
Differential Gene Expression Induced by Different TLR Agonists in A549 Lung Epithelial Cells Is Modulated by CRISPR Activation of TLR10

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
A549 Challenge
2.2. sgRNAs and dCas9 Vectors
2.2.1. sgRNA Design
2.2.2. Validation of the sgRNAs
2.2.3. Plasmids and Cloning
2.3. Overexpression of the Endogenous TLR10 and Challenge of the Cells with Putative TLR10 Ligands
2.4. RNA Extraction and Reverse Transcription into Single-Stranded cDNA
2.5. Gene Expression Analyses
2.5.1. Quantitative PCR (qPCR) Array Screening
2.5.2. qPCR Expression Profiling
2.6. Protein Quantification
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
3.1. CRISPR Activation of the TLR10
3.2. Differential Expression of Immune Associated Genes in the Challenged A549 Cells with Native Gene Expression
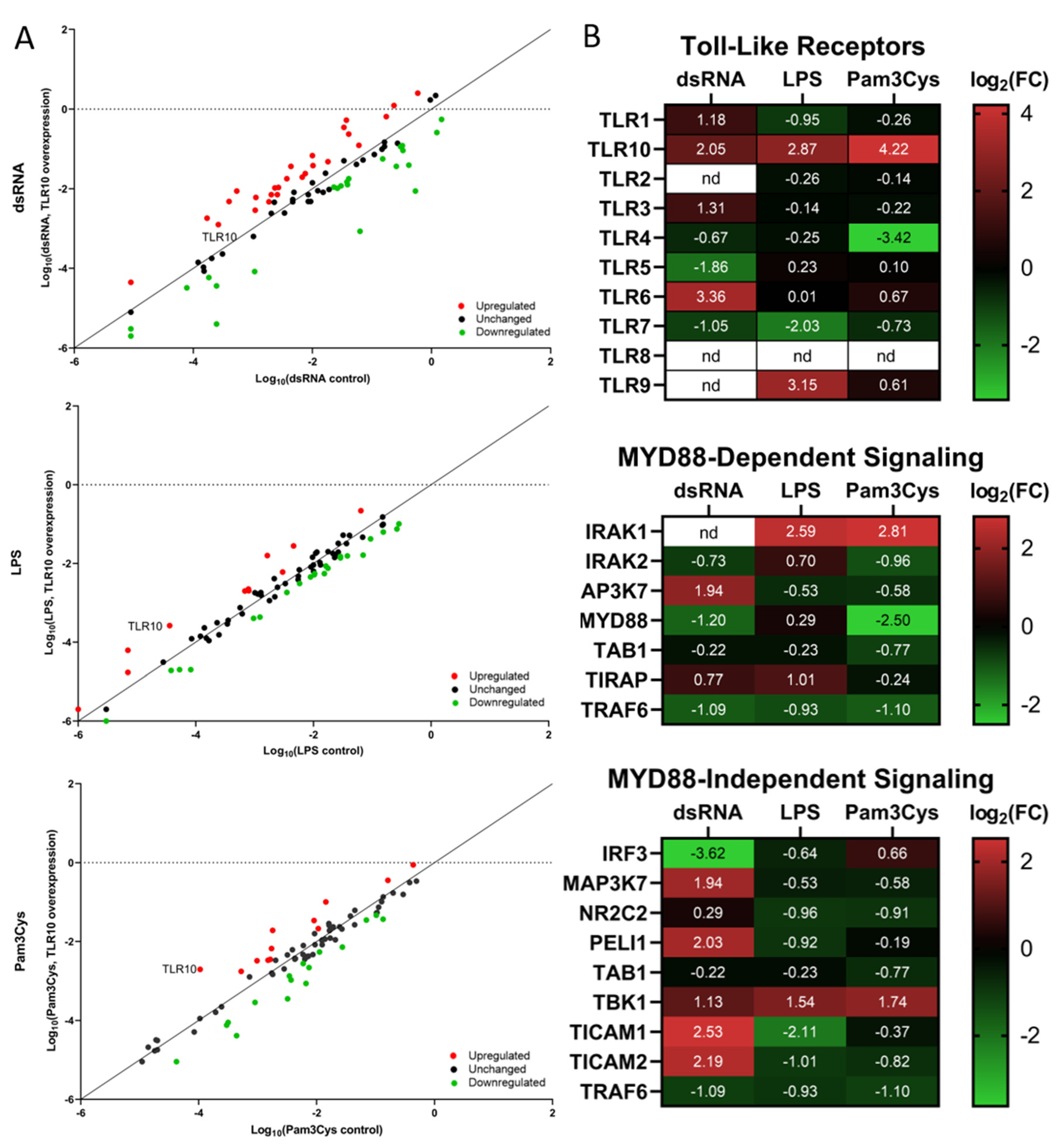
3.3. Differential Expression of Immune Associated Genes in the Challenged A549 Cells Overexpressing TLR10
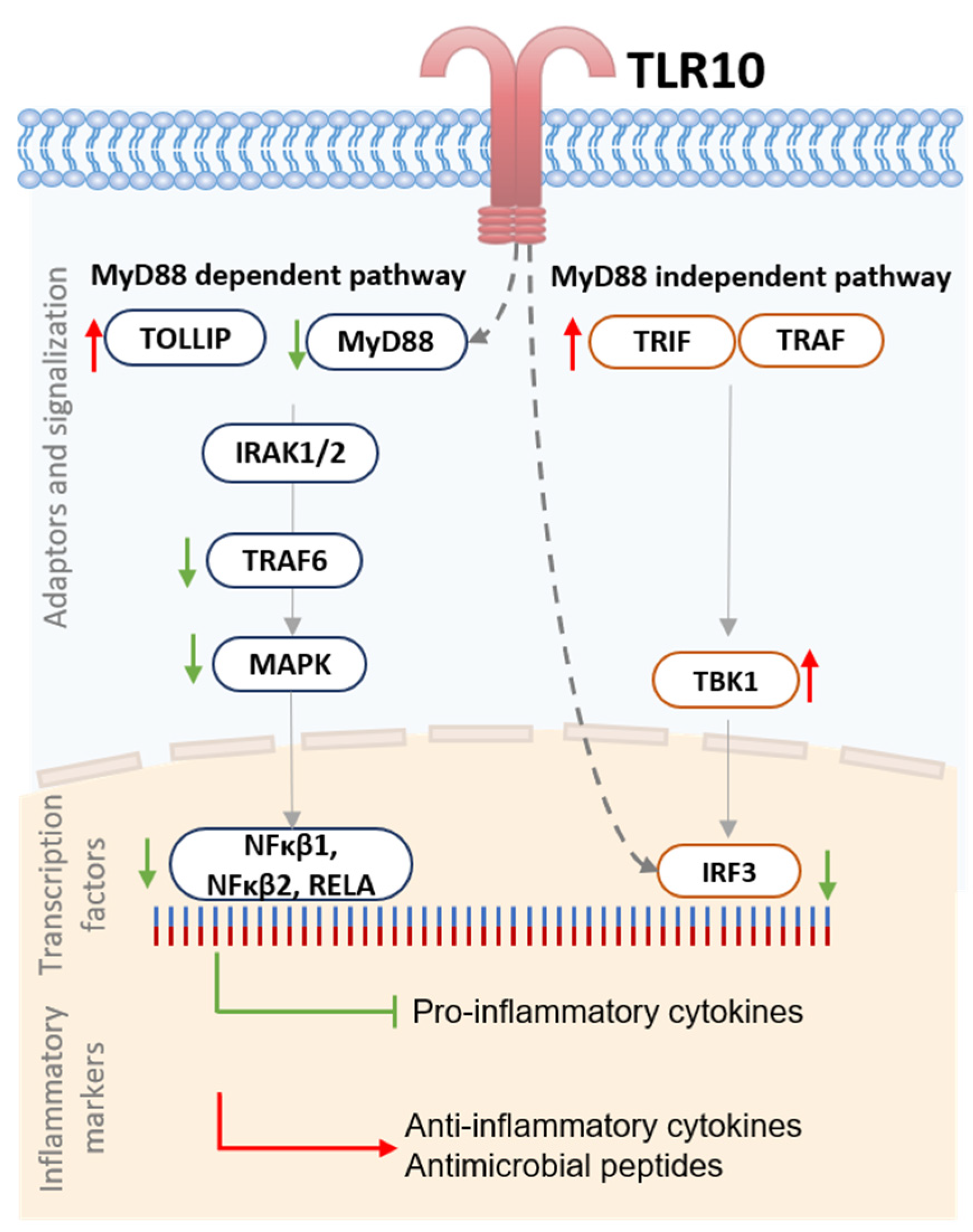
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.H.; Ulevitch, R.J. Identification of hTLR10: A novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta Gene Struct. Expr. 2001, 1518, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Fore, F.; Indriputri, C.; Mamutse, J.; Nugraha, J. TLR10 and its unique anti-inflammatory properties and potential use as a target in therapeutics. Immune Netw. 2020, 20, e21. [Google Scholar] [CrossRef] [PubMed]
- Henrick, B.M.; Yao, X.D.; Zahoor, M.A.; Abimiku, A.; Osawe, S.; Rosenthal, K.L. TLR10 senses HIV-1 proteins and significantly enhances HIV-1 infection. Front. Immunol. 2019, 10, 482. [Google Scholar] [CrossRef]
- Lee, S.M.Y.; Kok, K.H.; Jaume, M.; Cheung, T.K.W.; Yip, T.F.; Lai, J.C.C.; Guan, Y.; Webster, R.G.; Jin, D.Y.; Malik Peiris, J.S. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3793–3798. [Google Scholar] [CrossRef] [Green Version]
- Regan, T.; Nally, K.; Carmody, R.; Houston, A.; Shanahan, F.; MacSharry, J.; Brint, E. Identification of TLR10 as a Key Mediator of the Inflammatory Response to Listeria monocytogenes in Intestinal Epithelial Cells and Macrophages. J. Immunol. 2013, 191, 6084–6092. [Google Scholar] [CrossRef] [Green Version]
- Hess, N.J.; Jiang, S.; Li, X.; Guan, Y.; Tapping, R.I. TLR10 Is a B Cell Intrinsic Suppressor of Adaptive Immune Responses. J. Immunol. 2017, 198, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Oosting, M.; Cheng, S.C.; Bolscher, J.M.; Vestering-Stenger, R.; Plantinga, T.S.; Verschueren, I.C.; Arts, P.; Garritsen, A.; Van Eenennaam, H.; Sturm, P.; et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc. Natl. Acad. Sci. USA 2014, 111, E4478–E4484. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Li, X.; Hess, N.J.; Guan, Y.; Tapping, R.I. TLR10 Is a Negative Regulator of Both MyD88-Dependent and -Independent TLR Signaling. J. Immunol. 2016, 196, 3834–3841. [Google Scholar] [CrossRef] [Green Version]
- Hasan, U.; Chaffois, C.; Gaillard, C.; Saulnier, V.; Merck, E.; Tancredi, S.; Guiet, C.; Brie, F.; Vlach, J.; Lebecque, S.; et al. Human TLR10 Is a Functional Receptor, Expressed by B Cells. J Immunol 2005, 174, 2942–2950. [Google Scholar] [CrossRef]
- Van Le, H.; Kim, J.Y. Stable toll-like receptor 10 knockdown in THP-1 cells reduces TLR-ligand-induced proinflammatory cytokine expression. Int. J. Mol. Sci. 2016, 17, 859. [Google Scholar]
- Nagashima, H.; Cruz, M.; Abreu, J.A.J.; Uchida, T.; Mahachai, V.; Vilaichone, R.K.; Graham, D.Y.; Yamaoka, Y. Toll-like Receptor 10 in Helicobacter pylori Infection. J. Infect. Dis. 2015, 212, 1666–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.M.Y.; Yip, T.F.; Yan, S.; Jin, D.Y.; Wei, H.L.; Guo, R.T.; Peiris, J.S.M. Recognition of double-stranded RNA and regulation of interferon pathway by toll-like receptor 10. Front. Immunol. 2018, 9, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindaraj, R.G.; Manavalan, B.; Lee, G.; Choi, S. Molecular modeling-based evaluation of hTLR10 and identification of potential ligands in toll-like receptor signaling. PLoS ONE 2010, 5, e12713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Ranoa, D.R.E.; Jiang, S.; Mutha, S.K.; Li, X.; Baudry, J.; Tapping, R.I. Human TLRs 10 and 1 Share Common Mechanisms of Innate Immune Sensing but Not Signaling. J. Immunol. 2010, 184, 5094–5103. [Google Scholar] [CrossRef] [Green Version]
- Sindhu, S.; Akhter, N.; Kochumon, S.; Thomas, R.; Wilson, A.; Shenouda, S.; Tuomilehto, J.; Ahmad, R. Increased expression of the innate immune receptor TLR10 in obesity and type-2 Diabetes: Association with ROS-mediated oxidative stress. Cell. Physiol. Biochem. 2018, 45, 572–590. [Google Scholar] [CrossRef]
- Ge, L.; Xu, L.; Lu, S.; Yan, H. Expression and Function of Toll-Like Receptor 10 (TLR10) in Diffuse Large B Cell Lymphoma, Acute Myeloid Leukemia, and Glioma. Med. Sci. Monit. 2020, 26, e921500. [Google Scholar] [CrossRef]
- Fan, Y.; Yang, L.; Wei, Q.; Ding, Y.; Tang, Z.; Tan, P.; Lin, T.; Guo, D.; Qiu, S. Toll-like receptor 10 (TLR10) exhibits suppressive effects on inflammation of prostate epithelial cells. Asian J. Androl. 2019, 21, 393–399. [Google Scholar]
- Torices, S.; Julia, A.; Muñoz, P.; Varela, I.; Balsa, A.; Marsal, S.; Fernández-Nebro, A.; Blanco, F.; López-Hoyos, M.; Martinez-Taboada, V.; et al. A functional variant of TLR10 modifies the activity of NFkB and may help predict a worse prognosis in patients with rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 1–9. [Google Scholar] [CrossRef]
- Bulat-Kardum, L.J.; Etokebe, G.E.; Lederer, P.; Balen, S.; Dembic, Z. Genetic Polymorphisms in the Toll-like Receptor 10, Interleukin (IL)17A and IL17F Genes Differently Affect the Risk for Tuberculosis in Croatian Population. Scand. J. Immunol. 2015, 82, 63–69. [Google Scholar] [CrossRef]
- Törmänen, S.; Korppi, M.; Lauhkonen, E.; Koponen, P.; Teräsjärvi, J.; Vuononvirta, J.; Helminen, M.; He, Q.; Nuolivirta, K. Toll-like receptor 1 and 10 gene polymorphisms are linked to postbronchiolitis asthma in adolescence. Acta Paediatr. Int. J. Paediatr. 2018, 107, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Veltkamp, M.; Van Moorsel, C.H.M.; Rijkers, G.T.; Ruven, H.J.T.; Grutters, J.C. Genetic variation in the Toll-like receptor gene cluster (TLR10-TLR1-TLR6) influences disease course in sarcoidosis. Tissue Antigens 2012, 79, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.Y.T.; Chang, R.Y.K.; Chan, H.K. Inhalation delivery technology for genome-editing of respiratory diseases. Adv. Drug Deliv. Rev. 2021, 168, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Wiegand, D.J.; Ter-ovanesyan, D.; Braff, J.L.; Davidsohn, N. Highly-efficient Cas9-mediated transcriptional programming. Nat Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Akinci, E.; Hamilton, M.C.; Khowpinitchai, B.; Sherwood, R.I. Using CRISPR to understand and manipulate gene regulation. Development 2021, 148, 18266. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef]
- Boonstra, A.; Rajsbaum, R.; Holman, M.; Marques, R.; Asselin-Paturel, C.; Pereira, J.P.; Bates, E.E.M.; Akira, S.; Vieira, P.; Liu, Y.-J.; et al. Macrophages and Myeloid Dendritic Cells, but Not Plasmacytoid Dendritic Cells, Produce IL-10 in Response to MyD88- and TRIF-Dependent TLR Signals, and TLR-Independent Signals. J. Immunol. 2006, 177, 7551–7558. [Google Scholar] [CrossRef] [Green Version]
- Birchler, T.; Seibl, R.; Bchner, K.; Loeliger, S.; Seger, R.; Hossle, J.P.; Aguzzi, A.; Lauener, R.P. Human Toll-like receptor 2 mediates induction of the antimicrobial peptide human beta-defensin 2 in response to bacterial lipoprotein. Eur. J. Immunol. 2001, 31, 3131–3137. [Google Scholar] [CrossRef]
- Thorley, A.J.; Goldstraw, P.; Young, A.; Tetley, T.D. Primary human alveolar type II epithelial cell CCL20 (macrophage inflammatory protem-3α)-induced dendritic cell migration. Am. J. Respir. Cell Mol. Biol. 2005, 32, 262–267. [Google Scholar] [CrossRef]
- L’Heureux, M.; Kashiouris, M.; Fowler, A.; Fisher, B. Association Between Il-10 and Mortality in Sepsis-Induced Ards. Chest 2020, 158, A621. [Google Scholar] [CrossRef]
- Silva, B.S.A.; Lira, F.S.; Ramos, D.; Uzeloto, J.S.; Rossi, F.E.; Freire, A.P.C.F.; Silva, R.N.; Trevisan, I.B.; Gobbo, L.A.; Ramos, E.M.C. Severity of COPD and its relationship with IL-10. Cytokine 2018, 106, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfield, T.L.; Konstan, M.W.; Burfeind, P.; Panuska, J.R.; Hilliard, J.B.; Berger, M. Normal Bronchial Epithelial Cells Constitutively Produce the Anti-inflammatory Cytokine Interleukin-10, Which Is Downregulated in Cystic Fibrosis. Gastroenterology 1977, 72, 193. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, T.; Nakazato, M.; Date, Y.; Ashitani, J.I.; Minematsu, T.; Chino, N.; Matsukura, S. Identification of human β-defensin-2 in respiratory tract and plasma and its increase in bacterial pneumonia. Biochem. Biophys. Res. Commun. 1998, 249, 943–947. [Google Scholar] [CrossRef]
- Zhang, L.; Ghosh, S.K.; Basavarajappa, S.C.; Chen, Y.; Shrestha, P.; Penfield, J.; Brewer, A.; Ramakrishnan, P.; Buck, M.; Weinberg, A. HBD-2 binds SARS-CoV-2 RBD and blocks viral entry: Strategy to combat COVID-19. iScience 2022, 25, 103856. [Google Scholar] [CrossRef]
- Wang, B.; Shi, L.; Sun, X.; Wang, L.; Wang, X.; Chen, C. Production of CCL20 from lung cancer cells induces the cell migration and proliferation through PI3K pathway. J. Cell. Mol. Med. 2016, 20, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Nyman, T.; Stenmark, P.; Flodin, S.; Johansson, I.; Hammarström, M.; Nordlund, P.R. The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J. Biol. Chem. 2008, 283, 11861–11865. [Google Scholar] [CrossRef] [Green Version]
- Nimma, S.; Ve, T.; Williams, S.J.; Kobe, B. Towards the structure of the TIR-domain signalosome. Curr. Opin. Struct. Biol. 2017, 43, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Zhao, W. TANK-binding kinase 1 as a novel therapeutic target for viral diseases. Expert Opin. Ther. Targets 2019, 23, 437–446. [Google Scholar] [CrossRef]
- Thoresen, D.; Wang, W.; Galls, D.; Guo, R.; Xu, L.; Pyle, A.M. The molecular mechanism of RIG-I activation and signaling. Immunol. Rev. 2021, 304, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Palchetti, S.; Starace, D.; De Cesaris, P.; Filippini, A.; Ziparo, E.; Riccioli, A. Transfected poly(I:C) activates different dsRNA receptors, leading to apoptosis or immunoadjuvant response in androgen-independent prostate cancer cells. J. Biol. Chem. 2015, 290, 5470–5483. [Google Scholar] [CrossRef] [PubMed]
- Patil, J.S.; Sarasija, S. Pulmonary drug delivery strategies: A concise, systematic review. Lung India 2012, 29, 44–49. [Google Scholar]
- Margulieux, K.R.; Fox, J.W.; Nakamoto, R.K.; Hughes, M.A. CXCL10 acts as a bifunctional antimicrobial molecule against Bacillus anthracis. MBio 2016, 7, e00334-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martineau, A.R.; Jolliffe, D.A.; Greenberg, L.; Aloia, J.F.; Bergman, P.; Dubnov-Raz, G.; Esposito, S.; Ganmaa, D.; Ginde, A.A.; Goodall, E.C.; et al. Vitamin D supplementation to prevent acute respiratory infections: Individual participant data meta-analysis. Health Technol. Assess. (Rockv). 2019, 23, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.G.; Chang, K.; Osborne, D.; Walton, A.H.; Ghosh, S.; Dunne, W.M.; Hotchkiss, R.S.; Muenzer, J.T. TLR3 agonist improves survival to secondary pneumonia in a double injury model. J. Surg. Res. 2013, 182, 270–276. [Google Scholar] [CrossRef]
- Evans, S.E.; Xu, Y.; Tuvim, M.J.; Dickey, B.F. Inducible innate resistance of lung epithelium to infection. Annu. Rev. Physiol. 2009, 72, 413–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowdish, D.M.E.; Davidson, D.J.; Scott, M.G.; Hancock, R.E.W. Immunomodulatory activities of small host defense peptides. Antimicrob. Agents Chemother. 2005, 49, 1727–1732. [Google Scholar] [CrossRef] [Green Version]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 2013, 497, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bahamondez-Canas, T.F.; Zhang, Y.; Leal, J.; Smyth, H.D.C. PEGylated Chitosan for Nonviral Aerosol and Mucosal Delivery of the CRISPR/Cas9 System in Vitro. Mol. Pharm. 2018, 15, 4814–4826. [Google Scholar] [CrossRef]
- Maxwell, A.J.; Ding, J.; You, Y.; Dong, Z.; Chehade, H.; Alvero, A.; Mor, Y.; Draghici, S.; Mor, G. Identification of key signaling pathways induced by SARS-CoV2 that underlie thrombosis and vascular injury in COVID-19 patients. J. Leukoc. Biol. 2021, 109, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 724, 718–724. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knez, Š.; Narat, M.; Ogorevc, J. Differential Gene Expression Induced by Different TLR Agonists in A549 Lung Epithelial Cells Is Modulated by CRISPR Activation of TLR10. Biomolecules 2023, 13, 19. https://doi.org/10.3390/biom13010019
Knez Š, Narat M, Ogorevc J. Differential Gene Expression Induced by Different TLR Agonists in A549 Lung Epithelial Cells Is Modulated by CRISPR Activation of TLR10. Biomolecules. 2023; 13(1):19. https://doi.org/10.3390/biom13010019
Chicago/Turabian StyleKnez, Špela, Mojca Narat, and Jernej Ogorevc. 2023. "Differential Gene Expression Induced by Different TLR Agonists in A549 Lung Epithelial Cells Is Modulated by CRISPR Activation of TLR10" Biomolecules 13, no. 1: 19. https://doi.org/10.3390/biom13010019