

Modus operandi of ClC-K2 Cl− Channel in the Collecting Duct Intercalated Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
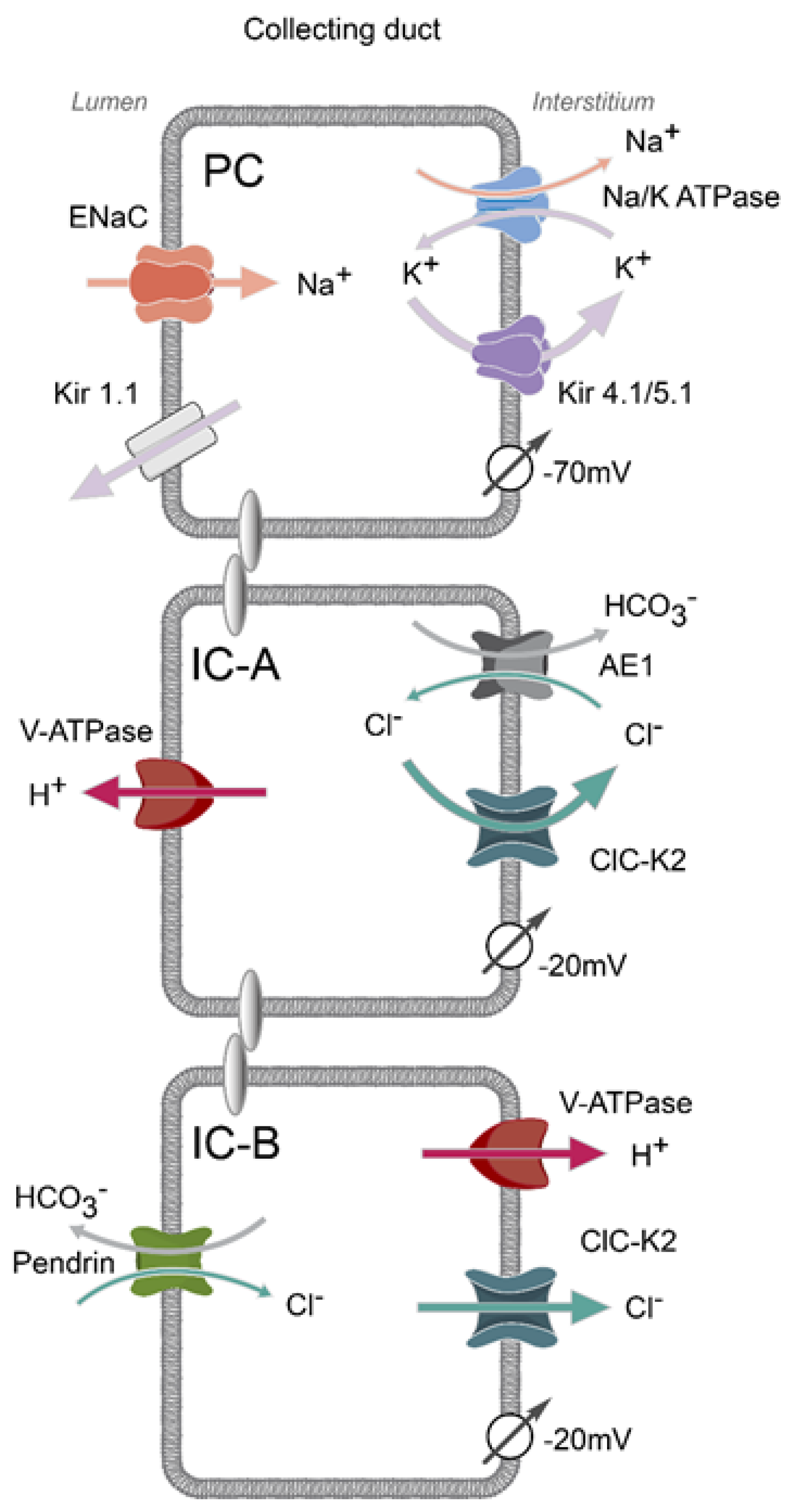
:1. Mosaic Architecture of the Collecting Duct
2. Basolateral Electrogenic Cl− Conductance Is Mediated by ClC-K2 in Intercalated Cells
3. Structural and Biophysical Properties of ClC-K2 Channel
4. Expression Sites and Physiological Relevance of ClC-K2
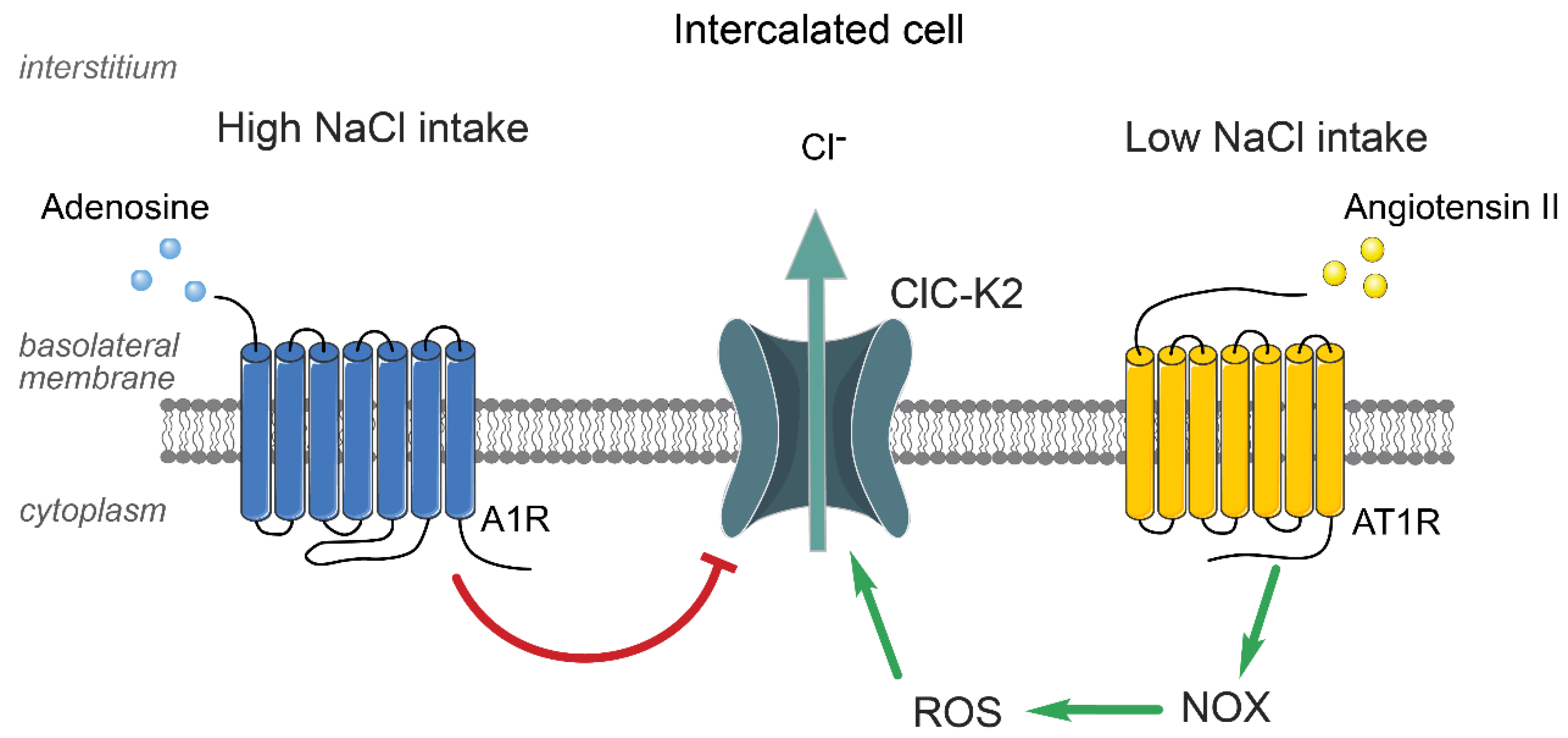
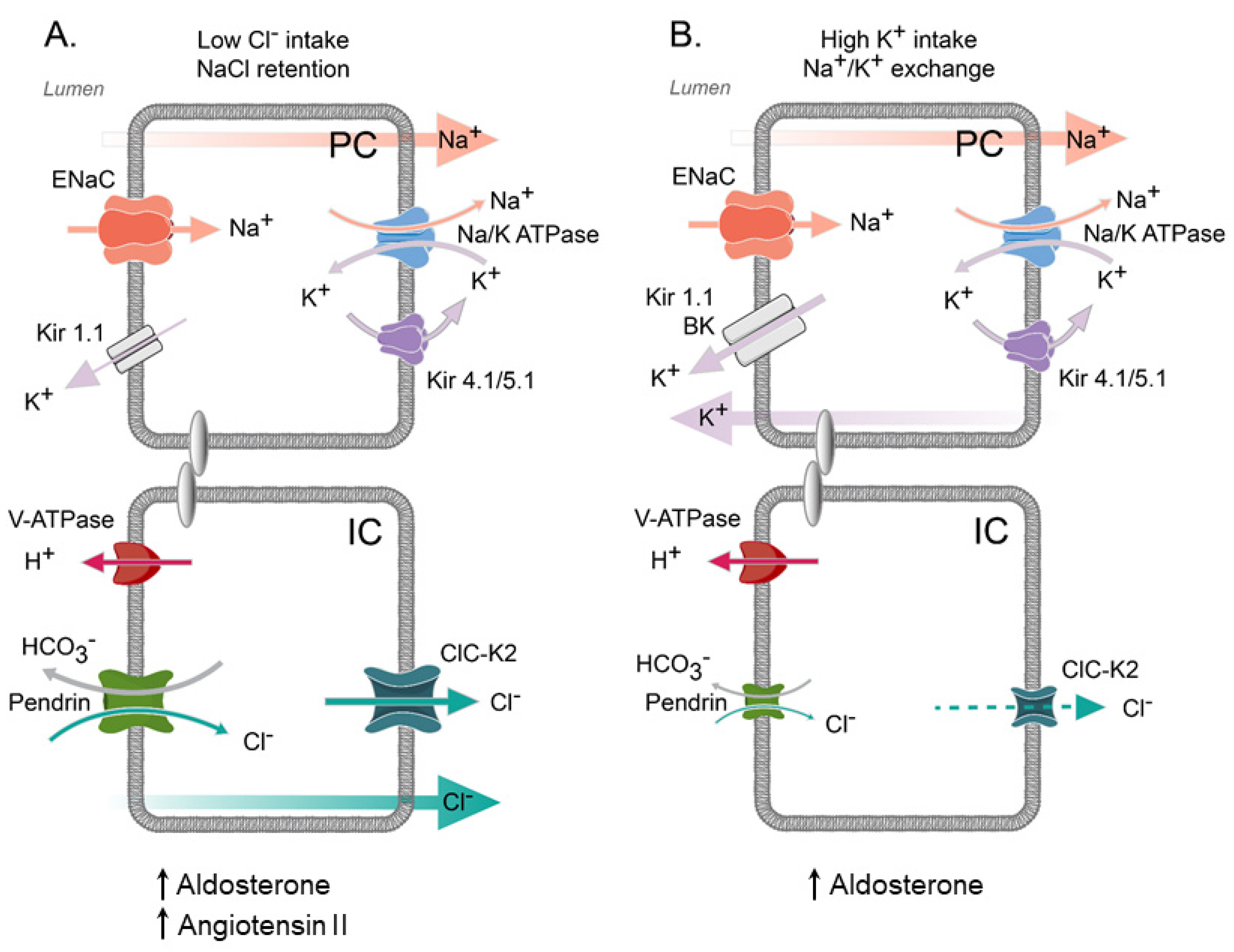
5. Regulation of ClC-K2 in the Collecting Duct by Systemic and Local Factors
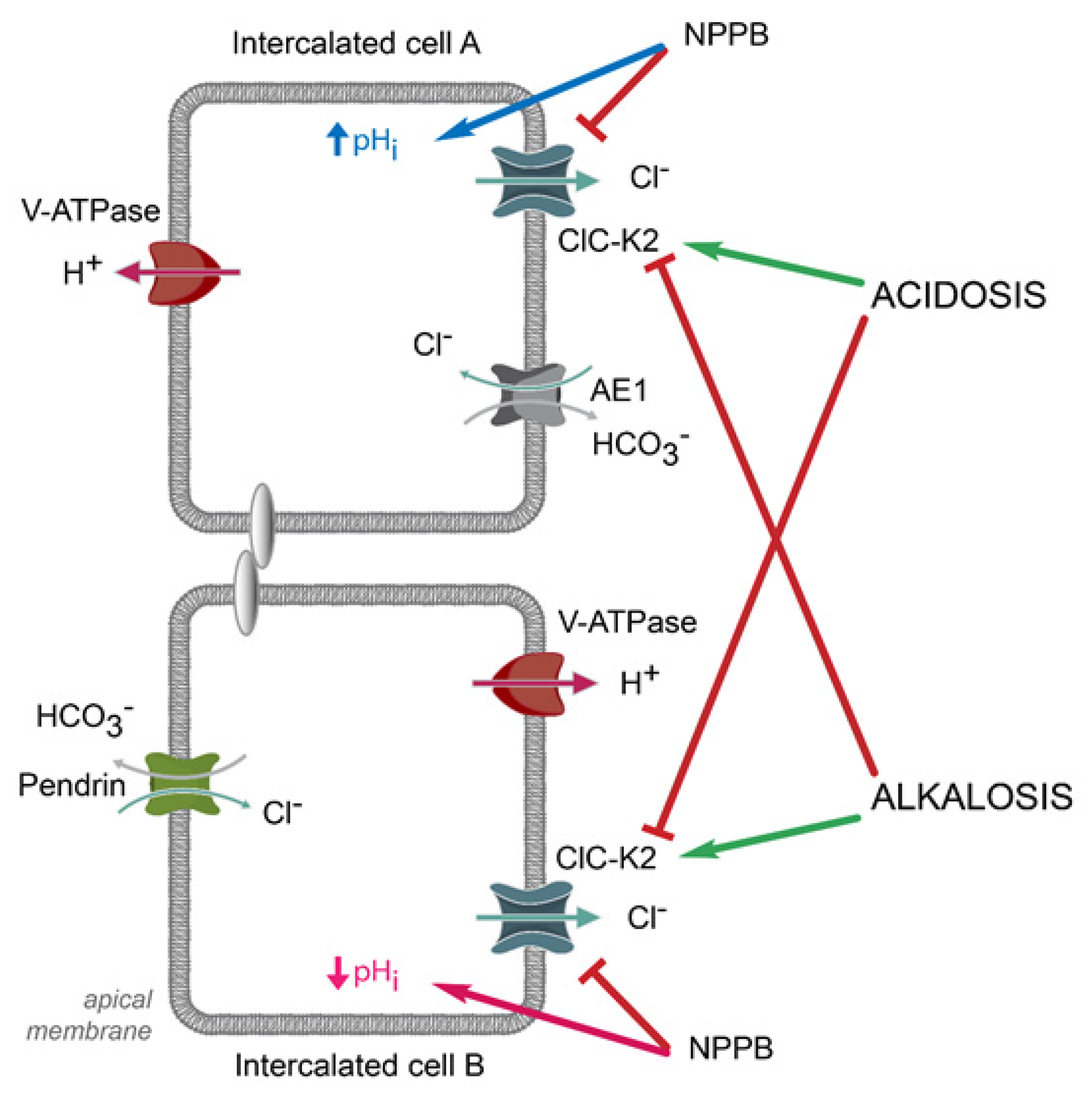
6. Contribution of ClC-K2 to H+ and HCO3− Secretion by Intercalated Cells
7. Final Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Tabibzadeh, N.; Crambert, G. Mechanistic insights into the primary and secondary alterations of renal ion and water transport in the distal nephron. J. Intern. Med. 2022, 293, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting Duct Principal Cell Transport Processes and Their Regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, S.M.; Verlander, J.W.; Romero, C.A. The Renal Physiology of Pendrin-Positive Intercalated Cells. Physiol. Rev. 2020, 100, 1119–1147. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Al-Bataineh, M.M.; Pastor-Soler, N.M. Collecting Duct Intercalated Cell Function and Regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 305–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrshev, K.; Khayyat, N.H.; Stavniichuk, A.; Tomilin, V.N.; Zaika, O.; Ramkumar, N.; Pochynyuk, O. ClC-K2 Cl− channel allows identification of A- and B-type of intercalated cells in split-opened collecting ducts. FASEB J. 2022, 36, e22275. [Google Scholar] [CrossRef]
- Schuster, V.L.; Stokes, J.B. Chloride transport by the cortical and outer medullary collecting duct. Am. J. Physiol. Physiol. 1987, 253, F203–F212. [Google Scholar] [CrossRef]
- Warden, D.H.; Schuster, V.L.; Stokes, J.B. Characteristics of the paracellular pathway of rabbit cortical collecting duct. Am. J. Physiol. Physiol. 1988, 255, F720–F727. [Google Scholar] [CrossRef]
- Monzon, C.M.; Garvin, J.L. Nitric oxide decreases the permselectivity of the paracellular pathway in thick ascending limbs. Hypertension 2015, 65, 1245–1250. [Google Scholar] [CrossRef] [Green Version]
- Pratt, J.H. Central Role for ENaC in Development of Hypertension. J. Am. Soc. Nephrol. 2005, 16, 3154–3159. [Google Scholar] [CrossRef] [Green Version]
- Hummler, E. Implication of ENaC in salt-sensitive hypertension. J. Steroid Biochem. Mol. Biol. 1999, 69, 385–390. [Google Scholar] [CrossRef]
- Bhalla, V.; Hallows, K.R. Mechanisms of ENaC Regulation and Clinical Implications. J. Am. Soc. Nephrol. 2008, 19, 1845–1854. [Google Scholar] [CrossRef] [Green Version]
- Hummler, E. Epithelial sodium channel, salt intake, and hypertension. Curr. Hypertens. Rep. 2003, 5, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Meneton, P.; Jeunemaitre, X.; De Wardener, H.E.; MacGregor, G.A. Links Between Dietary Salt Intake, Renal Salt Handling, Blood Pressure, and Cardiovascular Diseases. Physiol. Rev. 2005, 85, 679–715. [Google Scholar] [CrossRef] [PubMed]
- Appel, L.J. The Effects of Dietary Factors on Blood Pressure. Cardiol. Clin. 2017, 35, 197–212. [Google Scholar] [CrossRef]
- Graudal, N.A.; Hubeck-Graudal, T.; Jurgens, G. Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst. Rev. 2017, 4, CD004022. [Google Scholar] [CrossRef] [PubMed]
- Covelli, M.M. A review of long-term effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol and triglyceride. Evid. Based Nurs. 2012, 15, 70–71. [Google Scholar] [CrossRef] [PubMed]
- McCallum, L.; Lip, S.; Padmanabhan, S. The hidden hand of chloride in hypertension. Pflügers Arch. Eur. J. Physiol. 2015, 467, 595–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotchen, T.A.; Galla, J.H.; Luke, R.G. Failure of NaHCO3 and KHCO3 to inhibit renin in the rat. Am. J. Physiol. 1976, 231, 1050–1056. [Google Scholar] [CrossRef] [Green Version]
- Kotchen, T.A.; Luke, R.G.; Ott, C.E.; Galla, J.H.; Whitescarver, S. Effect of Chloride on Renin and Blood Pressure Responses to Sodium Chloride. Ann. Intern. Med. 1983, 98, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.C.; Steinberg, H.; Ganten, U.; Meyer, D.; Gless, K.H.; Lang, R.E.; Fineberg, N.S.; Rascher, W.; Unger, T.; Ganten, D. Effect of sodium chloride and sodium bicarbonate on blood pressure in stroke-prone spontaneously hypertensive rats. Clin. Sci. 1988, 74, 577–585. [Google Scholar] [CrossRef]
- Zaika, O.; Mamenko, M.; Staruschenko, A.; Pochynyuk, O. Direct Activation of ENaC by Angiotensin II: Recent Advances and New Insights. Curr. Hypertens. Rep. 2013, 15, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamenko, M.; Zaika, O.; Pochynyuk, O. Direct regulation of ENaC by bradykinin in the distal nephron. Implications for renal sodium handling. Curr. Opin. Nephrol. Hypertens. 2014, 23, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.-T.; Yang, C.-L.; Ellison, D.H. Kidney Is Essential for Blood Pressure Modulation by Dietary Potassium. Curr. Cardiol. Rep. 2020, 22, 124. [Google Scholar] [CrossRef] [PubMed]
- Hadchouel, J.; Ellison, D.H.; Gamba, G. Regulation of Renal Electrolyte Transport by WNK and SPAK-OSR1 Kinases. Annu. Rev. Physiol. 2016, 78, 367–389. [Google Scholar] [CrossRef] [PubMed]
- Terker, A.S.; Zhang, C.; McCormick, J.A.; Lazelle, R.A.; Zhang, C.; Meermeier, N.P.; Siler, D.A.; Park, H.J.; Fu, Y.; Cohen, D.M.; et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015, 21, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Nissant, A.; Paulais, M.; Lachheb, S.; Lourdel, S.; Teulon, J. Similar chloride channels in the connecting tubule and cortical collecting duct of the mouse kidney. Am. J. Physiol. Physiol. 2006, 290, F1421–F1429. [Google Scholar] [CrossRef] [Green Version]
- Lachheb, S.; Cluzeaud, F.; Bens, M.; Genete, M.; Hibino, H.; Lourdel, S.; Kurachi, Y.; Vandewalle, A.; Teulon, J.; Paulais, M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am. J. Physiol. Physiol. 2008, 294, F1398–F1407. [Google Scholar] [CrossRef]
- Tomilin, V.N.; Zaika, O.; Subramanya, A.R.; Pochynyuk, O. Dietary K+ and Cl− independently regulate basolateral conductance in principal and intercalated cells of the collecting duct. Pflügers Arch. Eur. J. Physiol. 2017, 470, 339–353. [Google Scholar] [CrossRef]
- Palygin, O.; Pochynyuk, O.; Staruschenko, A. Role and mechanisms of regulation of the basolateral Kir4.1/Kir5.1K+ channels in the distal tubules. Acta Physiol. 2016, 219, 260–273. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.-H.; Duan, X.-P.; Zheng, J.-Y.; Wang, W.-H. Role of inwardly rectifying K+ channel 5.1 (Kir5.1) in the regulation of renal membrane transport. Curr. Opin. Nephrol. Hypertens. 2022, 31, 479–485. [Google Scholar] [CrossRef]
- Muto, S.; Yasoshima, K.; Yoshitomi, K.; Imai, M.; Asano, Y. Electrophysiological identification of alpha- and beta-intercalated cells and their distribution along the rabbit distal nephron segments. J. Clin. Investig. 1990, 86, 1829–1839. [Google Scholar] [CrossRef]
- Zaika, O.; Palygin, O.; Tomilin, V.; Mamenko, M.; Staruschenko, A.; Pochynyuk, O. Insulin and IGF-1 activate Kir4.1/5.1 channels in cortical collecting duct principal cells to control basolateral membrane voltage. Am. J. Physiol. Physiol. 2016, 310, F311–F321. [Google Scholar] [CrossRef] [Green Version]
- Zaika, O.; Mamenko, M.; Boukelmoune, N.; Pochynyuk, O. IGF-1 and insulin exert opposite actions on ClC-K2 activity in the cortical collecting ducts. Am. J. Physiol. Physiol. 2015, 308, F39–F48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaika, O.L.; Mamenko, M.; Palygin, O.; Boukelmoune, N.; Staruschenko, A.; Pochynyuk, O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. Am. J. Physiol. Physiol. 2013, 305, F1277–F1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennings, J.C.; Andrini, O.; Picard, N.; Paulais, M.; Huebner, A.K.; Cayuqueo, I.K.L.; Bignon, Y.; Keck, M.; Cornière, N.; Böhm, D.; et al. The ClC-K2 Chloride Channel Is Critical for Salt Handling in the Distal Nephron. J. Am. Soc. Nephrol. 2017, 28, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, H.; Silva, T. Cloning and localization of KCC4 in rabbit kidney: Expression in distal convoluted tubule. Am. J. Physiol. Physiol. 2003, 285, F49–F58. [Google Scholar] [CrossRef] [Green Version]
- Boettger, T.; Hübner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Ferdaus, M.Z.; Terker, A.S.; Koumangoye, R.; Delpire, E. KCC3a, a Strong Candidate Pathway for K+ Loss in Alkalemia. Front. Cell Dev. Biol. 2022, 10, 931326. [Google Scholar] [CrossRef]
- Melo, Z.; Cruz-Rangel, S.; Bautista, R.; Vázquez, N.; Castañeda-Bueno, M.; Mount, D.B.; Pasantes-Morales, H.; Mercado, A.; Gamba, G. Molecular evidence for a role for K+-Cl− cotransporters in the kidney. Am. J. Physiol. Physiol. 2013, 305, F1402–F1411. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.B.; Faulhaber, J.; Budack, M.K.; Pfeffer, C.; Maritzen, T.; Didié, M.; Beck, F.-X.; Boettger, T.; Schubert, R.; Ehmke, H.; et al. Neurogenic Mechanisms Contribute to Hypertension in Mice With Disruption of the K-Cl Cotransporter KCC3. Circ. Res. 2006, 98, 549–556. [Google Scholar] [CrossRef]
- Adragna, N.C.; Di Fulvio, M.; Lauf, P.K. Regulation of K-Cl cotransport: From function to genes. J. Membr. Biol. 2004, 201, 109–137. [Google Scholar] [CrossRef] [PubMed]
- López-Cayuqueo, K.I.; Planells-Cases, R.; Pietzke, M.; Oliveras, A.; Kempa, S.; Bachmann, S.; Jentsch, T.J. Renal Deletion of LRRC8/VRAC Channels Induces Proximal Tubulopathy. J. Am. Soc. Nephrol. 2022, 33, 1528–1545. [Google Scholar] [CrossRef]
- L’Hoste, S.; Diakov, A.; Andrini, O.; Genete, M.; Pinelli, L.; Grand, T.; Keck, M.; Paulais, M.; Beck, L.; Korbmacher, C.; et al. Characterization of the mouse ClC-K1/Barttin chloride channel. Biochim. et Biophys. Acta (BBA) Biomembr. 2013, 1828, 2399–2409. [Google Scholar] [CrossRef] [Green Version]
- Estévez, R.; Boettger, T.; Stein, V.; Birkenhäger, R.; Otto, E.; Hildebrandt, F.; Jentsch, T.J. Barttin is a Cl- channel β-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 2001, 414, 558–561. [Google Scholar] [CrossRef]
- Waldegger, S.; Jeck, N.; Barth, P.; Peters, M.; Vitzthum, H.; Wolf, K.; Kurtz, A.; Konrad, M.; Seyberth, H.W. Barttin increases surface expression and changes current properties of ClC-K channels. Pflügers Arch. Eur. J. Physiol. 2002, 444, 411–418. [Google Scholar] [CrossRef]
- Park, E.; Campbell, E.B.; MacKinnon, R. Structure of a CLC chloride ion channel by cryo-electron microscopy. Nature 2017, 541, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, T.J. CLC Chloride Channels and Transporters: From Genes to Protein Structure, Pathology and Physiology. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 3–36. [Google Scholar] [CrossRef]
- Andrini, O.; Keck, M.; L’Hoste, S.; Briones, R.; Mansour-Hendili, L.; Grand, T.; Sepúlveda, F.V.; Blanchard, A.; Lourdel, S.; Vargas-Poussou, R.; et al. CLCNKB mutations causing mild Bartter syndrome profoundly alter the pH and Ca2+ dependence of ClC-Kb channels. Pflügers Arch. Eur. J. Physiol. 2014, 466, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Lourdel, S.; Paulais, M.; Marvao, P.; Nissant, A.; Teulon, J. A chloride channel at the basolateral membrane of the distal-convoluted tubule: A candidate ClC-K channel. J. Gen. Physiol. 2003, 121, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Dutzler, R.; Campbell, E.B.; MacKinnon, R. Gating the selectivity filter in ClC chloride channels. Science 2003, 300, 108–112. [Google Scholar] [CrossRef]
- Waldegger, S.; Jentsch, T.J. Functional and Structural Analysis of ClC-K Chloride Channels Involved in Renal Disease. J. Biol. Chem. 2000, 275, 24527–24533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradogna, A.; Babini, E.; Picollo, A.; Pusch, M. A regulatory calcium-binding site at the subunit interface of CLC-K kidney chloride channels. J. Gen. Physiol. 2010, 136, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Campbell, E.B.; MacKinnon, R. Molecular mechanism of proton transport in CLC Cl−/H+ exchange transporters. Proc. Natl. Acad. Sci. USA 2012, 109, 11699–11704. [Google Scholar] [CrossRef] [Green Version]
- Tajima, M.; Hayama, A.; Rai, T.; Sasaki, S.; Uchida, S. Barttin binds to the outer lateral surface of the ClC-K2 chloride channel. Biochem. Biophys. Res. Commun. 2007, 362, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Hayama, A.; Rai, T.; Sasaki, S.; Uchida, S. Molecular mechanisms of Bartter syndrome caused by mutations in the BSND gene. Histochem. Cell Biol. 2003, 119, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Steinke, K.V.; Gorinski, N.; Wojciechowski, D.; Todorov, V.; Guseva, D.; Ponimaskin, E.; Fahlke, C.; Fischer, M. Human CLC-K Channels Require Palmitoylation of Their Accessory Subunit Barttin to Be Functional. J. Biol. Chem. 2015, 290, 17390–17400. [Google Scholar] [CrossRef] [Green Version]
- Kieferle, S.; Fong, P.; Bens, M.; Vandewalle, A.; Jentsch, T.J. Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc. Natl. Acad. Sci. USA 1994, 91, 6943–6947. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Sasaki, S.; Furukawa, T.; Hiraoka, M.; Imai, T.; Hirata, Y.; Marumo, F. Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J. Biol. Chem. 1993, 268, 3821–3824. [Google Scholar] [CrossRef]
- Sage, C.L.; Marcus, D.C. Immunolocalization of ClC-K chloride channel in strial marginal cells and vestibular dark cells. Hear. Res. 2001, 160, 1–9. [Google Scholar] [CrossRef]
- Kobayashi, K.; Uchida, S.; Mizutani, S.; Sasaki, S.; Marumo, F. Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J. Am. Soc. Nephrol. 2001, 12, 1327–1334. [Google Scholar] [CrossRef]
- Uchida, S.; Sasaki, S.; Nitta, K.; Uchida, K.; Horita, S.; Nihei, H.; Marumo, F. Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J. Clin. Investig. 1995, 95, 104–113. [Google Scholar] [CrossRef]
- Matsumura, Y.; Uchida, S.; Kondo, Y.; Miyazaki, H.; Ko, S.B.; Hayama, A.; Morimoto, T.; Liu, W.; Arisawa, M.; Sasaki, S.; et al. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 1999, 21, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Andrini, O.; Keck, M.; Briones, R.; Lourdel, S.; Vargas-Poussou, R.; Teulon, J. ClC-K chloride channels: Emerging pathophysiology of Bartter syndrome type 3. Am. J. Physiol. Physiol. 2015, 308, F1324–F1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleta, R.; Bockenhauer, D. Bartter Syndromes and Other Salt-Losing Tubulopathies. Nephron Physiol. 2006, 104, 73–80. [Google Scholar] [CrossRef]
- Seyberth, H.W. An improved terminology and classification of Bartter-like syndromes. Nat. Clin. Pract. Nephrol. 2008, 4, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Guay-Woodford, L.M. Bartter syndrome: Unraveling the pathophysiologic enigma. Am. J. Med. 1998, 105, 151–161. [Google Scholar] [CrossRef]
- Ellison, D.H. Divalent cation transport by the distal nephron: Insights from Bartter’s and Gitelman’s syndromes. Am. J. Physiol. Physiol. 2000, 279, F616–F625. [Google Scholar] [CrossRef] [Green Version]
- Grill, A.; Schießl, I.M.; Gess, B.; Fremter, K.; Hammer, A.; Castrop, H. Salt-losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol. 2016, 218, 198–211. [Google Scholar] [CrossRef]
- Hebert, S.C. Bartter syndrome. Curr. Opin. Nephrol. Hypertens. 2003, 12, 527–532. [Google Scholar] [CrossRef]
- Lin, M.-H.; Chen, J.-C.; Tian, X.; Lee, C.-M.; Yu, I.-S.; Lo, Y.-F.; Uchida, S.; Huang, C.-L.; Chen, B.-C.; Cheng, C.-J. Impairment in renal medulla development underlies salt wasting in Clc-k2 channel deficiency. J. Clin. Investig. 2021, 6, e151039. [Google Scholar] [CrossRef]
- Birkenhäger, R.; Otto, E.; Schürmann, M.J.; Vollmer, M.; Ruf, E.-M.; Maier-Lutz, I.; Beekmann, F.; Fekete, A.; Omran, H.; Feldmann, D.; et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001, 29, 310–314. [Google Scholar] [CrossRef]
- De Baaij, J.H.F.; Hoenderop, J.G.J.; Bindels, R.J. Magnesium in Man: Implications for Health and Disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Lambers, T.T.; Bindels, R.J.; Hoenderop, J.G.J. Coordinated control of renal Ca2+ handling. Kidney Int. 2006, 69, 650–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navar, L.G.; Lewis, L.; Hymel, A.; Braam, B.; Mitchell, K.D. Tubular fluid concentrations and kidney contents of angiotensins I and II in anesthetized rats. J. Am. Soc. Nephrol. 1994, 5, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Khayyat, N.H.; Zaika, O.; Tomilin, V.N.; Pyrshev, K.; Pochynyuk, O. Angiotensin II increases activity of the ClC-K2 Cl− channel in collecting duct intercalated cells by stimulating production of reactive oxygen species. J. Biol. Chem. 2021, 296, 100347. [Google Scholar] [CrossRef] [PubMed]
- Mamenko, M.; Zaika, O.; Ilatovskaya, D.V.; Staruschenko, A.; Pochynyuk, O. Angiotensin II Increases Activity of the Epithelial Na+ Channel (ENaC) in Distal Nephron Additively to Aldosterone. J. Biol. Chem. 2012, 287, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, S.M.; Kim, Y.H.; Stanley, L.; Glapion, D.M.; Everett, L.A.; Green, E.D.; Verlander, J.W. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: Role in Cl− conservation. Hypertension 2004, 44, 982–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verlander, J.W.; Kim, Y.H.; Shin, W.; Pham, T.D.; Hassell, K.A.; Beierwaltes, W.H.; Green, E.D.; Everett, L.; Matthews, S.W.; Wall, S.M. Dietary Cl− restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am. J. Physiol. Physiol. 2006, 291, F833–F839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pech, V.; Kim, Y.H.; Weinstein, A.M.; Everett, L.A.; Pham, T.D.; Wall, S.M. Angiotensin II increases chloride absorption in the cortical collecting duct in mice through a pendrin-dependent mechanism. Am. J. Physiol. Physiol. 2007, 292, F914–F920. [Google Scholar] [CrossRef] [Green Version]
- Wall, S.M.; Weinstein, A.M. Cortical distal nephron Cl− transport in volume homeostasis and blood pressure regulation. Am. J. Physiol. Physiol. 2013, 305, F427–F438. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.P.; Ronzaud, C.; Lagnaz, D.; Staub, O.; Gamba, G. Aldosterone Paradox: Differential Regulation of Ion Transport in Distal Nephron. Physiology 2011, 26, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Eaton, D.C.; Malik, B.; Saxena, N.C.; Al-Khalili, O.K.; Yue, G. Mechanisms of Aldosterone’s Action on Epithelial Na + Transport. J. Membr. Biol. 2001, 184, 313–319. [Google Scholar] [CrossRef]
- Pham, T.D.; Verlander, J.W.; Wang, Y.; Romero, C.A.; Yue, Q.; Chen, C.; Thumova, M.; Eaton, D.C.; Lazo-Fernandez, Y.; Wall, S.M. Aldosterone Regulates Pendrin and Epithelial Sodium Channel Activity through Intercalated Cell Mineralocorticoid Receptor-Dependent and -Independent Mechanisms over a Wide Range in Serum Potassium. J. Am. Soc. Nephrol. 2020, 31, 483–499. [Google Scholar] [CrossRef]
- Shibata, S.; Rinehart, J.; Zhang, J.; Moeckel, G.; Castañeda-Bueno, M.; Stiegler, A.L.; Boggon, T.J.; Gamba, G.; Lifton, R.P. Mineralocorticoid Receptor Phosphorylation Regulates Ligand Binding and Renal Response to Volume Depletion and Hyperkalemia. Cell Metab. 2013, 18, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, L.; Klein-Robbenhaar, G.; Giebisch, G.; Smith, D.; DeFronzo, R. Effect of insulin on renal potassium metabolism. Am. J. Physiol. Physiol. 1987, 252, F60–F64. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Yeh, L.; Lansink, A.O.; Vogelzang, M.; Stegeman, C.A.; Rodgers, M.G.G.; van der Horst, I.C.C.; Wietasch, G.; Zijlstra, F.; Nijsten, M.W.N. Determinants of renal potassium excretion in critically ill patients: The role of insulin therapy. Crit. Care Med. 2012, 40, 762–765. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Levchenko, V.; Brands, M.W.; Pavlov, T.S.; Staruschenko, A.; Irsik, D.L.; Nizar, J.M.; Bhalla, V.; Blazer-Yost, B.L.; Zaika, O.; et al. Cross-talk between insulin and IGF-1 receptors in the cortical collecting duct principal cells: Implication for ENaC-mediated Na+ reabsorption. Am. J. Physiol. Physiol. 2015, 308, F713–F719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staruschenko, A.; Pochynyuk, O.; Vandewalle, A.; Bugaj, V.; Stockand, J.D. Acute Regulation of the Epithelial Na+ Channel by Phosphatidylinositide 3-OH Kinase Signaling in Native Collecting Duct Principal Cells. J. Am. Soc. Nephrol. 2007, 18, 1652–1661. [Google Scholar] [CrossRef] [Green Version]
- Kamenicky, P.; Blanchard, A.; Frank, M.; Salenave, S.; Letierce, A.; Azizi, M.; Lombès, M.; Chanson, P. Body Fluid Expansion in Acromegaly Is Related to Enhanced Epithelial Sodium Channel (ENaC) Activity. J. Clin. Endocrinol. Metab. 2011, 96, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Kamenicky, P.; Viengchareun, S.; Blanchard, A.; Meduri, G.; Zizzari, P.; Imbert-Teboul, M.; Doucet, A.; Chanson, P.; Lombes, M. Epithelial sodium channel is a key mediator of growth hormone-induced sodium retention in acromegaly. Endocrinology 2008, 149, 3294–3305. [Google Scholar] [CrossRef]
- Zou, A.-P.; Wu, F.; Li, P.-L.; Cowley, A.W., Jr. Effect of chronic salt loading on adenosine metabolism and receptor expression in renal cortex and medulla in rats. Hypertension 1999, 33, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Zaika, O.; Tomilin, V.N.; Pochynyuk, O. Adenosine inhibits the basolateral Cl− ClC-K2/b channel in collecting duct intercalated cells. Am. J. Physiol. Physiol. 2020, 318, F870–F877. [Google Scholar] [CrossRef]
- Schild, L. The ENaC channel as the primary determinant of two human diseases: Liddle syndrome and pseudohypoaldosteronism. Nephrologie 1996, 17, 395–400. [Google Scholar]
- Jacques, T.; Picard, N.; Miller, R.L.; Riemondy, K.A.; Houillier, P.; Sohet, F.; Ramakrishnan, S.K.; Büsst, C.J.; Jayat, M.; Cornière, N.; et al. Overexpression of Pendrin in Intercalated Cells Produces Chloride-Sensitive Hypertension. J. Am. Soc. Nephrol. 2013, 24, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Jeck, N.; Waldegger, P.; Doroszewicz, J.; Seyberth, H.; Waldegger, S. A common sequence variation of the CLCNKB gene strongly activates ClC-Kb chloride channel activity. Kidney Int. 2004, 65, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sile, S.; Velez, D.R.; Gillani, N.B.; Narsia, T.; Moore, J.H.; George, A.L., Jr.; Vanoye, C.G.; Williams, S.M. CLCNKB-T481S and essential hypertension in a Ghanaian population. J. Hypertens. 2009, 27, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.J.; Tsuruoka, S.; Vijayakumar, S.; Petrovic, S.; Mian, A.; Al-Awqati, Q. Acid incubation reverses the polarity of intercalated cell transporters, an effect mediated by hensin. J. Clin. Investig. 2002, 109, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Lynch, I.J.; Greenlee, M.M.; Gumz, M.L.; Rudin, A.; Xia, S.-L.; Wingo, C.S. Heterogeneity of H-K-ATPase-mediated acid secretion along the mouse collecting duct. Am. J. Physiol. Physiol. 2010, 298, F408–F415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.B.; Bindra, R.S.; Mansfield, T.A.; Nelson-Williams, C.; Mendonca, E.; Stone, R.; Schurman, S.; Nayir, A.; Alpay, H.; Bakkaloglu, A.; et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 1997, 17, 171–178. [Google Scholar] [CrossRef]
- Atkins, J.L.; Burg, M.B. Bicarbonate transport by isolated perfused rat collecting ducts. Am. J. Physiol. Physiol. 1985, 249, F485–F489. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.J.; Barasch, J.; Al-Awqati, Q. Plasticity of functional epithelial polarity. Nature 1985, 318, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Tsuruoka, S.; Schwartz, G.J. Adaptation of rabbit cortical collecting duct HCO3- transport to metabolic acidosis in vitro. J. Clin. Investig. 1996, 97, 1076–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-H.; Kwon, T.-H.; Frische, S.; Kim, J.; Tisher, C.C.; Madsen, K.M.; Nielsen, S. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am. J. Physiol. Physiol. 2002, 283, F744–F754. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stavniichuk, A.; Pyrshev, K.; Tomilin, V.N.; Kordysh, M.; Zaika, O.; Pochynyuk, O. Modus operandi of ClC-K2 Cl− Channel in the Collecting Duct Intercalated Cells. Biomolecules 2023, 13, 177. https://doi.org/10.3390/biom13010177
Stavniichuk A, Pyrshev K, Tomilin VN, Kordysh M, Zaika O, Pochynyuk O. Modus operandi of ClC-K2 Cl− Channel in the Collecting Duct Intercalated Cells. Biomolecules. 2023; 13(1):177. https://doi.org/10.3390/biom13010177
Chicago/Turabian StyleStavniichuk, Anna, Kyrylo Pyrshev, Viktor N. Tomilin, Mariya Kordysh, Oleg Zaika, and Oleh Pochynyuk. 2023. "Modus operandi of ClC-K2 Cl− Channel in the Collecting Duct Intercalated Cells" Biomolecules 13, no. 1: 177. https://doi.org/10.3390/biom13010177