
NRF2 in the Epidermal Pigmentary System

1
Department of Dermatology, Faculty of Medicine, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8575, Japan
2
Department of Dermatology, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita 565-0871, Japan
*
Author to whom correspondence should be addressed.
Biomolecules 2023, 13(1), 20; https://doi.org/10.3390/biom13010020
Submission received: 8 November 2022
/
Revised: 20 December 2022
/
Accepted: 21 December 2022
/
Published: 22 December 2022
(This article belongs to the Special Issue Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches II)
Abstract
:Melanogenesis is a major part of the environmental responses and tissue development of the integumentary system. The balance between reduction and oxidation (redox) governs pigmentary responses, for which coordination among epidermal resident cells is indispensable. Here, we review the current understanding of melanocyte biology with a particular focus on the “master regulator” of oxidative stress responses (i.e., the Kelch-like erythroid cell-derived protein with cap‘n’collar homology-associated protein 1-nuclear factor erythroid-2-related factor 2 system) and the autoimmune pigment disorder vitiligo. Our investigation revealed that the former is essential in pigmentogenesis, whereas the latter results from unbalanced redox homeostasis and/or defective intercellular communication in the interfollicular epidermis (IFE). Finally, we propose a model in which keratinocytes provide a “niche” for differentiated melanocytes and may “imprint” IFE pigmentation.
1. Introduction
Oxidative stress results from exposure to reactive oxygen intermediates, such as superoxide anions (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (HO), and can damage proteins, nucleic acids, and cell membranes, leading to mutagenesis or cell death [1]. Reactive oxygen species (ROS) are produced by mitochondria and peroxisomes during cellular physiological metabolism. ROS-induced cumulative damage eventually causes numerous diseases. The skin serves as the interface between an organism and its external environment, protecting against xenobiotics or ultraviolet (UV) radiation, which can cause ROS-mediated tissue damage. Melanocytes produce pigmentation in hair and the interfollicular epidermis (IFE), thereby contributing to photoprotection and thermoregulation [2]. Vitiligo is an autoimmune-driven chronic depigmenting disease in which oxidative stress plays important roles [3,4]. The Kelch-like erythroid cell-derived protein with cap‘n’collar homology-associated protein 1 (KEAP1)-nuclear factor erythroid-2-related factor 2 (NRF2) system is a major antioxidative apparatus whose activation largely depends on the oxidation of cysteine residues on the actin-anchored KEAP1 protein [5]. NRF2 not only affects melanocyte proliferation/differentiation [6] but also helps melanocytes survive the cytotoxic immune responses in vitiligo [7]. Thus, because reduction and oxidation (redox) governs myriad biological processes, NRF2 appears to have broad and significant roles in melanocyte biology. Here, we sought to summarize the significant role of the KEAP1-NRF2 system, whose activation is tightly controlled by the redox status of thiol and disulfide.
2. The KEAP1-NRF2 System as a Master Regulator of Redox Homeostasis
Redox refers to the give-and-take of electrons between molecules (and/or their moieties). Oxidants deprive the target of electrons and are thus called electrophiles. Reductants provide the target with electrons and are therefore called electron donors. Therefore, “antioxidants” are substances that rescue organisms from excessive oxidation, directly or indirectly. The former, such as the glutathione (GSH) precursor N-acetyl cysteine, replenishes the intracellular GSH pool, and the latter is often electrophilic and strongly induces cellular antioxidative defenses (such as sulforaphane), resulting in the production of antioxidative effectors (e.g., glutamate-cysteine ligase catalytic subunit [GCLC]) from cells in stress.
KEAP1 is an actin-anchored cysteine-rich protein that senses extra- or intracellular electrophilic milieus causing “oxidative damage,” and NRF2 is a ubiquitous transcription factor [5]. In the inactivated state, NRF2 is polyubiquitinated and degraded by proteasomes. During periods of stress, NRF2 exerts counter-responses. For instance, NRF2 transactivates cystine/glutamate transporter (solute carrier family 7 member 11 [SLC7A11]) [8] and upregulates GSH-synthesizing enzymes (GCLC and glutamate-cysteine ligase modifier subunit [GCLM]) and replenishes intracellular GSH storage [5]. Since the identification of the cis-acting antioxidant (electrophile) response element, many genes have been identified as downstream effectors of the KEAP1-NRF2 system, including ROS quenchers (thioredoxin reductase 1 [TXNRD1] and peroxiredoxin 1 [PRDX1]), phase II detoxification mediators (GSH S-transferase [GST], NAD(P)H quinone dehydrogenase 1 [NQO1], and heme oxygenase-1 [HMOX1]), transmembrane drug transporters (multidrug resistance-associated protein 1 [MRP1]), and structural proteins related to keratinization (small proline-rich proteins [SPRRs] and late cornified envelopes [LCEs]) [5,9].
3. Melanocyte Biology
Melanocytes originate from the neural crest and localize in the hair follicles (HFs) and IFE, endowing the body surface with pigmentation [2]. Human melanocytes exist in the IFE and HFs, whereas murine melanocytes exclusively reside in pelage HFs (exceptions are the ears, nose, paws, tail, etc.). Approximately 1200 melanocytes reside per square millimeter of human skin, regardless of race [10]. Epidermal melanocytes exist in the basal IFE layer and form the epidermal melanin unit, in which one melanocyte communicates with 30–40 IFE keratinocytes (KCs) [10]. Melanocytes adhere to KCs via adhesion molecules such as E-cadherin (Ecad) and desmoglein 1 (DSG1) [11]. There are two distinct melanocytic populations in HFs: melanocyte stem cells (McSCs) and their differentiated progeny. McSCs reside in the hair bulge and secondary hair germ (the lower permanent portion of the HFs) and show cyclic activity in parallel with HF stem cells [12].
The crosstalk between melanocytes and KCs through secreted factors (e.g., stem cell factor [SCF], basic fibroblast growth factor [bFGF], granulocyte macrophage-colony stimulating factor, endothelin 1, α-melanocyte-stimulating hormone [α-MSH], prostaglandin E2, prostaglandin F2α, and nerve growth factor) and cell–cell interaction controls melanocyte proliferation, differentiation, melanogenesis, and dendritogenesis [10,13,14]. Melanocytes are also controlled by dermal fibroblasts via secreted factors (e.g., SCF, bFGF, and hepatocyte growth factor) [10,13,14].
Melanocytes exhibit immunological characteristics. They can express major histocompatibility complex classes I and II, immunomodulatory adhesion molecules (e.g., intercellular adhesion molecule-1 and vascular cell adhesion molecule-1), and the costimulatory receptor CD40 [15,16,17,18]. In addition, melanocytes can produce several cytokines such as interleukin (IL)-1, IL-6, IL-8, and transforming growth factor-β1 [19,20,21].
3.1. Melanogenesis
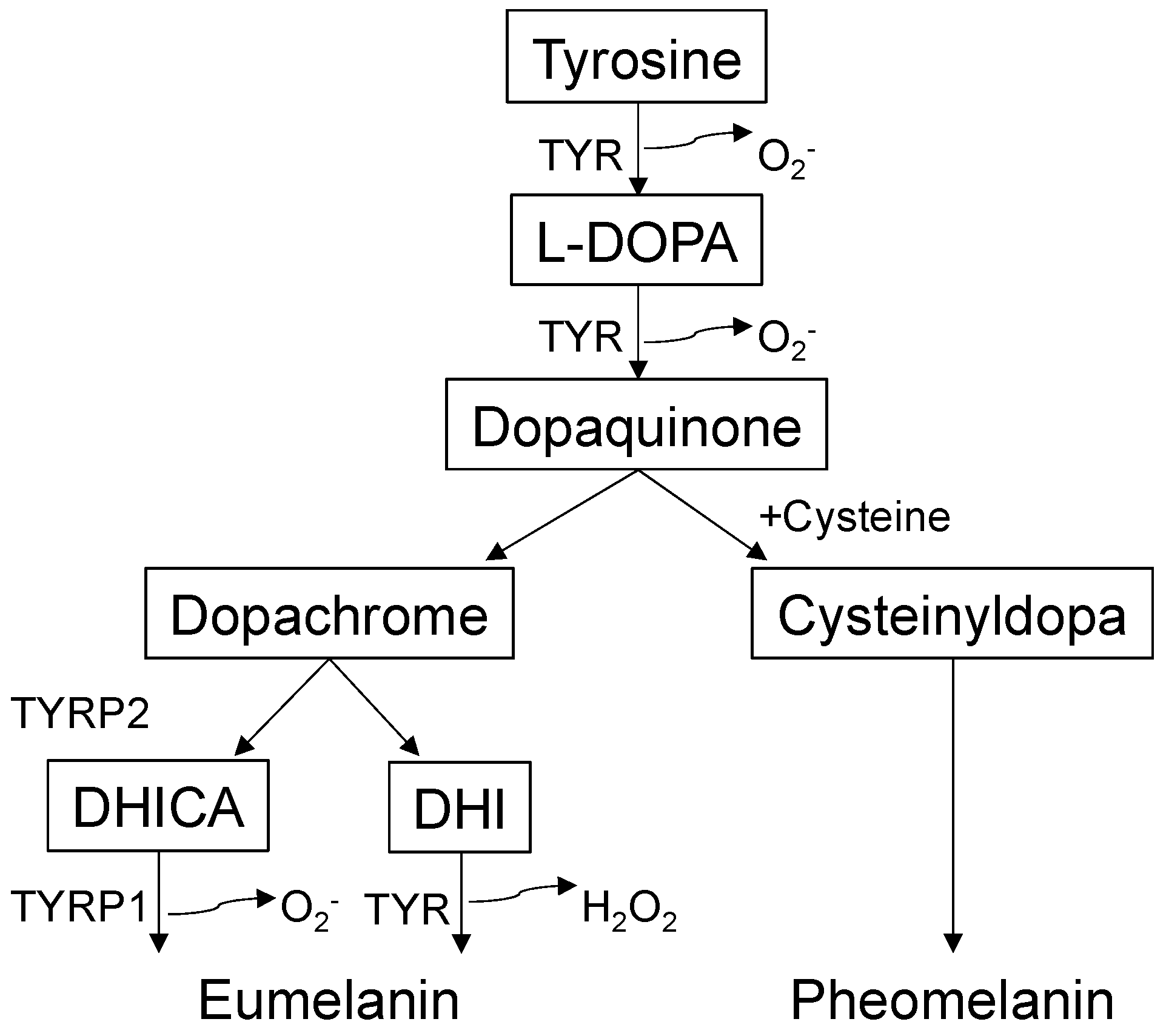
Melanogenesis (melanin synthesis) is a complex biosynthetic process [22] that occurs predominantly in a lysosome-like organelle called the melanosome [23] (Figure 1). Melanosomes are exported from melanocytes to adjacent KCs, and pigmentation differences occur owing to variations in the size, composition, and distribution of melanosomes [2]. Melanin has many biological functions, including UV light absorption/scattering, free radical scavenging, coupled redox reactions, and ion storage [10]. Melanin comes in two forms: yellow to red pheomelanin and brown to black eumelanin. The metabolic cascade of melanogenesis begins with the hydroxylation of the amino acid tyrosine to L-3,4-dihydroxyphenylalanine, which is then converted into dopaquinone by tyrosinase (TYR) [10]. This series of reactions involve physical contact between the melanosome and mitochondria [24], inherently generating ROS, such as O2− or H2O2 [25]. Dopaquinone oxidation produces dopachrome, which is decarboxylated into 5,6-dihydroxyindole or tautomerized to 5,6-dihydroxyindole-2-carboxylic acid by tyrosinase-related protein 2 (dopachrome tautomerase or TYRP2). TYR and tyrosinase-related protein 1 (TYRP1) catalyze further reactions to synthesize eumelanin [26]. The relative abundance of GSH/cysteine promotes the conversion of dopaquinone into cysteinyldopa, preferentially generating pheomelanin. Conversely, the relative abundance of glutathione disulfide (GSSG)/cystine (two cysteines bridged via disulfide) promotes melanosome maturation, yielding stable, polymerized eumelanin. In summary, like other biological processes, the redox milieu appears to play central roles in melanogenesis and thus skin pigmentation/depigmentation.
3.2. Redox “Switch” in Skin Pigmentation
In general, the pheomelanin synthetic pathway requires cysteine/GSH, whereas the eumelanin pathway is expedited in the absence of thiols (high cystine/cysteine or GSSG/GSH ratios) [27,28]. Compared with pheomelanin, eumelanin is more polymerized and structurally stable, and is better at protecting against ROS. Thus, darkly pigmented eumelanin serves as a superior antioxidant/photoprotector on the skin surface [10]. Melanogenesis consumes high energy, produces O2− or H2O2 from mitochondria [24,25], and generates a pro-oxidant state, potentially sensitizing the epidermis to oxidative stress [22] (Figure 1). An unbalanced extracellular redox milieu can also perturb cellular fate decisions, leading to cell death (apoptosis) [29] or proliferation (melanomagenesis) [30]. A rather perplexing fact is that pheomelanin has a potent photosensitizing capacity [31]. Pheomelanin toxicity likely causes premature aging and cutaneous tumorigenesis in fair-skinned individuals. In contrast, eumelanin can enhance permeability barrier function by lowering the surface pH of darkly pigmented human [32]/mouse [33] skin.
A proper antioxidative response induced by changes in the intra-/extracellular redox milieu appears to give an important direction regarding the trajectory of the pigmentary pathways. Defective transmembrane cystine transport caused by the subtle gray (sut) mutation in the Slc7a11 gene reduces intracellular GSH levels and increases GSSG levels (thus, the GSH/GSSG ratio decreases). The failure in response against extracellular oxidative signals skews melanogenesis toward the eumelanin synthesis pathway [34]. The mitochondrial redox-regulating enzyme nicotinamide nucleotide transhydrogenase (NNT) controls melanosomal maturation (and eumelanogenesis) [35]. This inner mitochondrial membrane protein regulates NAD(P)+/NAD(P)H homeostasis by mediating electron transfer [36]. Although NNT can bring about an intracellular oxidative milieu in certain circumstances [37], NNT increases GSH/GSSG ratios and negatively regulates eumelanogenesis [35], similar to what has been observed for NRF2 [6]/Slc7a11 (sut mice) [8]. However, the most striking facts are that single nucleotide polymorphisms (SNPs) in the NNT gene clearly differentiate skin pigmentary phenotypes, and inhibition takes place at the post-transcriptional levels [35]. Existing evidence argues against the classic pigmentation pathway dependent on the UV-cyclic adenosine monophosphate-microphthalmia-associated transcription factor (MITF) axis, which in turn transactivates TYRP1 and TYRP2 [35]. We should note that sut melanocytes, which mount suboptimal counter-responses against an extracellular oxidative milieu [8], also exhibit abnormal proliferation and differentiation in vitro [34]. Thus, redox milieus (intracellular or extracellular) can profoundly affect melanocyte biological behaviors and fate decisions [38]. In summary, investigating the redox “switch” in skin pigmentary pathways not only could lead to a profound understanding of skin pigmentation biology but also may pave a way toward repurposing small molecule inhibitors for diverse pigmentary disorders [35].
4. Vitiligo
Vitiligo is an acquired chronic pigmentary disorder that affects 0.5% to 2% of the world’s population without a clear preference for race or sex [39]. The US population-based prevalence estimate of vitiligo in adults was between 0.76% and 1.11% [40]. Vitiligo results from selective melanocyte loss, which leads to pigment dilution in the affected skin and mucosa. Typical vitiligo lesions present as milky-white nonscaly macules with distinct margins. Generally, vitiligo is clinically diagnosed, and no laboratory tests or biopsies are required. Two major forms of the disease are well-recognized according to the distribution of lesions: segmental vitiligo (SV) and non-segmental vitiligo (NSV) [41]. NSV includes acrofacial, mucosal, generalized, universal, mixed, and rare variants. Distinguishing SV from other types of vitiligo is important because of its prognostic implications [42].
Vitiligo pathogenesis involves multiple factors, such as genetic background, metabolic abnormalities, oxidative stress, generation of inflammatory mediators, autoimmune responses, and decreased melanocyte adhesiveness [43]. These multiple mechanisms may function collectively, leading to melanocyte destruction [42].
4.1. Genetic Background of Vitiligo
Multiple studies have revealed the genetic background of vitiligo development. Approximately 50 different genetic loci that contribute to the risk of vitiligo have been discovered, principally in European-derived whites and Chinese [44]. A genome-wide association study identified the susceptibility loci for autoimmunity (e.g., HLA classes 1 and 2, PTPN22, IL2R α, GZMB, FOXP3, BACH2, CD80, and CCR6) and melanocyte-specific gene TYR in patients with vitiligo [45]. In addition, altered NALP1 (the gene encoding NACHT leucine-rich repeat protein 1), a regulator of innate immunity, was found to be a risk factor for vitiligo [46]. Recently, polymorphic expression of MTHFR (the gene encoding methylene tetrahydrofolate reductase), which regulates homocysteine levels, has been identified in patients with vitiligo [47]. XBP1P1 (the gene encoding X-box binding protein 1) has also been associated with vitiligo. It is pivotal in attenuating the unfolded protein response and driving stress-induced inflammation in vivo [48].
4.2. Oxidative Stress in Vitiligo
In vivo and in vitro investigations have confirmed widespread alterations of the antioxidant system in the skin and blood of patients with vitiligo [7,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63], suggesting important roles of oxidative stress in vitiligo onset and progression [4,64]. Melanocytic oxidative stress can cause a local inflammatory response and activate innate immunity through damage-associated molecular patterns (DAMPs), generating melanocyte-specific cytotoxic immune responses in genetically predisposed individuals [43]. Vitiligo melanocytes have decreased Ecad and increased expression of the anti-adhesion molecule tenascin [65,66] and are susceptible to oxidative stress [55] or UVB [67]. Given that melanocyte-KC interaction is vital for skin pigmentation, oxidative stress-driven melanocyte dysadhesion may represent an initial pathogenic event, which further precipitates oxidative damage [55,67,68] and leads to senescence (degeneration) [43]. Alterations in TYRP1 synthesis/processing impair eumelanogenesis and hamper melanosome maturation, increasing melanocyte oxidative damage [67]. Although mitochondria-derived ROS mediates aging or apoptosis in healthy cells [43], it is the culprit for vitiligo melanocyte dysfunction [69,70]. Oxidative stress impairs the function of membrane lipids and cellular proteins in melanocytes [67,68]. Biopterin synthesis and recycling are altered (i.e., increased production of 6-tetrahydrobiopterin and 7-tetrahydrobiopterin), leading to further oxidative stress and cell damage [71]. Oxidative stress can also affect McSCs in HFs, which might lead to a higher incidence of early hair graying in patients with vitiligo [72].
Recently, a molecular mechanism involved in oxidative stress-induced melanocyte degeneration has been proposed. Oxeiptosis is an apoptosis-like, nonclassical, ROS-induced cell death pathway [73]. Because H2O2 induces vitiligo melanocyte cell death, oxeiptosis may contribute to vitiligo pathogenesis [74]. The microRNA (miRNA) miR-25 suppresses MITF levels in melanocytes and SCF and bFGF expression in KCs, thereby contributing to melanocyte degeneration [75].
4.3. Immune Activation in Vitiligo
Autoimmunity has been implicated in vitiligo pathogenesis [76]. This is supported by the presence of antibodies against melanocytes, association with polymorphism at immune loci, prominent T cell infiltration in perilesional areas, cytokine expression, and association with other autoimmune diseases (e.g., autoimmune thyroiditis and type 1 diabetes mellitus) [43].
Oxidative stress connects innate immunity and adaptive immunity [77,78]. The activation of innate immune cells occurs upon sensing exogenously or endogenous stress signals, mainly from melanocytes [43]. ROS overproduction causes melanocytes to release exosomes [79] containing melanocyte-specific antigens, miRNAs, heat shock proteins, and other proteins that act as DAMPs [42]. These exosomes activate nearby dendritic cells and induce their maturation [80,81,82], followed by cytokine- and chemokine-driven T helper 17 cell activation [83,84,85] and regulatory T cell dysfunction [86,87]. Cytotoxic CD8+ T cells that target melanocytes are responsible for melanocyte destruction [88]. CD8+ T cells produce several cytokines, including interferon (IFN)-γ [88,89]. IFN-γ signaling activates the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway, leading to CXC chemokine ligand 9 (CXCL9) and CXCL10 secretion in the skin [90]. CXCL9 promotes the bulk recruitment of melanocyte-specific CD8+ cytotoxic T cells (CTLs) to the skin [90,91], whereas CXCL10 promotes the effector function of CD8+ T cells and their localization within the epidermis [90,91].
4.4. NRF2 in Vitiligo Pathology
NRF2 protects against cellular oxidative damage [5]. This principle makes the idea of supporting vitiligo melanocyte survival legitimate. NRF2 overexpression via transfection of NRF2 gene-containing plasmids (pCMV6-XL5-Nrf2) protects the immortalized human melanocyte cell line PIG1 against H2O2-induced oxidative stress [92]. α-MSH induces NRF2 and protects normal human epidermal melanocytes (NHEMs) against UVB-induced oxidative stress [93]. NRF2 activation by small interfering RNA-mediated KEAP1 knockdown protects NHEMs against toxicity induced by the depigmenting agent monobenzone (monobenzyl ether of hydroquinone [MBEH]) [94]. Augmented NRF2 signaling could replenish the cellular GSH pool (thus increasing the GSH/GSSG ratio) [5]. Accordingly, adenovirus-mediated NRF2 overexpression inhibits melanogenesis in NHEMs [6]. Conversely, knockdown of NRF2 or its downstream genes, such as NQO1 and PRDX6 reduces cultured melanocyte viability [94]. Melanocytes isolated from sut mice that harbor the mutation in an NRF2-target Slc7a11 gene [8] exhibit reduced rates of melanocyte viability/proliferation [34]. Moreover, sut mice exhibit reduced hair pheomelanin content, whereas that of eumelanin is barely affected although its effect on IFE pigmentation (tanning response) has not been assessed [34]. Furthermore, H2O2-induced oxidative stress promotes cytoplasmic translocation/release of high mobility group box 1 from NHEMs, resulting in indirect activation of NRF2 and its target genes (HMOX1, NQO1, GCLC, and GCLM) [95].
Evidence indicates that antioxidative response aberrations are central to vitiligo pathogenesis. SNPs in the NRF2 promoter may increase vitiligo risk [96], suggesting that aberrant antioxidative responses can be genetically determined. The epidermis of patients with vitiligo harbors increased H2O2 levels [58,61], and the lesional epidermis exhibits higher NRF2, NQO1, GCLC, and GCLM expression levels compared with the non-lesional epidermis [97]. An enhanced oxidative damage may increase the vulnerability of melanocytes to oxidative damage [7,94], which can be counteracted by local HO-1 augmentation using psoralen plus UVA (PUVA) treatment [98]. However, IL-2-induced expansion of circulating melanocyte-specific CD8+ cytotoxic T cells (CTLs) [99] may bring about a reductive environment (thus reduces GSH/GSSG ratios) [100], decreasing serum HO-1 levels [7]. Collectively, the vitiligo lesional epidermis appears to suffer from high oxidative damage, which may in turn dampen normal antioxidative responses. This notion is further supported by the attenuated induction of phase II detoxification genes in the lesional skin after in vitro and ex vivo treatment with the electrophilic compounds curcumin and santalol [97]. In this meticulous experimental setting, isolated KCs were found to be more susceptible to apoptosis, whereas melanocytes were relatively resistant against apoptosis [97]. These results suggest that melanocyte-KC communication sustains overall redox balance in the epidermis [38] as well as circulation [7].
Augmenting the antioxidative responses of melanocytes and supporting their survival by pharmacological means could be attractive measures. Several compounds can protect melanocytes against oxidative stress through NRF2 activation (Table 1). Melatonin and its metabolites (6-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine, N-acetylserotonin, and 5-methoxytryptamine) [101], vitamins (folic acid [102], methylcobalamin [103], and vitamin D [104]), natural compounds (4-octyl itaconate [105], ginsenoside Rk1 [106], Cistanche deserticola polysaccharides [107], glycyrrhizin [108], Lycium barbarum polysaccharides [109], paeoniflorin [110], 6-shogaol [111], paeonol [112], afzelin [113], apigenin [114], baicalein [115], vitexin [116], and berberine [117]), and therapeutic agents (aspirin [118], dimethyl fumarate [DMF] [94], simvastatin [29], molecular hydrogen [52], and cold atmospheric plasma [119]) are shown to protect melanocytes against oxidative damage caused by H2O2, UVB, or MBEH. DMF is the methyl ester of fumaric acid and is one of the most successful NRF2 activators. DMF has been approved by the United States Food and Drug Administration (FDA) for relapsing-remitting multiple sclerosis, a demyelinating autoimmune disease [120]. The European Medicines Agency has also approved DMF to treat moderate-to-severe plaque psoriasis [121]. DMF treatment enhances NRF2 nuclear localization and protects NHEMs and vitiligo melanocytes against MBEH-induced oxidative stress [94]. However, topical application of DMF can cause contact dermatitis [122]. This might be attributable to NRF2-enhanced skin sensitization [77,78]. Therefore, caution should be taken when applying potent electrophilic chemicals percutaneously, and refined drug delivery systems may be needed.
4.5. Management of Vitiligo
Vitiligo is not “just a cosmetic condition” but is psychologically devastating and stigmatizing [123]. The psychological impact on quality of life (QOL) is similar to that of other skin diseases, such as atopic dermatitis and psoriasis [124].
The aim of clinical management is to halt the autoimmune-driven depigmentation and restore the homeostatic pigmentation. Treatment options depend on several factors, such as disease subtype, extent, distribution, activity, patient age, phototype, effect on QOL, and motivation for treatment [42]. These treatments include topical therapies (e.g., corticosteroids and calcineurin inhibitors), phototherapies (e.g., photochemotherapies, narrowband UVB [NB-UVB], and excimer lasers or lamps], oral therapies (e.g., steroids and other immunosuppressants), surgery, and combination therapies [125]. Treatments are graded from first- to fourth-line options [125]. First-line treatment consists of topical therapy with corticosteroids and calcineurin inhibitors; second-line treatment, NB-UVB, PUVA, and systemic steroid therapy; third-line treatment, surgical grafting techniques; and fourth-line treatment, depigmentation therapies.
Although the abovementioned roles of oxidative stress in vitiligo rationalize antioxidant-based treatment, evidence of the efficacy of this treatment is quite limited [125]. To achieve repigmentation, pseudocatalase, vitamin E, vitamin C, ubiquinone, lipoic acid, Polypodium leucotomos, catalase/superoxide dismutase combination, and Ginkgo biloba may be administered with or without UV therapy [125]. Since the discovery of the role of the IFN-γ signaling axis, several clinical trials involving JAK inhibitors have been conducted [126]. JAK inhibitors, which target the type II IFN signaling pathway, have been shown to stimulate repigmentation in patients with vitiligo [127,128,129]. Tofacitinib, ruxolitinib, and baricitinib are the three major JAK inhibitors used for vitiligo. Ruxolitinib, an inhibitor of Janus kinase 1 (JAK1) and 2 (JAK2), was recently approved by the FDA to treat NSV in adult and pediatric patients aged ≥ 12 years. Ruxolitinib cream resulted in repigmentation through 52 weeks in phase 2 [130] and 3 [131] trials; however, its use is accompanied by acne and pruritus at the application site. Large-scale, long-term studies are required to elucidate the effects and risks of ruxolitinib cream application for vitiligo treatment.
4.6. Conclusions
Recent progress in vitiligo research has paved the way for disease pathway-based therapy. The IFN-γ-JAK-STAT pathway drives vitiligo pathogenesis, and JAK inhibitors, which presumably inhibit the effector function of CD49a+ cytotoxic epidermal resident CD8+ T cells efficiently [132], hold promise for better management of this emotionally devastating ailment. In this review, we initially aimed to examine the role of the KEAP1-NRF2 system in melanocyte biology/vitiligo pathogenesis. It has turned out, however, that this role [5] appears too far-reaching to be a disease-specific pathway. Systemic activation of the KEAP1-NRF2 system by the Keap1-null mutation not only augmented phase II detoxification but also led to uncontrolled keratinization of the squamous epithelium (SE) [9]. We and others have characterized the roles of the KEAP1-NRF2 system in skin diseases involving aberrations in inflammation/keratinization (reviewed in [9]). The aggregated evidence underscores the prominent roles of the KEAP1-NRF2 system in epidermal biology. In summary, the NRF2/KEAP1 system is important in vitiligo but far more specific than a therapeutic target.
5. Future Directions
Cutaneous pigmentary/depigmentary disorders, such as vitiligo or lentigines, do not necessarily accompany aberrant keratinization or acanthosis. Nonetheless, when considering the SE as a pigmentary “unit” [10], similar to classic immune cell components (the epidermal proliferation [differentiation] unit comprising KCs and Langerhans cells) [133], the epidermal “niche” being the ultimate determinant of cellular behavior may be evident [38]. Previous reports support this notion; unlike other minor epidermal residents, melanocytes express the desmosomal cadherin DSG1 [134], one of the critical commitment factors of IFE differentiation [135]. Loss of DSG1 in epidermal KCs may lead to melanocyte loss from the epithelium and promote invasive/metastatic growth of transformed melanocytes (melanoma) [134,136], suggesting that IFE KCs “imprint” (or instruct) melanocytic behaviors. This reasoning is further supported by the classic morphological changes in epidermal melanosomes following the topical application of the antipolymerization agent 4-tertiary butyl catechol [137], which could also augment the response of epidermal KCs to cellular distress (i.e., keratinization) [9]. This treatment blocks the eumelanin synthesis pathway, causing the appearance of immature pheomelanosomes in hairless mice [137]. Compared with pigmentation in the hair, tanning responses (IFE melanogenesis) depend on the nature of differentiated McSC-derived melanocytes; wet-surfaced SE (squamous mucosa), palmoplantar epidermis, or its apparatus (the nail) hardly experiences tanning responses (caused by the predominance of eumelanogenesis over pheomelanogenesis). The “niche” instruction or “structural imprinting” [138] aspect of pigmentogenesis would be further rationalized when IFE differentiation is analogized to sulfur metabolism; thiol groups of the proliferative layer are converted to disulfide polymerized keratins [139] (Figure 2). The principle, along with the possibility that the eumelanin synthetic pathway is regulated post-transcriptionally [35], tempted us to determine the “niche factor” within the IFE component. We have recently found that the IFE differentiating factor loricrin (LOR), which has a potent disulfide-linking capacity (reviewed in [138]), is indispensable for protection against UV radiation or electrophilic carcinogens (reviewed in [138]). Thus, we hypothesize that LOR could imprint the behaviors of IFE melanocytes and resident leukocytes [138]. The major effector of cornification (LOR) may act as a fate determinant of IFE melanocytes. Although further investigation and validation are required, revealing the hitherto unproven aspects of epidermal cell biology may lead to the development of mechanism-based skin pigmenting/depigmenting measures.
Author Contributions
Y.I.: conceptualization; T.O.: writing—original draft preparation; Y.I.: writing—review, final editing, artwork, and approval. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by the following grants: JSPS KAKENHI Grant, Grant-in-Aid for Scientific Research (C) (22K08404 to Y.I.), Lydia O’Leary Memorial Pias Dermatological Foundation (to Y.I.) and Shiseido basic medical research grant (to Y.I.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| α-MSH | α-Melanocyte-stimulating hormone |
| bFGF | Basic fibroblast growth factor |
| CTL | Cytotoxic T cell |
| CXCL9 | CXC chemokine ligand 9 |
| CXCL10 | CXC chemokine ligand 10 |
| CXCR3 | CXC receptor type 3 |
| DAMP | Damage-associated molecular pattern |
| DC | Dendritic cell |
| DHI | 5,6-Dihydroxyindole |
| DHICA | 5,6-Dihydroxyindole-2-carboxylic acid |
| DMF | Dimethyl fumarate |
| DSG | Desmoglein 1 |
| Ecad | E-cadherin |
| FDA | Food and Drug Administration |
| GCLC | Glutamate-cysteine ligase catalytic subunit |
| GCLM | Glutamate-cysteine ligase modifier subunit |
| gp100 | Glycoprotein 100 |
| GSH | Glutathione |
| GSK-3β | Glycogen synthase kinase-3β |
| GSSG | Glutathione disulfide |
| GST | Glutathione S-transferase |
| HF | Hair follicle |
| HIF-1α | Hypoxia-inducible factor-1α |
| HMGB1 | High mobility group box 1 |
| HO | Hydroxyl radicals |
| HO-1 | Heme oxygenase-1 |
| H2O2 | Hydrogen peroxide |
| IFE | Interfollicular epidermis |
| IFN-γ | Interferon-gamma |
| IL-1 | Interleukin-1 |
| IL-1β | Interleukin-1β |
| IL-17A | Interleukin-17A |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| JAK | Janus kinase |
| JAK1 | Janus kinase 1 |
| JAK2 | Janus kinase 2 |
| KC | Keratinocyte |
| KEAP1 | Kelch-like erythroid cell-derived protein with cap‘n’collar homology-associated protein 1 |
| LCE | Late cornified envelope |
| LDH | Lactate dehydrogenase |
| L-DOPA | L-3,4-Dihydroxyphenylalanine |
| LOR | Loricrin |
| MAPK | Mitogen-activated protein kinase |
| MBEH | Monobenzyl ether of hydroquinone |
| McSC | Melanocyte stem cell |
| miRNA | microRNA |
| MITF | Microphthalmia-associated transcription factor |
| NB-UVB | Narrowband ultraviolet B |
| NF-κB | Nuclear factor-κB |
| NHEMs | Normal human epidermal melanocytes |
| NLVMs | Non-lesional vitiligo melanocytes |
| NNT | Nicotinamide nucleotide transhydrogenase |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NRF2 | Nuclear factor erythroid-2-related factor 2 |
| NSV | Non-segmental vitiligo |
| O2− | Superoxide anions |
| PLVMs | Perilesional vitiligo melanocytes |
| PRDX1 | Peroxiredoxin 1 |
| PRDX6 | Peroxiredoxin 6 |
| PUVA | Psoralen plus ultraviolet A |
| QOL | Quality of life |
| ROS | Reactive oxygen species |
| SC | Stratum corneum |
| SE | Squamous epithelium |
| SG | Stratum granulosum |
| SLC7A11 | Solute carrier family 7 member 11 |
| SNP | Single nucleotide polymorphism |
| SPRR | Small proline-rich protein |
| SS | Stratum spinosum |
| STAT | Signal transducer and activator of transcription |
| sut | Subtle gray |
| SV | Segmental vitiligo |
| Treg | Regulatory T cell |
| TRM | Resident memory T cell |
| TXNRD1 | Thioredoxin reductase 1 |
| TYRP1 | Tyrosinase-related protein 1 |
| TYRP2 | Tyrosinase-related protein 2 |
| UV | Ultraviolet |
| UVA | Ultraviolet A |
| UVB | Ultraviolet B |
References
- Storz, G.; Imlayt, J.A. Oxidative stress. Curr. Opin. Microbiol. 1999, 2, 188–194. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Ezzedine, K.; Eleftheriadou, V.; Whitton, M.; van Geel, N. Vitiligo. Lancet 2015, 386, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Gani, A.R.; Ansarullah, M.; Ramachandran, A.V.; Dalai, S.; Begum, R. Vitiligo: Interplay between oxidative stress and immune system. Exp. Dermatol. 2013, 22, 245–250. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.M.; Kim, M.Y.; Sohn, K.C.; Jung, S.Y.; Lee, H.E.; Lim, J.W.; Kim, S.; Lee, Y.H.; Im, M.; Seo, Y.J.; et al. Nrf2 negatively regulates melanogenesis by modulating PI3K/Akt signaling. PLoS ONE 2014, 9, e96035. [Google Scholar] [CrossRef] [Green Version]
- Jian, Z.; Li, K.; Song, P.; Zhu, G.; Zhu, L.; Cui, T.; Liu, B.; Tang, L.; Wang, X.; Wang, G.; et al. Impaired activation of the Nrf2-ARE signaling pathway undermines H2O2-induced oxidative stress response: A possible mechanism for melanocyte degeneration in vitiligo. J. Investig. Dermatol. 2014, 134, 2221–2230. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [Green Version]
- Ishitsuka, Y.; Ogawa, T.; Roop, D. The KEAP1/NRF2 Signaling Pathway in Keratinization. Antioxidants 2020, 9, 751. [Google Scholar] [CrossRef]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin melanocytes: Biology and development. Postepy Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef]
- Haass, N.K.; Smalley, K.S.; Li, L.; Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef]
- Rabbani, P.; Takeo, M.; Chou, W.; Myung, P.; Bosenberg, M.; Chin, L.; Taketo, M.M.; Ito, M. Coordinated activation of Wnt in epithelial and melanocyte stem cells initiates pigmented hair regeneration. Cell 2011, 145, 941–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirobe, T. How are proliferation and differentiation of melanocytes regulated? Pigment Cell Melanoma Res. 2011, 24, 462–478. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Brenner, M.; Hearing, V.J. The regulation of skin pigmentation. J. Biol. Chem. 2007, 282, 27557–27561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- al Badri, A.M.; Foulis, A.K.; Todd, P.M.; Gariouch, J.J.; Gudgeon, J.E.; Stewart, D.G.; Gracie, J.A.; Goudie, R.B. Abnormal expression of MHC class II and ICAM-1 by melanocytes in vitiligo. J. Pathol. 1993, 169, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhu, W.Y.; Tan, C.; Yu, G.H.; Gu, J.X. Melanocytes are potential immunocompetent cells: Evidence from recognition of immunological characteristics of cultured human melanocytes. Pigment Cell Res. 2002, 15, 454–460. [Google Scholar] [CrossRef]
- Smit, N.; Le Poole, I.; van den Wijngaard, R.; Tigges, A.; Westerhof, W.; Das, P. Expression of different immunological markers by cultured human melanocytes. Arch. Dermatol. Res. 1993, 285, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Yohn, J.J.; Critelli, M.; Lyons, M.B.; Norris, D.A. Modulation of melanocyte intercellular adhesion molecule-1 by immune cytokines. J. Investig. Dermatol. 1990, 95, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Poole, I.C.; Boyce, S.T. Keratinocytes suppress transforming growth factor-beta1 expression by fibroblasts in cultured skin substitutes. Br. J. Dermatol. 1999, 140, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Swope, V.B.; Sauder, D.N.; McKenzie, R.C.; Sramkoski, R.M.; Krug, K.A.; Babcock, G.F.; Nordlund, J.J.; Abdel-Malek, Z.A. Synthesis of interleukin-1 alpha and beta by normal human melanocytes. J. Investig. Dermatol. 1994, 102, 749–753. [Google Scholar] [CrossRef]
- Zachariae, C.O.; Thestrup-Pedersen, K.; Matsushima, K. Expression and secretion of leukocyte chemotactic cytokines by normal human melanocytes and melanoma cells. J. Investig. Dermatol. 1991, 97, 593–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Seiji, M.; Fitzpatrick, T.B. The reciprocal relationship between melanization and tyrosinase activity in melanosomes (melanin granules). J. Biochem. 1961, 49, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Daniele, T.; Hurbain, I.; Vago, R.; Casari, G.; Raposo, G.; Tacchetti, C.; Schiaffino, M.V. Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr. Biol. 2014, 24, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Investig. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Wakamatsu, K. Diversity of human hair pigmentation as studied by chemical analysis of eumelanin and pheomelanin. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1369–1380. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Human hair melanins: What we have learned and have not learned from mouse coat color pigmentation. Pigment Cell Melanoma Res. 2011, 24, 63–74. [Google Scholar] [CrossRef]
- Benedetto, J.P.; Ortonne, J.P.; Voulot, C.; Khatchadourian, C.; Prota, G.; Thivolet, J. Role of thiol compounds in mammalian melanin pigmentation: Part I. Reduced and oxidized glutathione. J. Investig. Dermatol. 1981, 77, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Li, S.; Guo, W.; Yang, Y.; Zhang, W.; Zhang, Q.; He, Y.; Yi, X.; Cui, T.; An, Y.; et al. Simvastatin Protects Human Melanocytes from H2O2-Induced Oxidative Stress by Activating Nrf2. J. Investig. Dermatol. 2017, 137, 1286–1296. [Google Scholar] [CrossRef] [Green Version]
- Fried, L.; Arbiser, J.L. The reactive oxygen-driven tumor: Relevance to melanoma. Pigment Cell Melanoma Res. 2008, 21, 117–122. [Google Scholar] [CrossRef]
- Takeuchi, S.; Zhang, W.; Wakamatsu, K.; Ito, S.; Hearing, V.J.; Kraemer, K.H.; Brash, D.E. Melanin acts as a potent UVB photosensitizer to cause an atypical mode of cell death in murine skin. Proc. Natl. Acad. Sci. USA 2004, 101, 15076–15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunathilake, R.; Schurer, N.Y.; Shoo, B.A.; Celli, A.; Hachem, J.P.; Crumrine, D.; Sirimanna, G.; Feingold, K.R.; Mauro, T.M.; Elias, P.M. pH-regulated mechanisms account for pigment-type differences in epidermal barrier function. J. Investig. Dermatol. 2009, 129, 1719–1729. [Google Scholar] [CrossRef] [Green Version]
- Man, M.Q.; Lin, T.K.; Santiago, J.L.; Celli, A.; Zhong, L.; Huang, Z.M.; Roelandt, T.; Hupe, M.; Sundberg, J.P.; Silva, K.A.; et al. Basis for enhanced barrier function of pigmented skin. J. Investig. Dermatol. 2014, 134, 2399–2407. [Google Scholar] [CrossRef] [Green Version]
- Chintala, S.; Li, W.; Lamoreux, M.L.; Ito, S.; Wakamatsu, K.; Sviderskaya, E.V.; Bennett, D.C.; Park, Y.M.; Gahl, W.A.; Huizing, M.; et al. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc. Natl. Acad. Sci. USA 2005, 102, 10964–10969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allouche, J.; Rachmin, I.; Adhikari, K.; Pardo, L.M.; Lee, J.H.; McConnell, A.M.; Kato, S.; Fan, S.; Kawakami, A.; Suita, Y.; et al. NNT mediates redox-dependent pigmentation via a UVB- and MITF-independent mechanism. Cell 2021, 184, 4268–4283.e20. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Pagliarani, A. Nicotinamide Nucleotide Transhydrogenase as a Sensor of Mitochondrial Biology. Trends Cell Biol. 2020, 30, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Loffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef] [Green Version]
- De Luca, M.; D’Anna, F.; Bondanza, S.; Franzi, A.T.; Cancedda, R. Human epithelial cells induce human melanocyte growth in vitro but only skin keratinocytes regulate its proper differentiation in the absence of dermis. J. Cell Biol. 1988, 107, 1919–1926. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.; Ezzedine, K.; Hamzavi, I.; Pandya, A.G.; Harris, J.E. New discoveries in the pathogenesis and classification of vitiligo. J. Am. Acad. Dermatol. 2017, 77, 1–13. [Google Scholar] [CrossRef]
- Sehgal, V.N.; Srivastava, G. Vitiligo: Compendium of clinico-epidemiological features. Indian J. Dermatol. Venereol. Leprol. 2007, 73, 149–156. [Google Scholar] [CrossRef]
- Ezzedine, K.; Lim, H.W.; Suzuki, T.; Katayama, I.; Hamzavi, I.; Lan, C.C.; Goh, B.K.; Anbar, T.; Silva de Castro, C.; Lee, A.Y.; et al. Revised classification/nomenclature of vitiligo and related issues: The Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012, 25, E1–E13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A Review. Dermatology 2020, 236, 571–592. [Google Scholar] [CrossRef] [PubMed]
- Picardo, M.; Dell’Anna, M.L.; Ezzedine, K.; Hamzavi, I.; Harris, J.E.; Parsad, D.; Taieb, A. Vitiligo. Nat. Rev. Dis. Prim. 2015, 1, 15011. [Google Scholar] [CrossRef] [PubMed]
- Spritz, R.A.; Andersen, G.H. Genetics of Vitiligo. Dermatol. Clin. 2017, 35, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Gowan, K.; Riccardi, S.L.; Holland, P.J.; Mailloux, C.M.; Sufit, A.J.; Hutton, S.M.; Amadi-Myers, A.; et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N. Engl. J. Med. 2010, 362, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Mailloux, C.M.; Gowan, K.; Riccardi, S.L.; LaBerge, G.; Bennett, D.C.; Fain, P.R.; Spritz, R.A. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 2007, 356, 1216–1225. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.X.; Shi, Q.; Wang, X.W.; Guo, S.; Dai, W.; Li, K.; Song, P.; Wei, C.; Wang, G.; Li, C.Y.; et al. Genetic polymorphisms in the methylenetetrahydrofolate reductase gene (MTHFR) and risk of vitiligo in Han Chinese populations: A genotype-phenotype correlation study. Br. J. Dermatol. 2014, 170, 1092–1099. [Google Scholar] [CrossRef]
- Ren, Y.; Yang, S.; Xu, S.; Gao, M.; Huang, W.; Gao, T.; Fang, Q.; Quan, C.; Zhang, C.; Sun, L.; et al. Genetic variation of promoter sequence modulates XBP1 expression and genetic risk for vitiligo. PLoS Genet. 2009, 5, e1000523. [Google Scholar] [CrossRef]
- Agrawal, D.; Shajil, E.M.; Marfatia, Y.S.; Begum, R. Study on the antioxidant status of vitiligo patients of different age groups in Baroda. Pigment Cell Res. 2004, 17, 289–294. [Google Scholar] [CrossRef]
- Boissy, R.E.; Manga, P. On the etiology of contact/occupational vitiligo. Pigment Cell Res. 2004, 17, 208–214. [Google Scholar] [CrossRef]
- Bulut, H.; Pehlivan, M.; Alper, S.; Tomatir, A.G.; Onay, H.; Yüksel, S.E.; Ozkinay, F.; Pehlivan, S. Lack of association between catalase gene polymorphism (T/C exon 9) and susceptibility to vitiligo in a Turkish population. Genet. Mol. Res. 2011, 10, 4126–4132. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Tang, L.; Wang, G.; Lin, J.; Liao, W.; Pan, W.; Xu, J. Molecular Hydrogen Protects Human Melanocytes from Oxidative Stress by Activating Nrf2 Signaling. J. Investig. Dermatol. 2020, 140, 2230–2241.e9. [Google Scholar] [CrossRef] [PubMed]
- Kostyuk, V.A.; Potapovich, A.I.; Cesareo, E.; Brescia, S.; Guerra, L.; Valacchi, G.; Pecorelli, A.; Deeva, I.B.; Raskovic, D.; De Luca, C.; et al. Dysfunction of glutathione S-transferase leads to excess 4-hydroxy-2-nonenal and H2O2 and impaired cytokine pattern in cultured keratinocytes and blood of vitiligo patients. Antioxid. Redox Signal. 2010, 13, 607–620. [Google Scholar] [CrossRef]
- Liu, L.; Li, C.; Gao, J.; Li, K.; Zhang, R.; Wang, G.; Li, C.; Gao, T. Promoter variant in the catalase gene is associated with vitiligo in Chinese people. J. Investig. Dermatol. 2010, 130, 2647–2653. [Google Scholar] [CrossRef] [Green Version]
- Maresca, V.; Roccella, M.; Roccella, F.; Camera, E.; Del Porto, G.; Passi, S.; Grammatico, P.; Picardo, M. Increased sensitivity to peroxidative agents as a possible pathogenic factor of melanocyte damage in vitiligo. J. Investig. Dermatol. 1997, 109, 310–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrone, A.; Picardo, M.; de Luca, C.; Terminali, O.; Passi, S.; Ippolito, F. Catecholamines and vitiligo. Pigment Cell Res. 1992, 5, 65–69. [Google Scholar] [CrossRef]
- Ozturk, I.C.; Batcioglu, K.; Karatas, F.; Hazneci, E.; Genc, M. Comparison of plasma malondialdehyde, glutathione, glutathione peroxidase, hydroxyproline and selenium levels in patients with vitiligo and healthy controls. Indian J. Dermatol. 2008, 53, 106–110. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Gaze, D.C.; Tobin, D.J.; Marshall, H.S.; Panske, A.; Panzig, E.; Hibberts, N.A. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J. Investig. Dermatol. Symp. Proc. 1999, 4, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Schallreuter, K.U.; Salem, M.A.; Gibbons, N.C.; Maitland, D.J.; Marsch, E.; Elwary, S.M.; Healey, A.R. Blunted epidermal L-tryptophan metabolism in vitiligo affects immune response and ROS scavenging by Fenton chemistry, part 2: Epidermal H2O2/ONOO(-)-mediated stress in vitiligo hampers indoleamine 2,3-dioxygenase and aryl hydrocarbon receptor-mediated immune response signaling. FASEB J. 2012, 26, 2471–2485. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Wood, J.M.; Berger, J. Low catalase levels in the epidermis of patients with vitiligo. J. Investig. Dermatol. 1991, 97, 1081–1085. [Google Scholar] [CrossRef]
- Shalbaf, M.; Gibbons, N.C.; Wood, J.M.; Maitland, D.J.; Rokos, H.; Elwary, S.M.; Marles, L.K.; Schallreuter, K.U. Presence of epidermal allantoin further supports oxidative stress in vitiligo. Exp. Dermatol. 2008, 17, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Sravani, P.V.; Babu, N.K.; Gopal, K.V.; Rao, G.R.; Rao, A.R.; Moorthy, B.; Rao, T.R. Determination of oxidative stress in vitiligo by measuring superoxide dismutase and catalase levels in vitiliginous and non-vitiliginous skin. Indian J. Dermatol. Venereol. Leprol. 2009, 75, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, M.; Baysal, V.; Inaloz, H.S.; Can, M. The role of oxidants and antioxidants in generalized vitiligo at tissue level. J. Eur. Acad. Dermatol. Venereol. 2004, 18, 683–686. [Google Scholar] [CrossRef]
- Speeckaert, R.; Dugardin, J.; Lambert, J.; Lapeere, H.; Verhaeghe, E.; Speeckaert, M.M.; van Geel, N. Critical appraisal of the oxidative stress pathway in vitiligo: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1089–1098. [Google Scholar] [CrossRef]
- Le Poole, I.C.; van den Wijngaard, R.M.; Westerhof, W.; Das, P.K. Tenascin is overexpressed in vitiligo lesional skin and inhibits melanocyte adhesion. Br. J. Dermatol. 1997, 137, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Wagner, R.Y.; Luciani, F.; Cario-André, M.; Rubod, A.; Petit, V.; Benzekri, L.; Ezzedine, K.; Lepreux, S.; Steingrimsson, E.; Taieb, A.; et al. Altered E-Cadherin Levels and Distribution in Melanocytes Precede Clinical Manifestations of Vitiligo. J. Investig. Dermatol. 2015, 135, 1810–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimbow, K.; Chen, H.; Park, J.S.; Thomas, P.D. Increased sensitivity of melanocytes to oxidative stress and abnormal expression of tyrosinase-related protein in vitiligo. Br. J. Dermatol. 2001, 144, 55–65. [Google Scholar] [CrossRef]
- Dell’Anna, M.L.; Ottaviani, M.; Albanesi, V.; Vidolin, A.P.; Leone, G.; Ferraro, C.; Cossarizza, A.; Rossi, L.; Picardo, M. Membrane lipid alterations as a possible basis for melanocyte degeneration in vitiligo. J. Investig. Dermatol. 2007, 127, 1226–1233. [Google Scholar] [CrossRef]
- Dell’Anna, M.L.; Maresca, V.; Briganti, S.; Camera, E.; Falchi, M.; Picardo, M. Mitochondrial impairment in peripheral blood mononuclear cells during the active phase of vitiligo. J. Investig. Dermatol. 2001, 117, 908–913. [Google Scholar] [CrossRef] [Green Version]
- Dell’Anna, M.L.; Urbanelli, S.; Mastrofrancesco, A.; Camera, E.; Iacovelli, P.; Leone, G.; Manini, P.; D’Ischia, M.; Picardo, M. Alterations of mitochondria in peripheral blood mononuclear cells of vitiligo patients. Pigment Cell Res. 2003, 16, 553–559. [Google Scholar] [CrossRef]
- Hasse, S.; Gibbons, N.C.; Rokos, H.; Marles, L.K.; Schallreuter, K.U. Perturbed 6-tetrahydrobiopterin recycling via decreased dihydropteridine reductase in vitiligo: More evidence for H2O2 stress. J. Investig. Dermatol. 2004, 122, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Luo, L.F.; Liu, X.M.; Zhou, Q.; Xu, S.Z.; Lei, T.C. Premature graying as a consequence of compromised antioxidant activity in hair bulb melanocytes and their precursors. PLoS ONE 2014, 9, e93589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.A.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef]
- Kang, P.; Chen, J.; Zhang, W.; Guo, N.; Yi, X.; Cui, T.; Chen, J.; Yang, Y.; Wang, Y.; Du, P.; et al. Oxeiptosis: A novel pathway of melanocytes death in response to oxidative stress in vitiligo. Cell Death Discov. 2022, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, W.; Guo, S.; Jian, Z.; Li, S.; Li, K.; Ge, R.; Dai, W.; Wang, G.; Gao, T.; et al. Oxidative stress-induced overexpression of miR-25: The mechanism underlying the degeneration of melanocytes in vitiligo. Cell Death Differ. 2016, 23, 496–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongenae, K.; Van Geel, N.; Naeyaert, J.-M. Evidence for an Autoimmune Pathogenesis of Vitiligo. Pigment. Cell Res. 2003, 16, 90–100. [Google Scholar] [CrossRef]
- Ogawa, T.; Ishitsuka, Y. The Role of KEAP1-NRF2 System in Atopic Dermatitis and Psoriasis. Antioxidants 2022, 11, 1397. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Ishitsuka, Y.; Nakamura, Y.; Kubota, N.; Saito, A.; Fujisawa, Y.; Watanabe, R.; Okiyama, N.; Suga, Y.; Roop, D.R.; et al. NRF2 Augments Epidermal Antioxidant Defenses and Promotes Atopy. J. Immunol. 2020, 205, 907–914. [Google Scholar] [CrossRef]
- van den Boorn, J.G.; Picavet, D.I.; van Swieten, P.F.; van Veen, H.A.; Konijnenberg, D.; van Veelen, P.A.; van Capel, T.; Jong, E.C.; Reits, E.A.; Drijfhout, J.W.; et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J. Investig. Dermatol. 2011, 131, 1240–1251. [Google Scholar] [CrossRef] [Green Version]
- Al-Shobaili, H.A.; Rasheed, Z. Mitochondrial DNA acquires immunogenicity on exposure to nitrosative stress in patients with vitiligo. Hum. Immunol. 2014, 75, 1053–1061. [Google Scholar] [CrossRef]
- Kroll, T.M.; Bommiasamy, H.; Boissy, R.E.; Hernandez, C.; Nickoloff, B.J.; Mestril, R.; Caroline Le Poole, I. 4-Tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cell-mediated killing: Relevance to vitiligo. J. Investig. Dermatol. 2005, 124, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passeron, T.; Ortonne, J.P. Activation of the unfolded protein response in vitiligo: The missing link? J. Investig. Dermatol. 2012, 132, 2502–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassiouny, D.A.; Shaker, O. Role of interleukin-17 in the pathogenesis of vitiligo. Clin. Exp. Dermatol. 2011, 36, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Kotobuki, Y.; Tanemura, A.; Yang, L.; Itoi, S.; Wataya-Kaneda, M.; Murota, H.; Fujimoto, M.; Serada, S.; Naka, T.; Katayama, I. Dysregulation of melanocyte function by Th17-related cytokines: Significance of Th17 cell infiltration in autoimmune vitiligo vulgaris. Pigment Cell Melanoma Res. 2012, 25, 219–230. [Google Scholar] [CrossRef]
- Wang, C.Q.; Cruz-Inigo, A.E.; Fuentes-Duculan, J.; Moussai, D.; Gulati, N.; Sullivan-Whalen, M.; Gilleaudeau, P.; Cohen, J.A.; Krueger, J.G. Th17 cells and activated dendritic cells are increased in vitiligo lesions. PLoS ONE 2011, 6, e18907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Eby, J.M.; Al-Khami, A.A.; Soloshchenko, M.; Kang, H.-K.; Kaur, N.; Naga, O.S.; Murali, A.; Nishimura, M.I.; Caroline Le Poole, I.; et al. A Quantitative Increase in Regulatory T Cells Controls Development of Vitiligo. J. Investig. Dermatol. 2014, 134, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarquist, J.; Denman, C.J.; Hernandez, C.; Wainwright, D.J.; Strickland, F.M.; Overbeck, A.; Mehrotra, S.; Nishimura, M.I.; Le Poole, I.C. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. 2010, 23, 276–286. [Google Scholar] [CrossRef]
- van den Boorn, J.G.; Konijnenberg, D.; Dellemijn, T.A.; van der Veen, J.P.; Bos, J.D.; Melief, C.J.; Vyth-Dreese, F.A.; Luiten, R.M. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J. Investig. Dermatol. 2009, 129, 2220–2232. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.E.; Harris, T.H.; Weninger, W.; Wherry, E.J.; Hunter, C.A.; Turka, L.A. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8+ T-cell accumulation in the skin. J. Investig. Dermatol. 2012, 132, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Rashighi, M.; Agarwal, P.; Richmond, J.M.; Harris, T.H.; Dresser, K.; Su, M.W.; Zhou, Y.; Deng, A.; Hunter, C.A.; Luster, A.D.; et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci. Transl. Med. 2014, 6, 223ra223. [Google Scholar] [CrossRef]
- Harris, J.E. Cellular stress and innate inflammation in organ-specific autoimmunity: Lessons learned from vitiligo. Immunol. Rev. 2016, 269, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J. Investig. Dermatol. 2011, 131, 1420–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokot, A.; Metze, D.; Mouchet, N.; Galibert, M.D.; Schiller, M.; Luger, T.A.; Böhm, M. Alpha-melanocyte-stimulating hormone counteracts the suppressive effect of UVB on Nrf2 and Nrf-dependent gene expression in human skin. Endocrinology 2009, 150, 3197–3206. [Google Scholar] [CrossRef] [Green Version]
- Arowojolu, O.A.; Orlow, S.J.; Elbuluk, N.; Manga, P. The nuclear factor (erythroid-derived 2)-like 2 (NRF2) antioxidant response promotes melanocyte viability and reduces toxicity of the vitiligo-inducing phenol monobenzone. Exp. Dermatol. 2017, 26, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Mou, K.; Liu, W.; Miao, Y.; Cao, F.; Li, P. HMGB1 deficiency reduces H2O2-induced oxidative damage in human melanocytes via the Nrf2 pathway. J. Cell. Mol. Med. 2018, 22, 6148–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, C.P.; Zhou, M.N.; Xu, A.E.; Kang, K.F.; Liu, J.F.; Wei, X.D.; Li, Y.W.; Zhao, D.K.; Hong, W.S. The susceptibility to vitiligo is associated with NF-E2-related factor2 (Nrf2) gene polymorphisms: A study on Chinese Han population. Exp. Dermatol. 2008, 17, 1059–1062. [Google Scholar] [CrossRef]
- Natarajan, V.T.; Singh, A.; Kumar, A.A.; Sharma, P.; Kar, H.K.; Marrot, L.; Meunier, J.-R.; Natarajan, K.; Rani, R.; Gokhale, R.S. Transcriptional Upregulation of Nrf2-Dependent Phase II Detoxification Genes in the Involved Epidermis of Vitiligo Vulgaris. J. Investig. Dermatol. 2010, 130, 2781–2789. [Google Scholar] [CrossRef] [Green Version]
- Elassiuty, Y.E.; Klarquist, J.; Speiser, J.; Yousef, R.M.; EL Refaee, A.A.; Hunter, N.S.; Shaker, O.G.; Gundeti, M.; Nieuweboer-Krobotova, L.; Caroline Le Poole, I. Heme oxygenase-1 expression protects melanocytes from stress-induced cell death: Implications for vitiligo. Exp. Dermatol. 2011, 20, 496–501. [Google Scholar] [CrossRef]
- Maeda, Y.; Nishikawa, H.; Sugiyama, D.; Ha, D.; Hamaguchi, M.; Saito, T.; Nishioka, M.; Wing, J.B.; Adeegbe, D.; Katayama, I.; et al. Detection of self-reactive CD8+ T cells with an anergic phenotype in healthy individuals. Science 2014, 346, 1536–1540. [Google Scholar] [CrossRef]
- Angelini, G.; Gardella, S.; Ardy, M.; Ciriolo, M.R.; Filomeni, G.; Di Trapani, G.; Clarke, F.; Sitia, R.; Rubartelli, A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc. Natl. Acad. Sci. USA 2002, 99, 1491–1496. [Google Scholar] [CrossRef]
- Janjetovic, Z.; Jarrett, S.G.; Lee, E.F.; Duprey, C.; Reiter, R.J.; Slominski, A.T. Melatonin and its metabolites protect human melanocytes against UVB-induced damage: Involvement of NRF2-mediated pathways. Sci. Rep. 2017, 7, 1274. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Zhang, S.; Li, S.; Yang, Y.; Kang, P.; Chen, J.; Gao, T.; Li, C.; Zhang, Q.; Zhang, W. Folic Acid Protects Melanocytes from Oxidative Stress via Activation of Nrf2 and Inhibition of HMGB1. Oxidative Med. Cell. Longev. 2021, 2021, 1608586. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Li, D.; Dong, Y.; She, Q.; Zhou, T.; Nie, X.; Pan, R.; Deng, Y. Methylcobalamin Protects Melanocytes from H2O2-Induced Oxidative Stress by Activating the Nrf2/HO-1 Pathway. Drug Des. Devel. Ther. 2021, 15, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Fang, W.; Lin, J.; Li, J.; Wu, W.; Xu, J. Vitamin D protects human melanocytes against oxidative damage by activation of Wnt/β-catenin signaling. Lab. Investig. 2018, 98, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Chen, Z.; Wu, Z. Four-Octyl Itaconate Attenuates UVB-Induced Melanocytes and Keratinocytes Apoptosis by Nrf2 Activation-Dependent ROS Inhibition. Oxidative Med. Cell. Longev. 2022, 2022, 9897442. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Yang, J.; Yan, K.; Guo, J. Ginsenoside Rk1 protects human melanocytes from H2O2-induced oxidative injury via regulation of the PI3K/AKT/Nrf2/HO-1 pathway. Mol. Med. Rep. 2021, 24, 821. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, J.; Li, Y.; Jiang, L.; Ouyang, Y.; Li, Y.; Yang, L.; Zhao, X.; Huang, L.; Xiang, H.; et al. Cistanche deserticola polysaccharide induces melanogenesis in melanocytes and reduces oxidative stress via activating NRF2/HO-1 pathway. J. Cell. Mol. Med. 2020, 24, 4023–4035. [Google Scholar] [CrossRef]
- Mou, K.; Pan, W.; Han, D.; Wen, X.; Cao, F.; Miao, Y.; Li, P. Glycyrrhizin protects human melanocytes from H2O2-induced oxidative damage via the Nrf2-dependent induction of HO-1. Int. J. Mol. Med. 2019, 44, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Lu, Y.; Zhong, J.; Ke, Y.; Li, Y.; Liang, B.; Li, H.; Zhu, H.; Li, Z. Lycium barbarum polysaccharide promotes proliferation of human melanocytes via activating the Nrf2/p62 signaling pathway by inducing autophagy in vitro. J. Food Biochem. 2022, 46, e14301. [Google Scholar] [CrossRef]
- Yuan, J.; Lu, Y.; Wang, H.; Feng, Y.; Jiang, S.; Gao, X.-H.; Qi, R.; Wu, Y.; Chen, H.-D. Paeoniflorin Resists H2O2-Induced Oxidative Stress in Melanocytes by JNK/Nrf2/HO-1 Pathway. Front. Pharmacol. 2020, 11, 536. [Google Scholar] [CrossRef]
- Yang, L.; Yang, F.; Teng, L.; Katayama, I. 6-Shogaol Protects Human Melanocytes against Oxidative Stress through Activation of the Nrf2-Antioxidant Response Element Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 3537. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Q. Paeonol protects melanocytes against hydrogen peroxide-induced oxidative stress through activation of Nrf2 signaling pathway. Drug Dev. Res. 2021, 82, 861–869. [Google Scholar] [CrossRef]
- Jung, E.; Kim, J.H.; Kim, M.O.; Lee, S.Y.; Lee, J. Melanocyte-protective effect of afzelin is mediated by the Nrf2-ARE signalling pathway via GSK-3β inactivation. Exp. Dermatol. 2017, 26, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Zhao, G.; Lin, M.; Lang, Y.; Zhang, D.; Feng, D.; Tu, C. Apigenin protects human melanocytes against oxidative damage by activation of the Nrf2 pathway. Cell Stress Chaperones 2020, 25, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, S.; Zhu, L.; Guo, S.; Yi, X.; Cui, T.; He, Y.; Chang, Y.; Liu, B.; Li, C.; et al. Baicalein protects human vitiligo melanocytes from oxidative stress through activation of NF-E2-related factor2 (Nrf2) signaling pathway. Free Radic. Biol. Med. 2018, 129, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-S.; Tang, X.-Y.; Su, W.; Li, X. Vitexin protects melanocytes from oxidative stress via activating MAPK-Nrf2/ARE pathway. Immunopharmacol. Immunotoxicol. 2020, 42, 594–603. [Google Scholar] [CrossRef]
- Jiang, W.; Li, S.; Chen, X.; Zhang, W.; Chang, Y.; He, Y.; Zhang, S.; Su, X.; Gao, T.; Li, C.; et al. Berberine protects immortalized line of human melanocytes from H2O2-induced oxidative stress via activation of Nrf2 and Mitf signaling pathway. J. Dermatol. Sci. 2019, 94, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Jian, Z.; Tang, L.; Yi, X.; Liu, B.; Zhang, Q.; Zhu, G.; Wang, G.; Gao, T.; Li, C. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H2O2-induced oxidative stress. J. Cell. Mol. Med. 2016, 20, 1307–1318. [Google Scholar] [CrossRef]
- Zhai, S.; Xu, M.; Li, Q.; Guo, K.; Chen, H.; Kong, M.G.; Xia, Y. Successful Treatment of Vitiligo with Cold Atmospheric Plasma—Activated Hydrogel. J. Investig. Dermatol. 2021, 141, 2710–2719.e2716. [Google Scholar] [CrossRef]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef]
- Mrowietz, U.; Barker, J.; Boehncke, W.H.; Iversen, L.; Kirby, B.; Naldi, L.; Reich, K.; Tanew, A.; van de Kerkhof, P.C.M.; Warren, R.B. Clinical use of dimethyl fumarate in moderate-to-severe plaque-type psoriasis: A European expert consensus. J. Eur. Acad. Dermatol. Venereol. 2018, 32 (Suppl. 3), 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahti, A.; Maibach, H.I. Contact urticaria from diethyl fumarate. Contact Dermat. 1985, 12, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ezzedine, K.; Sheth, V.; Rodrigues, M.; Eleftheriadou, V.; Harris, J.E.; Hamzavi, I.H.; Pandya, A.G. Vitiligo is not a cosmetic disease. J. Am. Acad. Dermatol. 2015, 73, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Linthorst Homan, M.W.; Spuls, P.I.; de Korte, J.; Bos, J.D.; Sprangers, M.A.; van der Veen, J.P.W. The burden of vitiligo: Patient characteristics associated with quality of life. J. Am. Acad. Dermatol. 2009, 61, 411–420. [Google Scholar] [CrossRef]
- Taieb, A.; Alomar, A.; Böhm, M.; Dell’anna, M.L.; De Pase, A.; Eleftheriadou, V.; Ezzedine, K.; Gauthier, Y.; Gawkrodger, D.J.; Jouary, T.; et al. Guidelines for the management of vitiligo: The European Dermatology Forum consensus. Br. J. Dermatol. 2013, 168, 5–19. [Google Scholar] [CrossRef]
- Qi, F.; Liu, F.; Gao, L. Janus Kinase Inhibitors in the Treatment of Vitiligo: A Review. Front. Immunol. 2021, 12, 790125. [Google Scholar] [CrossRef]
- Joshipura, D.; Alomran, A.; Zancanaro, P.; Rosmarin, D. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib: A 32-week open-label extension study with optional narrow-band ultraviolet B. J. Am. Acad. Dermatol. 2018, 78, 1205–1207.e1201. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.Y.; Strassner, J.P.; Refat, M.A.; Harris, J.E.; King, B.A. Repigmentation in vitiligo using the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J. Am. Acad. Dermatol. 2017, 77, 675–682.e1. [Google Scholar] [CrossRef]
- Rothstein, B.; Joshipura, D.; Saraiya, A.; Abdat, R.; Ashkar, H.; Turkowski, Y.; Sheth, V.; Huang, V.; Au, S.C.; Kachuk, C.; et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J. Am. Acad. Dermatol. 2017, 76, 1054–1060.e1. [Google Scholar] [CrossRef]
- Rosmarin, D.; Pandya, A.G.; Lebwohl, M.; Grimes, P.; Hamzavi, I.; Gottlieb, A.B.; Butler, K.; Kuo, F.; Sun, K.; Ji, T.; et al. Ruxolitinib cream for treatment of vitiligo: A randomised, controlled, phase 2 trial. Lancet 2020, 396, 110–120. [Google Scholar] [CrossRef]
- Rosmarin, D.; Passeron, T.; Pandya, A.G.; Grimes, P.; Harris, J.E.; Desai, S.R.; Lebwohl, M.; Ruer-Mulard, M.; Seneschal, J.; Wolkerstorfer, A.; et al. Two Phase 3, Randomized, Controlled Trials of Ruxolitinib Cream for Vitiligo. N. Engl. J. Med. 2022, 387, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, S.; Schlums, H.; Gallais Serezal, I.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Matte-Martone, C.; Gonzalez, D.G.; Lathrop, E.A.; May, D.P.; Pineda, C.M.; Moore, J.L.; Boucher, J.D.; Marsh, E.; Schmitter-Sanchez, A.; et al. Skin-resident immune cells actively coordinate their distribution with epidermal cells during homeostasis. Nat. Cell. Biol. 2021, 23, 476–484. [Google Scholar] [CrossRef]
- Li, G.; Schaider, H.; Satyamoorthy, K.; Hanakawa, Y.; Hashimoto, K.; Herlyn, M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene 2001, 20, 8125–8135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getsios, S.; Simpson, C.L.; Kojima, S.; Harmon, R.; Sheu, L.J.; Dusek, R.L.; Cornwell, M.; Green, K.J. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J. Cell Biol. 2009, 185, 1243–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnette, C.R.; Roth-Carter, Q.R.; Koetsier, J.L.; Broussard, J.A.; Burks, H.E.; Cheng, K.; Amadi, C.; Gerami, P.; Johnson, J.L.; Green, K.J. Keratinocyte cadherin desmoglein 1 controls melanocyte behavior through paracrine signaling. Pigment Cell Melanoma Res. 2020, 33, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Gellin, G.A.; Hoshino, S.; Epstein, J.H.; Epstein, W.L.; Fukuyama, K. Ultrastructural demonstration of chemical modification of melanogenesis in hairless mouse skin. Anat. Rec. 1982, 202, 193–201. [Google Scholar] [CrossRef]
- Ishitsuka, Y.; Roop, D.R. Loricrin at the Boundary between Inside and Outside. Biomolecules 2022, 12, 673. [Google Scholar] [CrossRef]
- Van Scott, E.; Flesch, P. Sulfhydryl and disulfide in keratinization. Science 1954, 119, 70–71. [Google Scholar] [CrossRef]
Figure 1.
Overview of melanogenesis pathways. Tyrosinase (TYR), the rate-limiting enzyme for melanogenesis, oxidizes tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) and dopaquinone. Dopaquinone reacts with excess cysteine/glutathione and generates cysteinyldopa, giving rise to pheomelanin. With cystine/glutathione disulfide abundance, dopaquinone generates dopachrome. Dopachrome undergoes spontaneous decarboxylation and forms 5,6-dihydroxyindole (DHI). In the presence of tyrosinase-related protein 2 (TYRP2), dopachrome produces 5,6-dihydroxyindole-2-carboxylic acid (DHICA) through tautomerization. TYR and tyrosinase-related protein 1 (TYRP1) catalyze further conversions, yielding eumelanin. The catalytic activity of TYR or TYRP1 generates superoxide anions (O2−) and hydrogen peroxide (H2O2).
Figure 1.
Overview of melanogenesis pathways. Tyrosinase (TYR), the rate-limiting enzyme for melanogenesis, oxidizes tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) and dopaquinone. Dopaquinone reacts with excess cysteine/glutathione and generates cysteinyldopa, giving rise to pheomelanin. With cystine/glutathione disulfide abundance, dopaquinone generates dopachrome. Dopachrome undergoes spontaneous decarboxylation and forms 5,6-dihydroxyindole (DHI). In the presence of tyrosinase-related protein 2 (TYRP2), dopachrome produces 5,6-dihydroxyindole-2-carboxylic acid (DHICA) through tautomerization. TYR and tyrosinase-related protein 1 (TYRP1) catalyze further conversions, yielding eumelanin. The catalytic activity of TYR or TYRP1 generates superoxide anions (O2−) and hydrogen peroxide (H2O2).

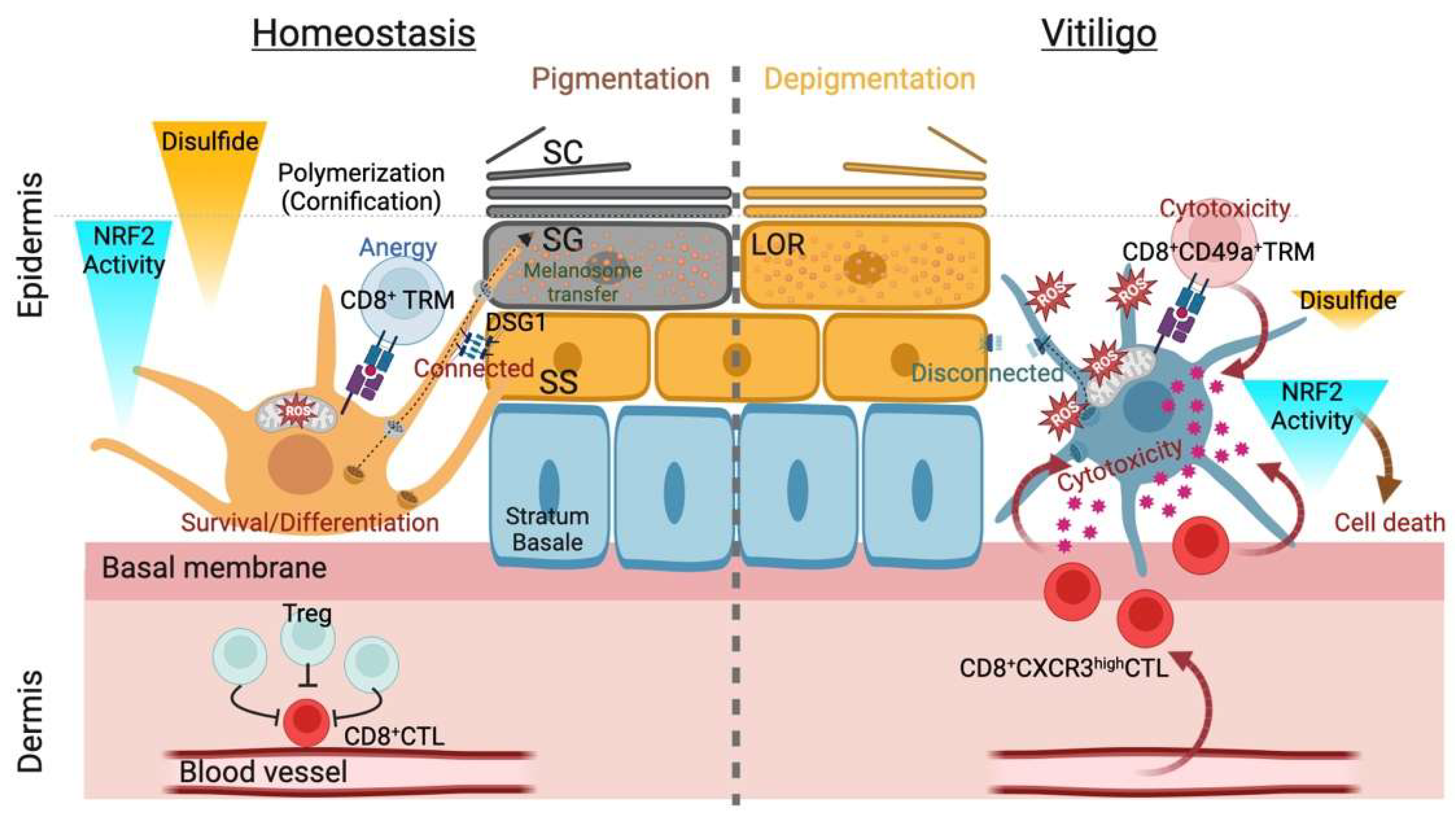
Figure 2.
Summary of thiols/disulfides in the epidermal pigmentary system. Interfollicular epidermis (IFE) pigmentation largely depends on the balance between reduction and oxidation (redox) status with internal or external causes. The former refers to mitochondria-derived reactive oxygen species (ROS) during melanogenesis, and the latter may correspond to epidermal differentiation in which keratinocyte (KC) structural proteins undergo extensive disulfide bridge formation upon the initiation of cornification (transition from the stratum granulosum [SG] to the stratum corneum [SC]). Successful cornification largely depends on the biochemical nature of the structural protein loricrin (LOR, indicated as granules). Differentiating layer (stratum spinosum [SS] and SG)-specific desmosomal cadherin desmoglein 1 (DSG1) appears indispensable for functional IFE pigmentogenesis: melanosome transfer and maturation (eumelanogenesis) (left). In vitiligo melanocytes, stress from environmental factors (e.g., toxic chemicals) can cause aberrations in the IFE antioxidant systems in genetically predisposed individuals. Breached immune tolerance recruits melanocyte antigen-specific CD8+ cytotoxic T cells (CTLs) expressing CXC receptor type 3 (CXCR3) or CD49+ resident memory T cells (TRMs) in vitiligo (right), whereas the autoreactive CTLs constitutively become anergic with the help of regulatory T cells (Tregs) in homeostasis (left). The homeostatic gradient of nuclear factor erythroid-2-related factor 2 (NRF2)-mediated epidermal antioxidative defense and ensuing cornification yield a polymerized/pigmented SC, protecting against oxidative damage. However, local clonal expansion of CTLs in the IFE eventually eliminates melanocytes from the IFE niche (the epidermal melanin unit), perturbing the xenobiotic metabolism coordinated by NRF2 and resulting in depigmentation (leukoderma).
Figure 2.
Summary of thiols/disulfides in the epidermal pigmentary system. Interfollicular epidermis (IFE) pigmentation largely depends on the balance between reduction and oxidation (redox) status with internal or external causes. The former refers to mitochondria-derived reactive oxygen species (ROS) during melanogenesis, and the latter may correspond to epidermal differentiation in which keratinocyte (KC) structural proteins undergo extensive disulfide bridge formation upon the initiation of cornification (transition from the stratum granulosum [SG] to the stratum corneum [SC]). Successful cornification largely depends on the biochemical nature of the structural protein loricrin (LOR, indicated as granules). Differentiating layer (stratum spinosum [SS] and SG)-specific desmosomal cadherin desmoglein 1 (DSG1) appears indispensable for functional IFE pigmentogenesis: melanosome transfer and maturation (eumelanogenesis) (left). In vitiligo melanocytes, stress from environmental factors (e.g., toxic chemicals) can cause aberrations in the IFE antioxidant systems in genetically predisposed individuals. Breached immune tolerance recruits melanocyte antigen-specific CD8+ cytotoxic T cells (CTLs) expressing CXC receptor type 3 (CXCR3) or CD49+ resident memory T cells (TRMs) in vitiligo (right), whereas the autoreactive CTLs constitutively become anergic with the help of regulatory T cells (Tregs) in homeostasis (left). The homeostatic gradient of nuclear factor erythroid-2-related factor 2 (NRF2)-mediated epidermal antioxidative defense and ensuing cornification yield a polymerized/pigmented SC, protecting against oxidative damage. However, local clonal expansion of CTLs in the IFE eventually eliminates melanocytes from the IFE niche (the epidermal melanin unit), perturbing the xenobiotic metabolism coordinated by NRF2 and resulting in depigmentation (leukoderma).


{kind=link}
{kind=link}
Table 1.
NRF2-targeted therapy in vitiligo.
| Treatment | Administration Route | Model | NRF2 Status | Effect | Reference |
|---|---|---|---|---|---|
| Hormone | |||||
| Melatonin | In vitro | UVB-treated NHEMs | Activation | Increases DNA repair and levels of p53 phosphorylated at serine 15 Reduces ROS generation | [101] |
| Vitamins | |||||
| Folic acid | In vitro | H2O2-treated PIG1 | Activation | Increases cell viability and proliferation Decreases apoptosis and ROS generation Inhibits HMGB1 | [102] |
| Methylcobalamin | In vitro | H2O2-treated PIG1 | Activation | Increases cell viability and melanogenesis Decreases apoptosis and ROS generation | [103] |
| Vitamin D | In vitro | H2O2-treated PIG1 H2O2-treated PIG3V | Activation | Increases cell viability, proliferation, and migration Decreases apoptosis and ROS generation Activates Wnt/β-catenin signaling | [104] |
| Organic compounds | |||||
| 4-Octyl itaconate | In vitro Intravenous | UVB-treated PIG1 UVB-treated HaCaT UVB-treated mice | Activation | Increases cell viability Decreases apoptosis and ROS generation Attenuates UVB-induced skin damage | [105] |
| Ginsenoside Rk1 | In vitro | H2O2-treated PIG1 | Activation | Increases cell viability Decreases apoptosis | [106] |
| Glycosides | |||||
| Cistanche deserticola polysaccharides | In vitro | H2O2-treated NHEMs H2O2-treated mouse melanoma B16F10 cells | Activation | Increase cell viability and melanogenesis Decrease cytotoxicity, apoptosis, and ROS generation | [107] |
| Glycyrrhizin | In vitro | H2O2-treated NHEMs | Activation | Increases cell viability Decreases apoptosis and ROS generation | [108] |
| Lycium barbarum polysaccharides | In vitro | H2O2-treated PIG1 | Activation | Increase cell proliferation and melanogenesis Decrease apoptosis Activate the NRF2/p62 signaling pathway and induce autophagy | [109] |
| Paeoniflorin | In vitro | H2O2-treated PIG1 H2O2-treated PIG3V | Activation | Increases cell viability Decreases apoptosis and ROS generation | [110] |
| Polyphenols | |||||
| 6-Shogaol | In vitro | H2O2-treated NHEMs | Activation | Increases cell viability and melanogenesis Inhibits apoptosis | [111] |
| Paeonol | In vitro | H2O2-treated PIG1 | Activation | Increases cell viability and melanogenesis Decreases ROS generation and lipid peroxidation | [112] |
| Flavonoids | |||||
| Afzelin | In vitro | H2O2-treated NHEMs | Activation | Increases cell proliferation and phosphorylation of GSK-3β Decreases apoptosis, ROS generation, and lipid peroxidation | [113] |
| Apigenin | In vitro | H2O2-treated PIG3V | Activation | Increases cell viability Decreases lipid peroxidation | [114] |
| Baicalein | In vitro | H2O2-treated PIG3V | Activation | Increases cell viability Decreases apoptosis and mitochondrial dysfunction | [115] |
| Vitexin | In vitro | H2O2-treated PIG1 | Activation | Increases cell viability and proliferation Decreases apoptosis, ROS generation, and IL-1β and IL-17A expression | [116] |
| Alkaloids | |||||
| Berberine | In vitro | H2O2-treated PIG1 | Activation | Increases cell viability and melanogenesis Decreases apoptosis, ROS generation, and NF-κB activation | [117] |
| Therapeutic agents | |||||
| Aspirin | In vitro | H2O2-treated NHEMs | Activation | Increases cell viability Decreases apoptosis, ROS generation, and LDH release | [118] |
| Dimethyl fumarate | In vitro | MBEH-treated NHEMs MBEH-treated NLVMs MBEH-treated PLVMs | Activation | Increases cell viability | [94] |
| Simvastatin | In vitro | H2O2-treated NHEMs | Activation | Increases cell viability Decreases apoptosis and ROS generation Activates the MAPK pathway and p62 | [29] |
| Molecular hydrogen | In vitro | Vitiligo epidermal cells H2O2-treated PIG1 H2O2-treated PIG3V H2O2-treated HaCaT | Activation | Increases cell viability, migration, melanogenesis, and mitochondrial function Decreases apoptosis, ROS generation, and lipid peroxidation | [52] |
| Cold atmospheric plasma | Topical | Vitiligo-like mouse model Vitiligo skin | Activation | Ameliorates vitiligo lesions in mice and patients Restores melanin distribution in mice and skin pigmentation in patients with vitiligo Decreases CD11c+ DC/CD3+ T cell/CD8+ T cell infiltration, CXCL10/IFN-γ/HIF-1α release, and inducible nitric oxide synthase in mice Increases gp100+ cells and decreases CD8+ T cells in patients | [119] |
DC, dendritic cell; gp100, glycoprotein 100; GSK-3β, glycogen synthase kinase-3β; HIF-1α, hypoxia-inducible factor-1α; HMGB1, high mobility group box 1; H2O2, hydrogen peroxide; IL-1β, interleukin-1 β; IL-17A, interleukin-17A; LDH, lactate dehydrogenase; MAPK, mitogen-activated protein kinase; MBEH, monobenzyl ether of hydroquinone; NF-κB, nuclear factor-κB; NHEMs, normal human epidermal melanocytes; NLVMs, non-lesional vitiligo melanocytes; PLVMs, perilesional vitiligo melanocytes; UVB, ultraviolet B.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ogawa, T.; Ishitsuka, Y. NRF2 in the Epidermal Pigmentary System. Biomolecules 2023, 13, 20. https://doi.org/10.3390/biom13010020
AMA Style
Ogawa T, Ishitsuka Y. NRF2 in the Epidermal Pigmentary System. Biomolecules. 2023; 13(1):20. https://doi.org/10.3390/biom13010020
Chicago/Turabian StyleOgawa, Tatsuya, and Yosuke Ishitsuka. 2023. "NRF2 in the Epidermal Pigmentary System" Biomolecules 13, no. 1: 20. https://doi.org/10.3390/biom13010020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.