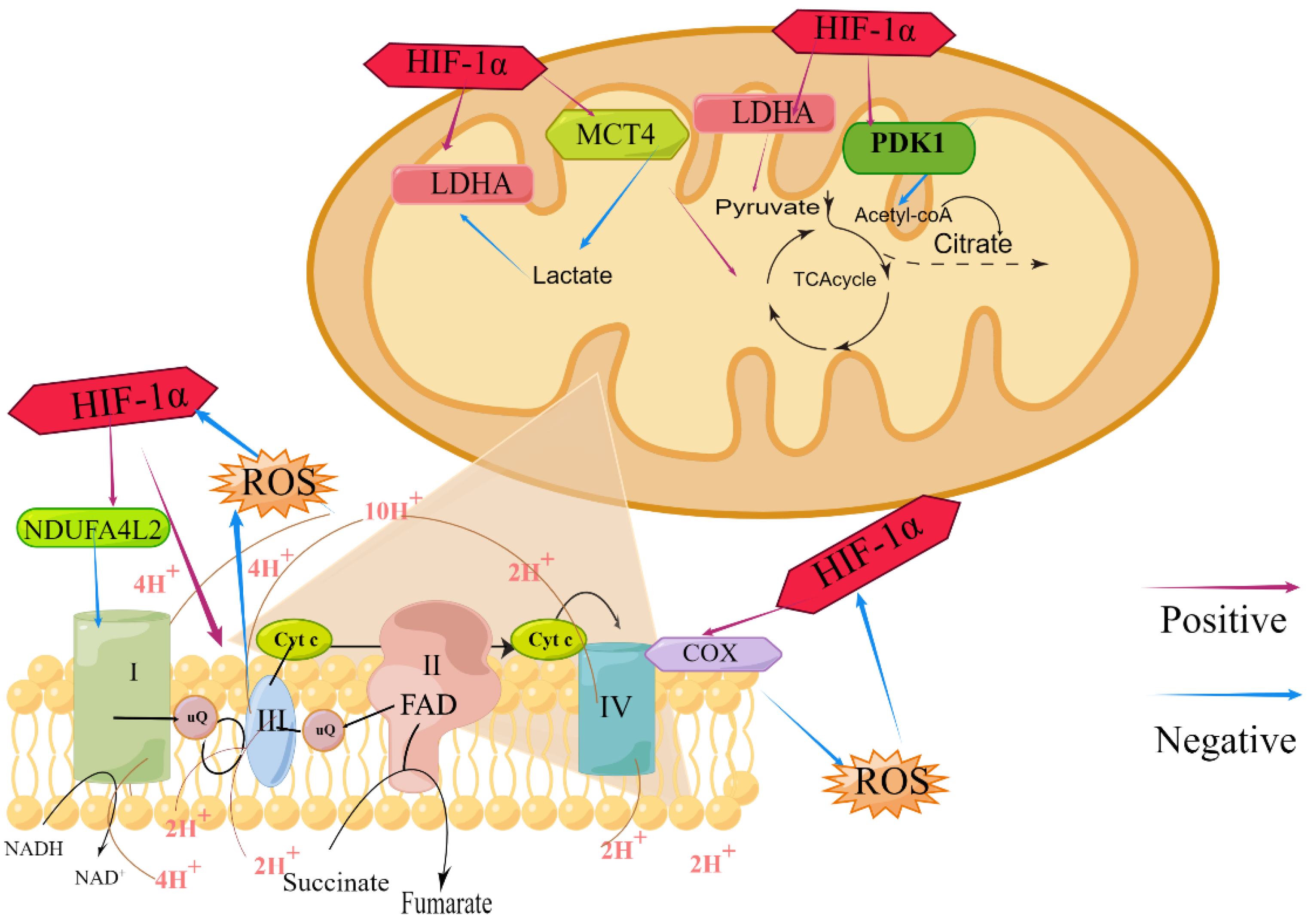{kind=link}
| Related Mitochondrial Function | Proteins or Receptors | Effects on HIF-1 | Ref. No. |
|---|---|---|---|
| Mitochondrial metabolic enzymes | Succinate dehydrogenase(SDH) Fumarate hydratase(FH) | Negative | [59,60,61,62,63,64,65] |
| Thioredoxin (Trx2) and Thioredoxin reductase (TrxR2) | Positive | [66] | |
| Mitochondrial respiratory chain | Complex I II III IV | Positive | [67,68,69] |
| ROS | Positive/Negative | [70,71,72,73,74,75,76,77,78,79,80,81,82,83,84] | |
| Uncoupling proteins | Rottlerin and franchise control chain project(FCCP) | Negative | [85] |
| UCP2 | Negative | [86,87] | |
| Mitochondrial ATP-sensitive potassium channels | Mito KATP channel | Positive | [88,89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2023, 13, 50. https://doi.org/10.3390/biom13010050
Huang X, Zhao L, Peng R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules. 2023; 13(1):50. https://doi.org/10.3390/biom13010050
Chicago/Turabian StyleHuang, Xiaochen, Li Zhao, and Ruiyun Peng. 2023. "Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection" Biomolecules 13, no. 1: 50. https://doi.org/10.3390/biom13010050