
Glutathione-S-Transferases as Potential Targets for Modulation of Nitric Oxide-Mediated Vasodilation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
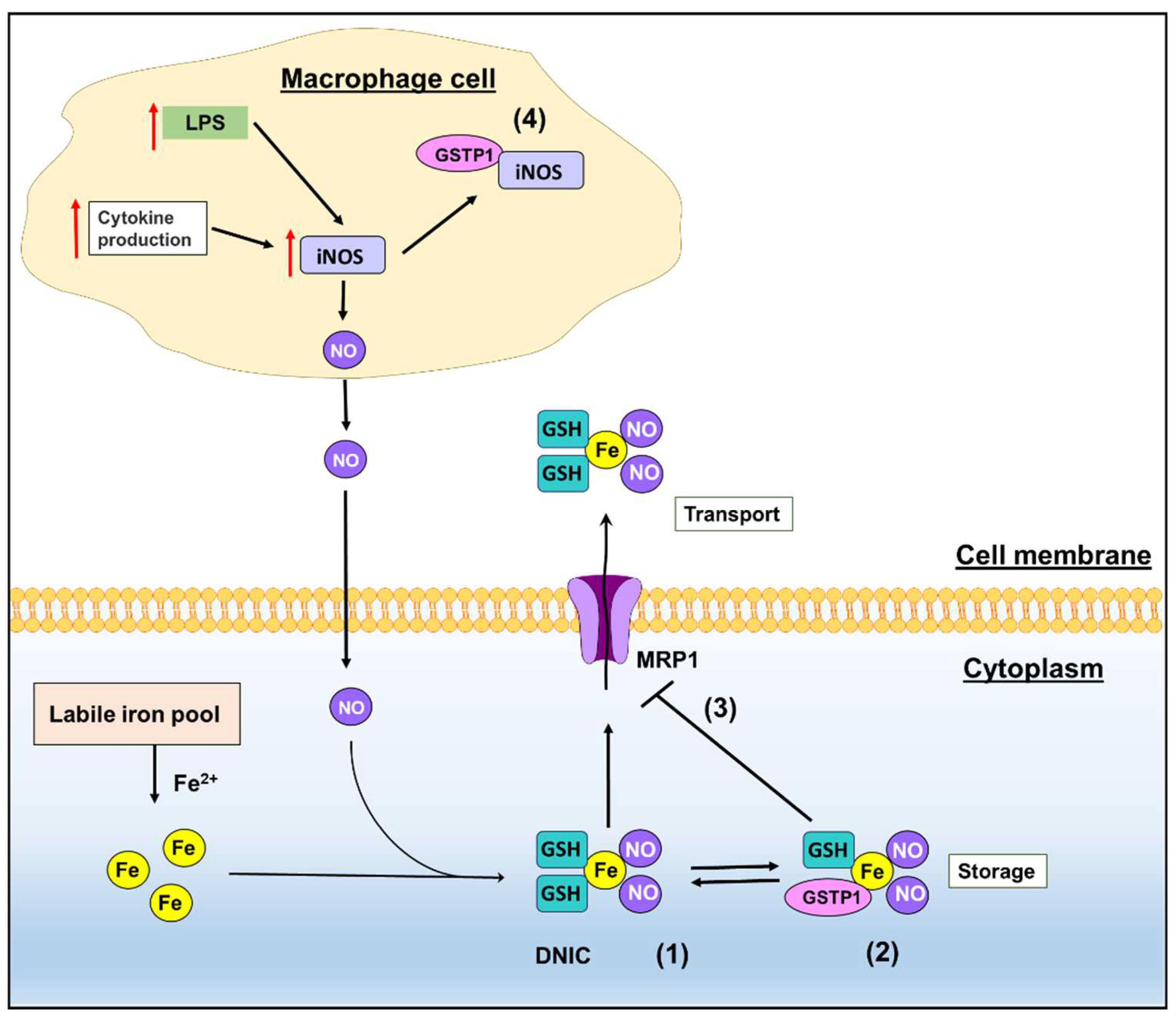
2. GSTs and Emerging Roles in NO Metabolism
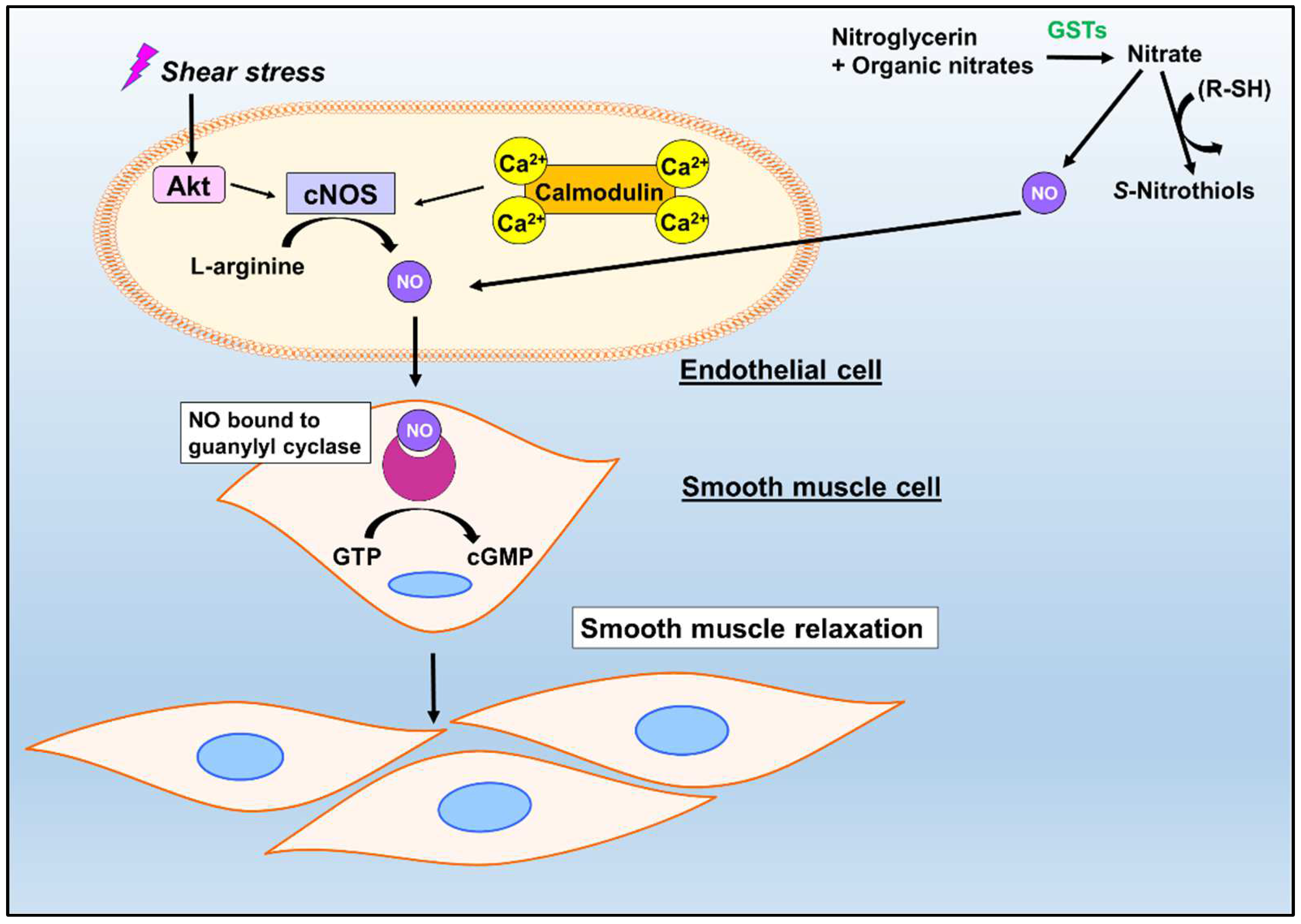
3. Nitric Oxide: A Hallmark Vasodilator
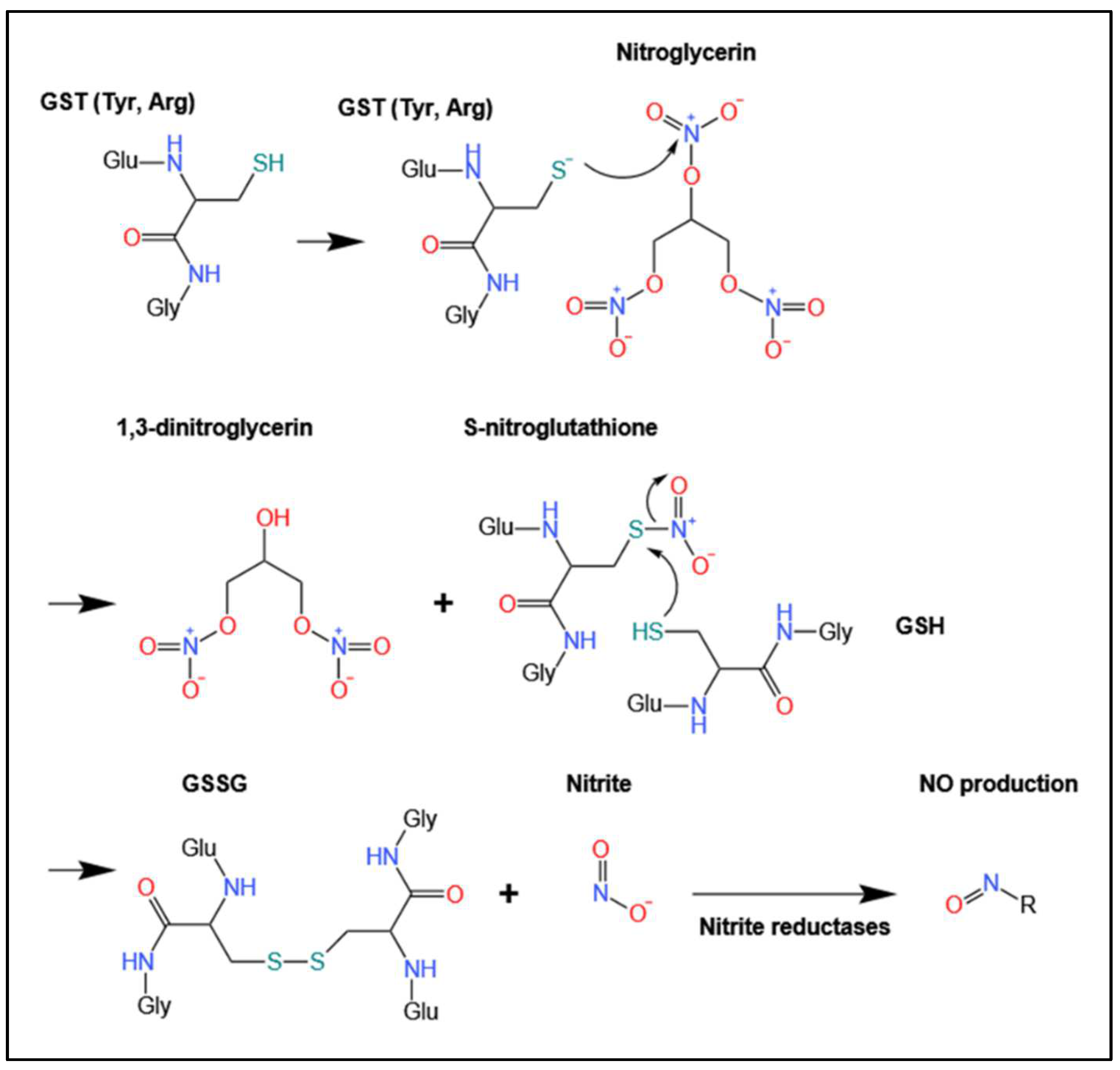
4. Biotransformation and Bioactivation via GSTs
4.1. Biotransformation of Organic Nitrates
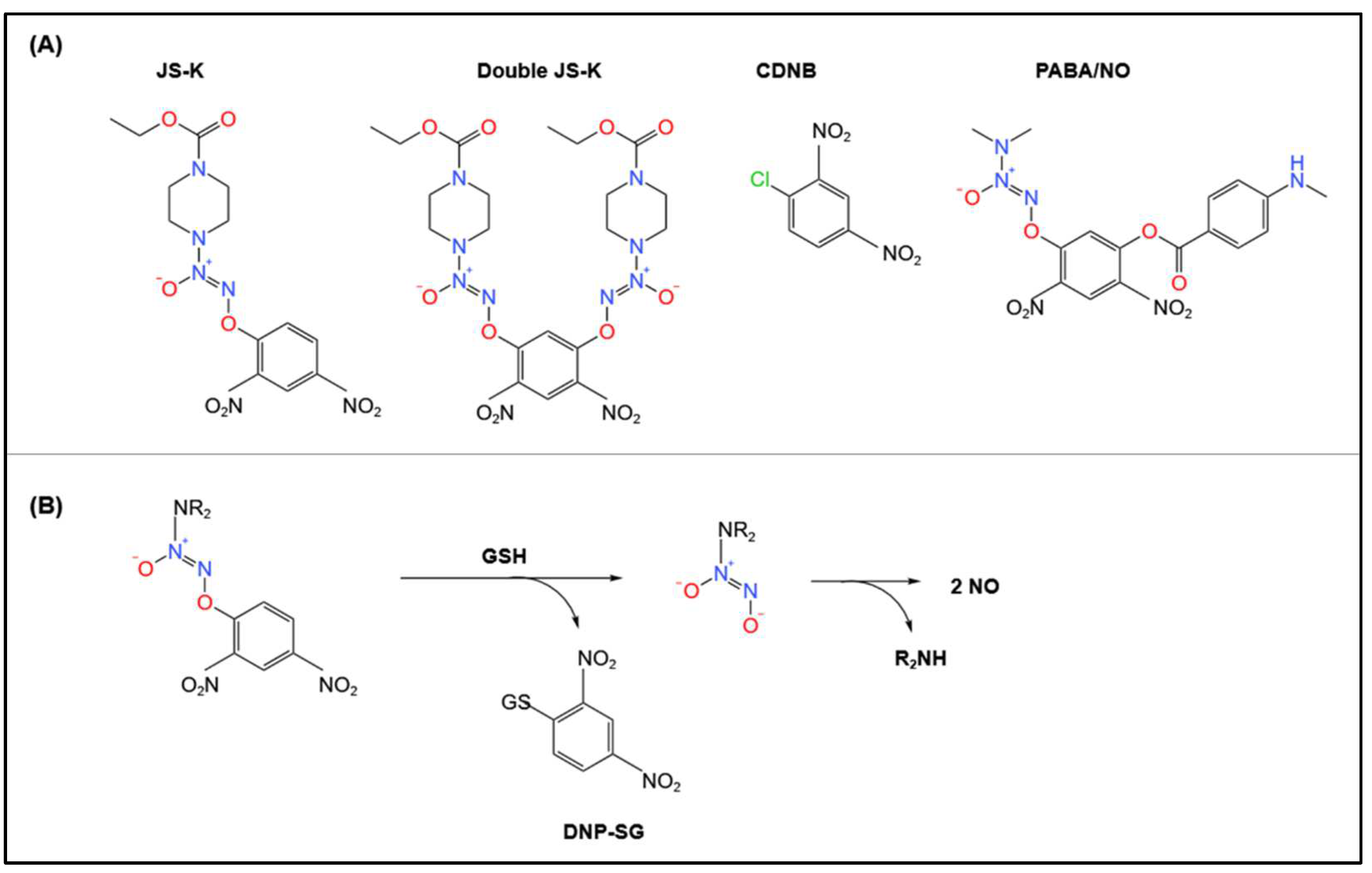
4.2. Biotransformation of Other Pro-NO Drugs by GSTs
5. GST Inhibitors Prevent Organic Nitrate-Induced Vasodilation
Impact of GST Inhibitors on the Half-Life of Nitroglycerin
6. Role of GST Isoform-Specific Biotransformation on Vasodilator Activity
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morrow, C.S.; Smitherman, P.K.; Townsend, A.J. Combined Expression of Multidrug Resistance Protein (MRP) and Glutathione S-Transferase P1-1 (GSTP1-1) in MCF7 Cells and High Level Resistance to the Cytotoxicities of Ethacrynic Acid but Not Oxazaphosphorines or Cisplatin. Biochem. Pharmacol. 1998, 56, 1013–1021. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J. The Glutathione S-Transferase Supergene Family: Regulation of GST and the Contribution of the Isoenzymes to Cancer Chemoprotection and Drug Resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–520. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B.; Board, P.G.; Hayes, J.D.; Listowsky, I.; Pearson, W.R. Nomenclature for Mammalian Soluble Glutathione Transferases. In Methods in Enzymology; Sies, H., Packer, L., Eds.; Gluthione Transferases and Gamma-Glutamyl Transpeptidases; Academic Press: Cambridge, MA, USA, 2005; Volume 401, pp. 1–8. [Google Scholar]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, Function and Evolution of Glutathione Transferases: Implications for Classification of Non-Mammalian Members of an Ancient Enzyme Superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Kong, X.; Zhou, Y.; Lan, L.; Luo, L.; Yin, Z. Glutathione S-Transferase P1 Suppresses INOS Protein Stability in RAW264.7 Macrophage-like Cells after LPS Stimulation. Free Radic. Res. 2015, 49, 1438–1448. [Google Scholar] [CrossRef]
- Cesareo, E.; Parker, L.J.; Pedersen, J.Z.; Nuccetelli, M.; Mazzetti, A.P.; Pastore, A.; Federici, G.; Caccuri, A.M.; Ricci, G.; Adams, J.J.; et al. Nitrosylation of Human Glutathione Transferase P1-1 with Dinitrosyl Diglutathionyl Iron Complex in Vitro and in Vivo. J. Biol. Chem. 2005, 280, 42172–42180. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Farrotti, A.; Stella, L.; Ketterman, A.J.; Pedersen, J.Z.; Allocati, N.; Lau, P.C.K.; Grosse, S.; Eltis, L.D.; et al. The Impact of Nitric Oxide Toxicity on the Evolution of the Glutathione Transferase Superfamily: A Proposal for an Evolutionart Driving Force. J. Biol. Chem. 2013, 288, 24936–24947. [Google Scholar] [CrossRef]
- Lok, H.C.; Sahni, S.; Jansson, P.J.; Kovacevic, Z.; Hawkins, C.L.; Richardson, D.R. A Nitric Oxide Storage and Transport System That Protects Activated Macrophages from Endogenous Nitric Oxide Cytotoxicity. J. Biol. Chem. 2016, 291, 27042–27061. [Google Scholar] [CrossRef]
- Lok, H.C.; Sahni, S.; Richardson, V.; Kalinowski, D.S.; Kovacevic, Z.; Lane, D.J.R.; Richardson, D.R. Glutathione S-Transferase and MRP1 Form an Integrated System Involved in the Storage and Transport of Dinitrosyl–Dithiolato Iron Complexes in Cells. Free Radic. Biol. Med. 2014, 75, 14–29. [Google Scholar] [CrossRef]
- Maria, F.D.; Pedersen, J.Z.; Caccuri, A.M.; Antonini, G.; Turella, P.; Stella, L.; Bello, M.L.; Federici, G.; Ricci, G. The Specific Interaction of Dinitrosyl-Diglutathionyl-Iron Complex, a Natural NO Carrier, with the Glutathione Transferase Superfamily: Suggestion for an Evolutionary Pressure in the Direction of the Storage of Nitric Oxide. J. Biol. Chem. 2003, 278, 42283–42293. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Sakanashi, M.; Nakasone, J.; Noguchi, K.; Miyagi, K.; Sakanashi, M.; Kukita, I.; Aniya, Y.; Sakanashi, M. Effects of Glutathione S-Transferase Inhibitors on Nitroglycerin Action in Pig Isolated Coronary Arteries. Clin. Exp. Pharmacol. 2002, 29, 1091–1095. [Google Scholar] [CrossRef]
- Tsuchida, S.; Maki, T.; Sato, K. Purification and Characterization of Glutathione Transferases with an Activity toward Nitroglycerin from Human Aorta and Heart. Multiplicity of the Human Class Mu Forms. J. Biol. Chem. 1990, 265, 7150–7157. [Google Scholar] [CrossRef]
- Kurz, M.A.; Boyer, T.D.; Whalen, R.; Peterson, T.E.; Harrison, D.G. Nitroglycerin Metabolism in Vascular Tissue: Role of Glutathione S-Transferases and Relationship between NO. and NO2– Formation. Biochem. J. 1993, 292, 545–550. [Google Scholar] [CrossRef]
- Gorren, A.C.F.; Russwurm, M.; Kollau, A.; Koesling, D.; Schmidt, K.; Mayer, B. Effects of Nitroglycerin/L-Cysteine on Soluble Guanylate Cyclase: Evidence for an Activation/Inactivation Equilibrium Controlled by Nitric Oxide Binding and Haem Oxidation. Biochem. J. 2005, 390, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Lok, H.C.; Rahmanto, Y.S.; Hawkins, C.L.; Kalinowski, D.S.; Morrow, C.S.; Townsend, A.J.; Ponka, P.; Richardson, D.R. Nitric Oxide Storage and Transport in Cells Are Mediated by Glutathione S-Transferase P1-1 and Multidrug Resistance Protein 1 via Dinitrosyl Iron Complexes. J. Biol. Chem. 2012, 287, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Richardson, D.R. Nitrogen Monoxide (NO) and Glucose: Unexpected Links between Energy Metabolism and NO-Mediated Iron Mobilization from Cells. J. Biol. Chem. 2001, 276, 4724–4732. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Richardson, D.R. Examination of the Mechanism of Action of Nitrogen Monoxide on Iron Uptake from Transferrin. J. Lab. Clin. Med. 2000, 136, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Richardson, D.R. The Mechanism of Nitrogen Monoxide (NO)-Mediated Iron Mobilization from Cells. Eur. J. Biochem. 2002, 269, 3383–3392. [Google Scholar] [CrossRef]
- Richardson, D.; Neumannova, V.; Nagy, E.; Ponka, P. The Effect of Redox-Related Species of Nitrogen Monoxide on Transferrin and Iron Uptake and Cellular Proliferation of Erythroleukemia (K562) Cells. Blood 1995, 86, 3211–3219. [Google Scholar] [CrossRef]
- Richardson, D.R.; Neumannova, V.; Ponka, P. Nitrogen Monoxide Decreases Iron Uptake from Transferrin but Does Not Mobilise Iron from Prelabelled Neoplastic Cells. Biochim. Biophys. Acta Mol. Cell Res. 1995, 1266, 250–260. [Google Scholar] [CrossRef]
- Pedersen, J.Z.; De Maria, F.; Turella, P.; Federici, G.; Mattei, M.; Fabrini, R.; Dawood, K.F.; Massimi, M.; Caccuri, A.M.; Ricci, G. Glutathione Transferases Sequester Toxic Dinitrosyl-Iron Complexes in Cells: A Portection Mechanism against Excess Nitric Oxide. J. Biol. Chem. 2007, 282, 6364–6371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.; Ponka, P. The Molecular Mechanisms of the Metabolism and Transport of Iron in Normal and Neoplastic Cells. Biochim. Biophys. Acta Mol. Cell Res. 1997, 1331, 1–40. [Google Scholar] [CrossRef]
- Richardson, D. DNICs and Intracellular Iron: Nitrogen Monoxide (NO)-Mediated Iron Release from Cells Is Linked to NO-Mediated Glutathione Efflux via MRP1. In Radicals for Life: The Various Forms of Nitric Oxide; Elsevier Press: Amsterdam, The Netherlands, 2007; pp. 97–118. [Google Scholar]
- Hickok, J.R.; Sahni, S.; Shen, H.; Arvind, A.; Antoniou, C.; Fung, L.W.M.; Thomas, D.D. Dinitrosyliron Complexes Are the Most Abundant Nitric Oxide-Derived Cellular Adduct: Biological Parameters of Assembly and Disappearance. Free Radic. Biol. Med. 2011, 51, 1558–1566. [Google Scholar] [CrossRef]
- Watts, R.N.; Hawkins, C.; Ponka, P.; Richardson, D.R. Nitrogen Monoxide (NO)-Mediated Iron Release from Cells Is Linked to NO-Induced Glutathione Efflux via Multidrug Resistance-Associated Protein 1. Proc. Natl. Acad. Sci. USA 2006, 103, 7670–7675. [Google Scholar] [CrossRef]
- Priviero, F.B.M.; Webb, R.C. Heme-Dependent and Independent Soluble Guanylate Cyclase Activators and Vasodilation. J. Cardiovasc. Pharmacol. 2010, 56, 229–233. [Google Scholar] [CrossRef]
- Hwang, T.L.; Wu, C.C.; Teng, C.M. Comparison of Two Soluble Guanylyl Cyclase Inhibitors, Methylene Blue and ODQ, on Sodium Nitroprusside-Induced Relaxation in Guinea-Pig Trachea. Br. J. Pharmacol. 1998, 125, 1158–1163. [Google Scholar] [CrossRef]
- Boerrigter, G.; Costello-Boerrigter, L.C.; Cataliotti, A.; Lapp, H.; Stasch, J.-P.; Burnett, J.C. Targeting Heme-Oxidized Soluble Guanylate Cyclase in Experimental Heart Failure. Hypertension 2007, 49, 1128–1133. [Google Scholar] [CrossRef]
- Watts, R.N.; Ponka, P.; Richardson, D.R. Effects of Nitrogen Monoxide and Carbon Monoxide on Molecular and Cellular Iron Metabolism: Mirror-Image Effector Molecules That Target Iron. Biochem. J. 2003, 369, 429–440. [Google Scholar] [CrossRef]
- Archer, S.L.; Huang, J.M.; Hampl, V.; Nelson, D.P.; Shultz, P.J.; Weir, E.K. Nitric Oxide and CGMP Cause Vasorelaxation by Activation of a Charybdotoxin-Sensitive K Channel by CGMP-Dependent Protein Kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 7583–7587. [Google Scholar] [CrossRef]
- Schmidt, H.H.H.W.; Lohmann, S.M.; Walter, U. The Nitric Oxide and CGMP Signal Transduction System: Regulation and Mechanism of Action. Biochim. Biophys. Acta Mol. Cell Res. 1993, 1178, 153–175. [Google Scholar] [CrossRef]
- Denninger, J.W.; Marletta, M.A. Guanylate Cyclase and the⋅NO/CGMP Signaling Pathway. Biochim. Biophys. Acta Mol. Cell Res. 1999, 1411, 334–350. [Google Scholar] [CrossRef] [Green Version]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.-X.; Allescher, H.-D.; Korth, M.; Wilm, M.; et al. Regulation of Intracellular Calcium by a Signalling Complex of IRAG, IP 3 Receptor and CGMP Kinase Iβ. Nature 2000, 404, 197–201. [Google Scholar] [CrossRef]
- Jurzik, L.; Froh, M.; Straub, R.H.; Schölmerich, J.; Wiest, R. Up-Regulation of NNOS and Associated Increase in Nitrergic Vasodilation in Superior Mesenteric Arteries in Pre-Hepatic Portal Hypertension. J. Hepatol. 2005, 43, 258–265. [Google Scholar] [CrossRef]
- Meredith, I.T.; Currie, K.E.; Anderson, T.J.; Roddy, M.A.; Ganz, P.; Creager, M.A. Postischemic Vasodilation in Human Forearm Is Dependent on Endothelium-Derived Nitric Oxide. Am. J. Physiol. Heart Circ. 1996, 270, H1435–H1440. [Google Scholar] [CrossRef]
- Gilligan, D.M.; Panza, J.A.; Kilcoyne, C.M.; Waclawiw, M.A.; Casino, P.R.; Quyyumi, A.A. Contribution of Endothelium-Derived Nitric Oxide to Exercise-Induced Vasodilation. Circulation 1994, 90, 2853–2858. [Google Scholar] [CrossRef]
- Kuchan, M.J.; Frangos, J.A. Role of Calcium and Calmodulin in Flow-Induced Nitric Oxide Production in Endothelial Cells. Am. J. Physiol. Cell Physiol. 1994, 266, C628–C636. [Google Scholar] [CrossRef]
- Schuh, K.; Uldrijan, S.; Telkamp, M.; Röthlein, N.; Neyses, L. The Plasmamembrane Calmodulin–Dependent Calcium Pump: A Major Regulator of Nitric Oxide Synthase I. J. Cell Biol. 2001, 155, 201–206. [Google Scholar] [CrossRef]
- Jordan, M.L.; Rominski, B.; Jaquins-Gerstl, A.; Geller, D.; Hoffman, R.A. Regulation of Inducible Nitric Oxide Production by Intracellular Calcium. Surgery 1995, 118, 138–146. [Google Scholar] [CrossRef]
- Williams, B.A.; Liu, C.; Deyoung, L.; Brock, G.B.; Sims, S.M. Regulation of Intracellular Ca2+ Release in Corpus Cavernosum Smooth Muscle: Synergism between Nitric Oxide and CGMP. Am. J. Physiol. Cell Physiol. 2005, 288, C650–C658. [Google Scholar] [CrossRef]
- Felbel, J.; Trockur, B.; Ecker, T.; Landgraf, W.; Hofmann, F. Regulation of Cytosolic Calcium by CAMP and CGMP in Freshly Isolated Smooth Muscle Cells from Bovine Trachea. J. Biol. Chem. 1988, 263, 16764–16771. [Google Scholar] [CrossRef]
- Lau, D.T.-W.; Benet, L.Z. Effects of Sulfobromophthalein and Ethacrynic Acid on Glyceryl Trinitrate Relaxation. Biochem. Pharmacol. 1992, 43, 2247–2254. [Google Scholar] [CrossRef]
- Yeates, R.A.; Schmid, M.; Leitold, M. Antagonism of Glycerol Trinitrate Activity by an Inhibitor of Glutathione S-Transferase. Biochem. Pharmacol. 1989, 38, 1749–1753. [Google Scholar] [CrossRef]
- Brien, J.F.; McLaughlin, B.E.; Breedon, T.H.; Bennett, B.M.; Nakatsu, K.; Marks, G.S. Biotransformation of Glyceryl Trinitrate Occurs Concurrently with Relaxation of Rabbit Aorta. J. Pharmacol. Exp. Ther. 1986, 237, 608–614. [Google Scholar]
- Nigam, R.; Anderson, D.J.; Lee, S.F.; Bennett, B.M. Isoform-Specific Biotransformation of Glyceryl Trinitrate by Rat Aortic Glutathione S-Transferases. J. Pharmacol. Exp. Ther. 1996, 279, 1527–1534. [Google Scholar]
- Lau, D.T.; Chan, E.K.; Benet, L.Z. Glutathione S-Transferase-Mediated Metabolism of Glyceryl Trinitrate in Subcellular Fractions of Bovine Coronary Arteries. Pharm. Res. 1992, 9, 1460–1464. [Google Scholar] [CrossRef]
- Bennett, B.M.; McDonald, B.J.; Nigam, R.; Craig Simon, W. Biotransformation of Organic Nitrates and Vascular Smooth Muscle Cell Function. Trends Pharmacol. Sci. 1994, 15, 245–249. [Google Scholar] [CrossRef]
- Cederqvist, B.; Persson, M.G.; Gustafsson, L.E. Direct Demonstration of No Formation in Vivo from Organic Nitrites and Nitrates, and Correlation to Effects on Blood Pressure and to in Vitro Effects. Biochem. Pharmacol. 1994, 47, 1047–1053. [Google Scholar] [CrossRef]
- Münzel, T.; Steven, S.; Daiber, A. Organic Nitrates: Update on Mechanisms Underlying Vasodilation, Tolerance and Endothelial Dysfunction. Vasc. Pharmacol. 2014, 63, 105–113. [Google Scholar] [CrossRef]
- França-Silva, M.S.; Balarini, C.M.; Cruz, J.C.; Khan, B.A.; Rampelotto, P.H.; Braga, V.A. Organic Nitrates: Past, Present and Future. Molecules 2014, 19, 15314–15323. [Google Scholar] [CrossRef]
- Kleschyov, A.L.; Oelze, M.; Daiber, A.; Huang, Y.; Mollnau, H.; Schulz, E.; Sydow, K.; Fichtlscherer, B.; Mülsch, A.; Münzel, T. Does Nitric Oxide Mediate the Vasodilator Activity of Nitroglycerin? Circ. Res. 2003, 93, e104–e112. [Google Scholar] [CrossRef]
- Núñez, C.; Víctor, V.M.; Tur, R.; Alvarez-Barrientos, A.; Moncada, S.; Esplugues, J.V.; D’Ocón, P. Discrepancies between Nitroglycerin and NO-Releasing Drugs on Mitochondrial Oxygen Consumption, Vasoactivity, and the Release of NO. Circ. Res. 2005, 97, 1063–1069. [Google Scholar] [CrossRef]
- Heppel, L.A.; Hilmoe, R.J. Metabolism of Inorganic Nitrite and Nitrate Esters: II. The Enzymatic Reduction of Nitroglycerin and Erythritol Tetranitrate by Glutathione. J. Biol. Chem. 1950, 183, 129–138. [Google Scholar] [CrossRef]
- Habig, W.H.; Keen, J.H.; Jakoby, W.B. Glutathione S-Transferase in the Formation of Cyanide from Organic Thiocyanates and as an Organic Nitrate Reductase. Biochem. Biophys. Res. Commun. 1975, 64, 501–506. [Google Scholar] [CrossRef]
- Keen, J.H.; Habig, W.H.; Jakoby, W.B. Mechanism for the Several Activities of the Glutathione S-Transferases. J. Biol. Chem. 1976, 251, 6183–6188. [Google Scholar] [CrossRef]
- Tsikas, D.; Surdacki, A. Biotransformation of Organic Nitrates by Glutathione S-Transferases and Other Enzymes: An Appraisal of the Pioneering Work by William B. Jakoby. Anal. Biochem. 2022, 644, 113993. [Google Scholar] [CrossRef]
- Castiglione, N.; Rinaldo, S.; Giardina, G.; Stelitano, V.; Cutruzzolà, F. Nitrite and Nitrite Reductases: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2012, 17, 684–716. [Google Scholar] [CrossRef]
- Angelucci, F.; Baiocco, P.; Brunori, M.; Gourlay, L.; Morea, V.; Bellelli, A. Insights into the Catalytic Mechanism of Glutathione S-Transferase: The Lesson from Schistosoma Haematobium. Structure 2005, 13, 1241–1246. [Google Scholar] [CrossRef]
- Sjödin, B.; Mannervik, B. Role of Human Glutathione Transferases in Biotransformation of the Nitric Oxide Prodrug JS-K. Sci. Rep. 2021, 11, 20765. [Google Scholar] [CrossRef]
- Weyerbrock, A.; Osterberg, N.; Psarras, N.; Baumer, B.; Kogias, E.; Werres, A.; Bette, S.; Saavedra, J.E.; Keefer, L.K.; Papazoglou, A. JS-K, a Glutathione S-Transferase-Activated Nitric Oxide Donor with Antineoplastic Activity in Malignant Gliomas. Neurosurgery 2012, 70, 497–510. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Li, J.; Tang, J.; Li, B.; Zhang, Y.; Gu, N.; Yang, F. Sphingosine 1-Phosphate Liposomes for Targeted Nitric Oxide Delivery to Mediate Anticancer Effects against Brain Glioma Tumors. Adv. Mater. 2021, 33, 2101701. [Google Scholar] [CrossRef]
- Kaur, I.; Terrazas, M.; Kosak, K.M.; Kern, S.E.; Boucher, K.M.; Shami, P.J. Cellular Distribution Studies of the Nitric Oxide-Generating Antineoplastic Prodrug O2-(2,4-Dinitrophenyl)1-((4-Ethoxycarbonyl)Piperazin-1-Yl)Diazen-1-Ium-1,2-Diolate Formulated in Pluronic P123 Micelles. J. Pharm. Pharmacol. 2013, 65, 1329–1336. [Google Scholar] [CrossRef]
- Saavedra, J.E.; Srinivasan, A.; Buzard, G.S.; Davies, K.M.; Waterhouse, D.J.; Inami, K.; Wilde, T.C.; Citro, M.L.; Cuellar, M.; Deschamps, J.R.; et al. PABA/NO as an Anticancer Lead: Analogue Synthesis, Structure Revision, Solution Chemistry, Reactivity toward Glutathione, and in Vitro Activity. J. Med. Chem. 2006, 49, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Hong, S.Y.; Maciag, A.E.; Saavedra, J.E.; Adamson, D.H.; Prud’homme, R.K.; Keefer, L.K.; Chakrapani, H. Stabilization of the Nitric Oxide (NO) Prodrugs and Anticancer Leads, PABA/NO and Double JS-K, through Incorporation into PEG-Protected Nanoparticles. Mol. Pharm. 2010, 7, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Findlay, V.J.; Fazilev, F.; Ogle, M.; Fraser, J.; Saavedra, J.E.; Ji, X.; Keefer, L.K.; Tew, K.D. A Glutathione S-Transferase π-Activated Prodrug Causes Kinase Activation Concurrent with S-Glutathionylation of Proteins. Mol. Pharmacol. 2006, 69, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Shami, P.J.; Saavedra, J.E.; Bonifant, C.L.; Chu, J.; Udupi, V.; Malaviya, S.; Carr, B.I.; Kar, S.; Wang, M.; Jia, L.; et al. Antitumor Activity of JS-K [O2-(2,4-Dinitrophenyl) 1-[(4-Ethoxycarbonyl)Piperazin-1-Yl]Diazen-1-Ium-1,2-Diolate] and Related O2-Aryl Diazeniumdiolates in Vitro and in Vivo. J. Med. Chem. 2006, 49, 4356–4366. [Google Scholar] [CrossRef]
- Nigam, R.; Whiting, T.; Bennett, B.M. Effect of Inhibitors of Glutathione S-Transferase on Glyceryl Trinitrate Activity in Isolated Rat Aorta. Can. J. Physiol. Pharmacol. 1993, 71, 179–184. [Google Scholar] [CrossRef]
- Kenkare, S.R.; Benet, L.Z. Effect of Ethacrynic Acid, a Glutathione-S-Transferase Inhibitor, on Nitroglycerin-Mediated CGMP Elevation and Vasorelaxation of Rabbit Aortic Strips. Biochem. Pharmacol. 1993, 46, 279–284. [Google Scholar] [CrossRef]
- Fulci, C.; Rotili, D.; De Luca, A.; Stella, L.; Morozzo della Rocca, B.; Forgione, M.; Di Paolo, V.; Mai, A.; Falconi, M.; Quintieri, L.; et al. A New Nitrobenzoxadiazole-Based GSTP1-1 Inhibitor with a Previously Unheard of Mechanism of Action and High Stability. J. Enzyme Inhib. Med. Chem. 2017, 32, 240–247. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK Signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef]
- Sandoval-Carrillo, A.; Aguilar-Duran, M.; Vázquez-Alaniz, F.; Castellanos-Juárez, F.X.; Barraza-Salas, M.; Sierra-Campos, E.; Téllez-Valencia, A.; La Llave-León, O.; Salas-Pacheco, J.M. Polymorphisms in the GSTT1 and GSTM1 Genes Are Associated with Increased Risk of Preeclampsia in the Mexican Mestizo Population. Genet. Mol. Res. 2014, 13, 2160–2165. [Google Scholar] [CrossRef]
- Akther, L.; Rahman, M.M.; Bhuiyan, M.E.S.; Hosen, M.B.; Nesa, A.; Kabir, Y. Role of GSTT1 and GSTM1 Gene Polymorphism for Development of Preeclampsia in Bangladeshi Women. FASEB J. 2018, 32, 538.6. [Google Scholar] [CrossRef]
- McBride, M.W.; Carr, F.J.; Graham, D.; Anderson, N.H.; Clark, J.S.; Lee, W.K.; Charchar, F.J.; Brosnan, M.J.; Dominiczak, A.F. Microarray Analysis of Rat Chromosome 2 Congenic Strains. Hypertension 2003, 41, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, J.C.; Tin, A.; Pourafshar, S.; Cechova, S.; Wang, Y.T.; Sung, S.J.; Bodonyi-Kovacs, G.; Cross, J.V.; Yang, G.; Nguyen, N.; et al. GSTM1 Deletion Exaggerates Kidney Injury in Experimental Mouse Models and Confers the Protective Effect of Cruciferous Vegetables in Mice and Humans. J. Am. Soc. Nephrol. 2020, 31, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Eslami, S.; Sahebkar, A. Glutathione-S-Transferase M1 and T1 Null Genotypes Are Associated with Hypertension Risk: A Systematic Review and Meta-Analysis of 12 Studies. Curr. Hypertens. Rep. 2014, 16, 432. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russell, T.M.; Richardson, D.R. Glutathione-S-Transferases as Potential Targets for Modulation of Nitric Oxide-Mediated Vasodilation. Biomolecules 2022, 12, 1292. https://doi.org/10.3390/biom12091292
Russell TM, Richardson DR. Glutathione-S-Transferases as Potential Targets for Modulation of Nitric Oxide-Mediated Vasodilation. Biomolecules. 2022; 12(9):1292. https://doi.org/10.3390/biom12091292
Chicago/Turabian StyleRussell, Tiffany M., and Des R. Richardson. 2022. "Glutathione-S-Transferases as Potential Targets for Modulation of Nitric Oxide-Mediated Vasodilation" Biomolecules 12, no. 9: 1292. https://doi.org/10.3390/biom12091292