
Biochemical and Structural Characterization of Chi-Class Glutathione Transferases: A Snapshot on the Glutathione Transferase Encoded by sll0067 Gene in the Cyanobacterium Synechocystis sp. Strain PCC 6803
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Mutagenesis, Expression and Purification
2.2. Crystallization, X-ray Data Collection, Processing and Refinement
2.3. Structure Analysis Based on Electron Density Distribution
2.4. Molecular Dynamics Simulation
2.5. Enzymatic Assays
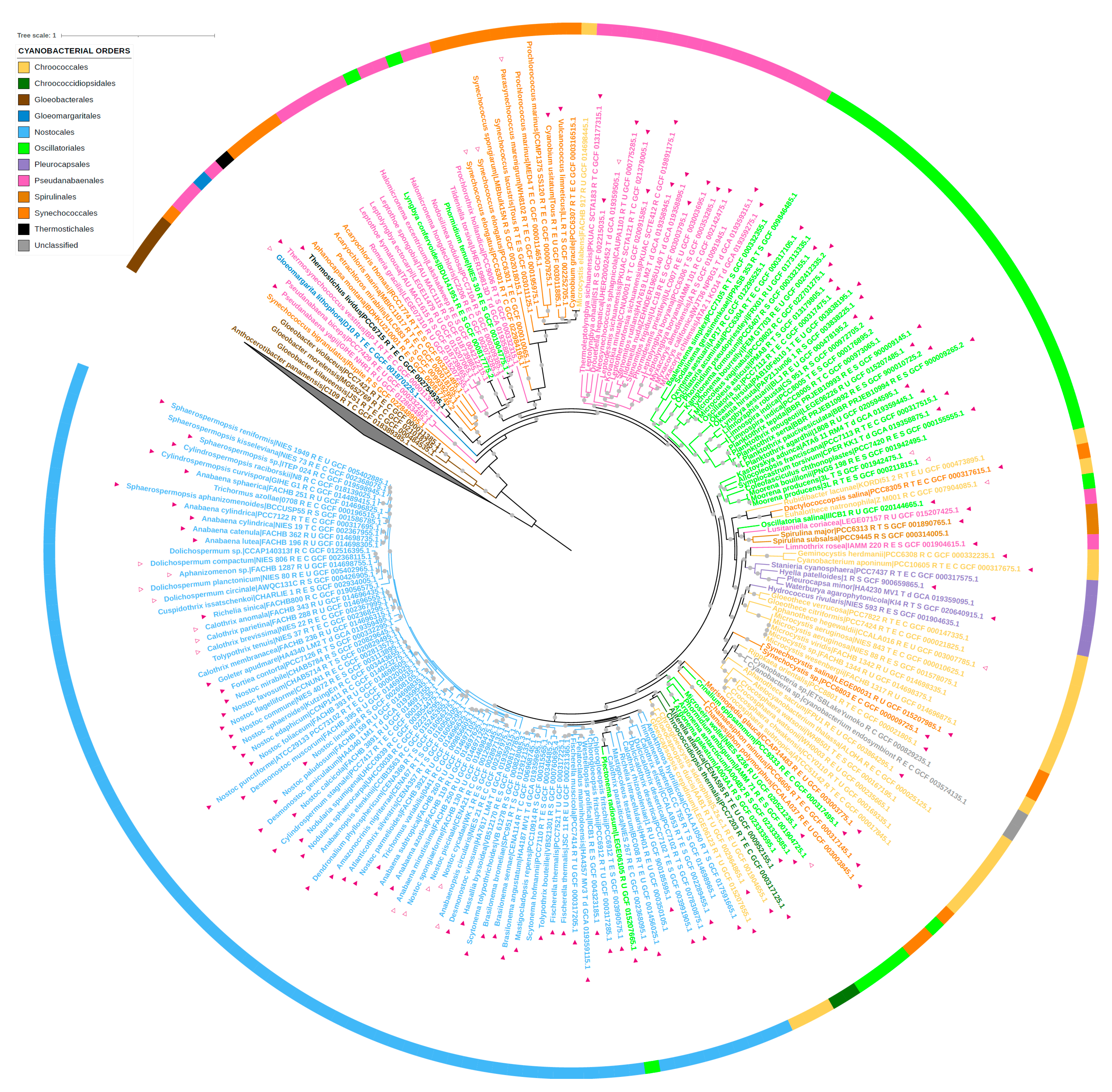
2.6. Phylogenetic Analysis
3. Results and Discussion
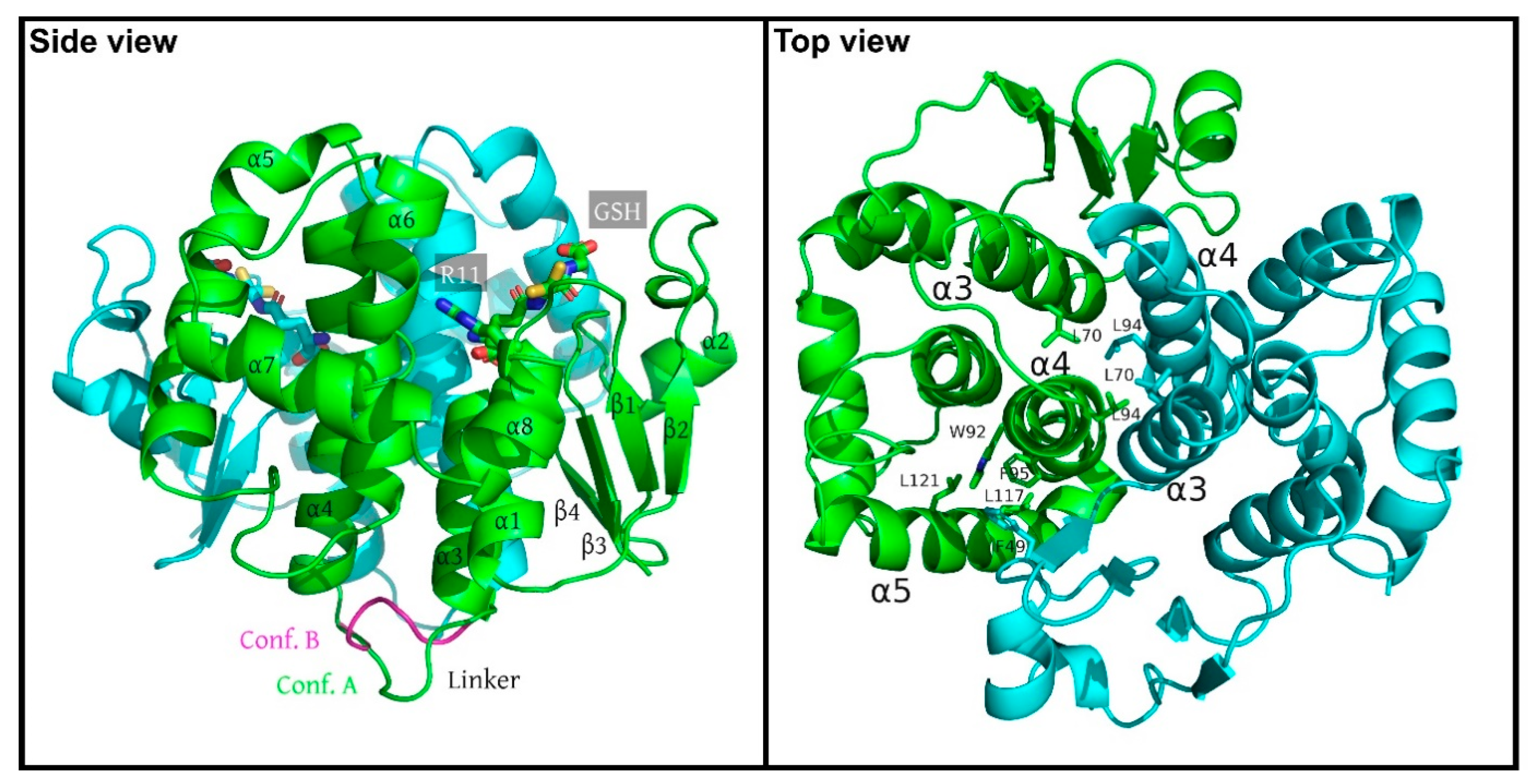
3.1. Crystal Structure of SynGSTC1
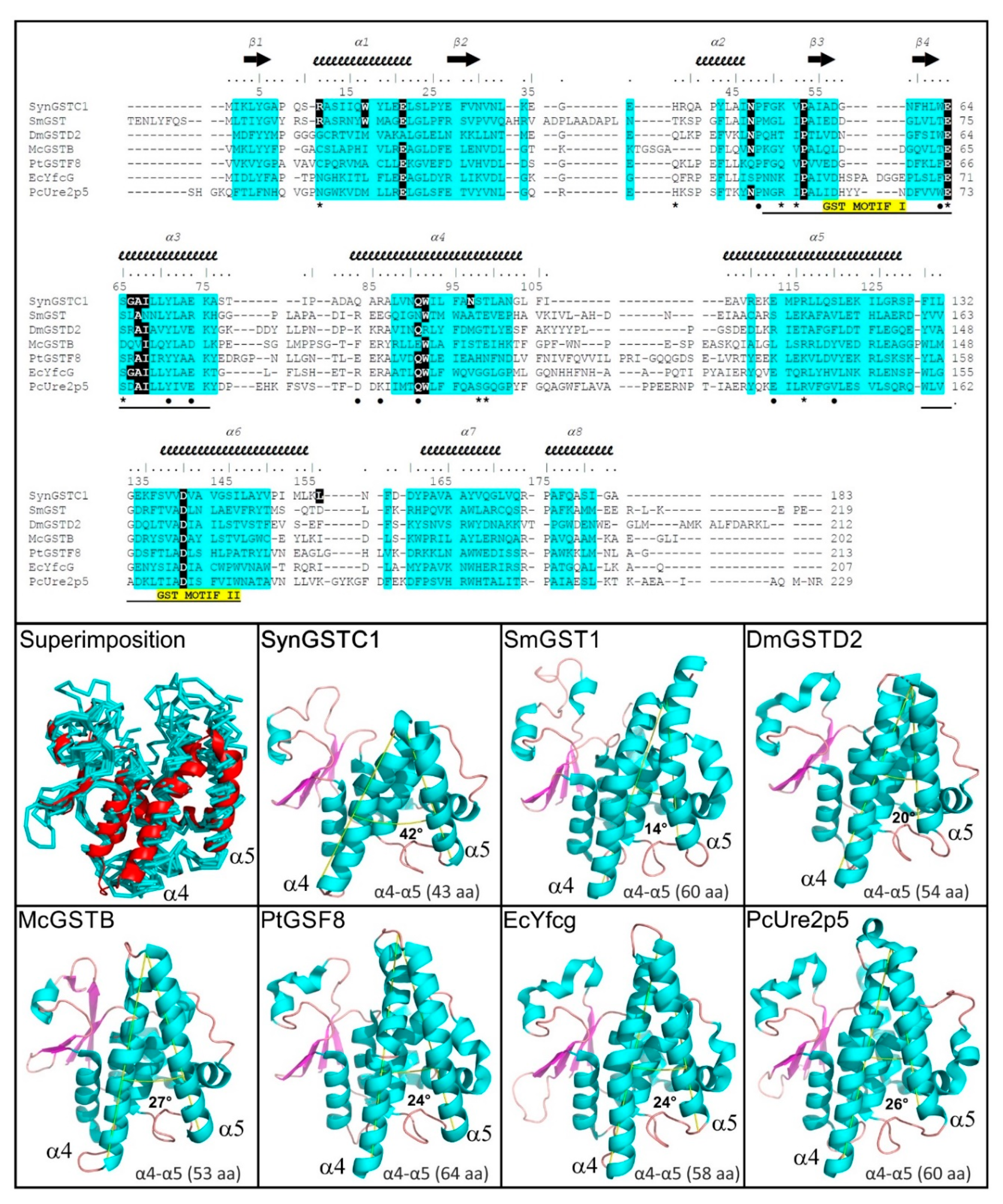
3.2. Structural Comparison
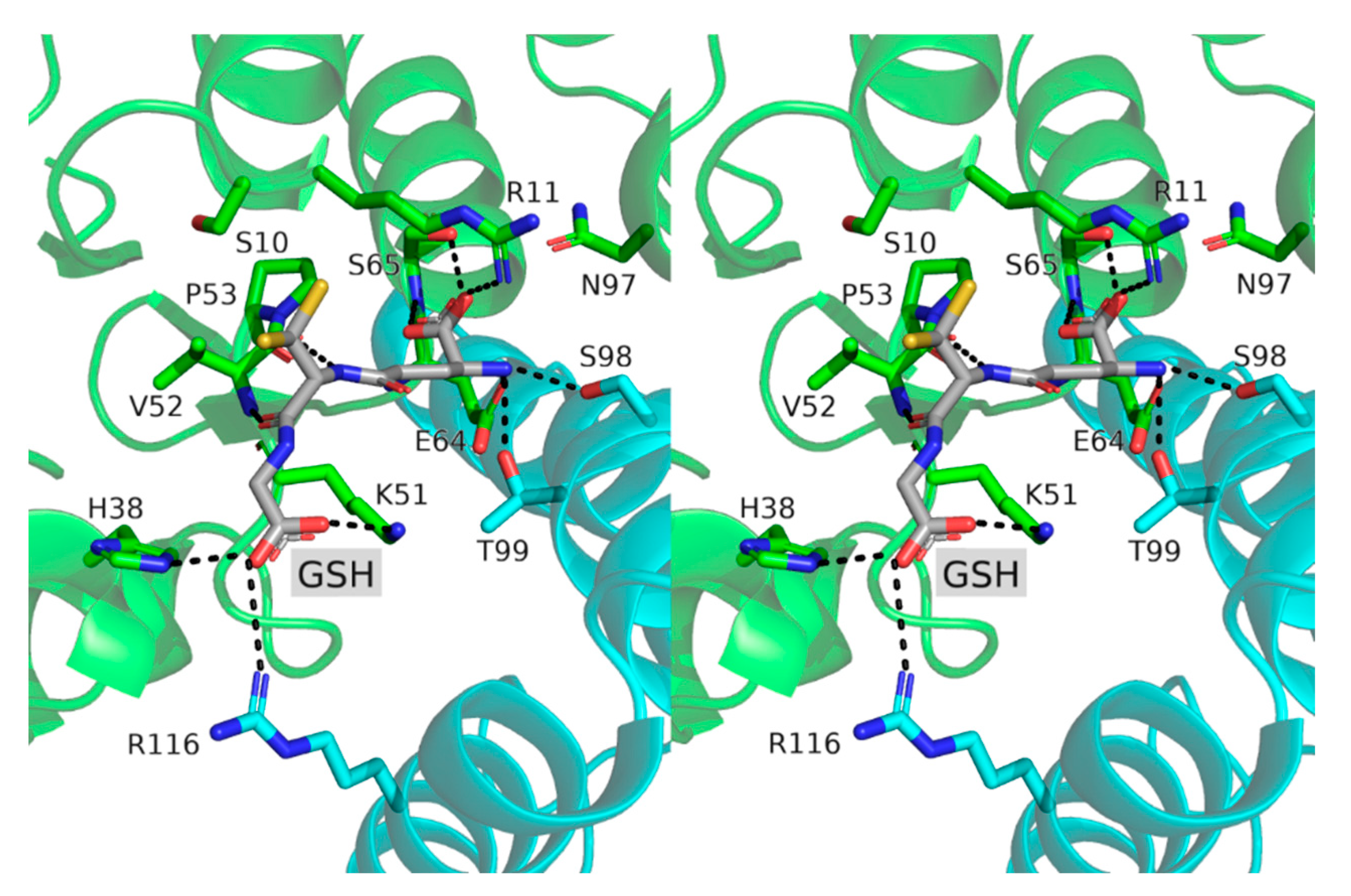
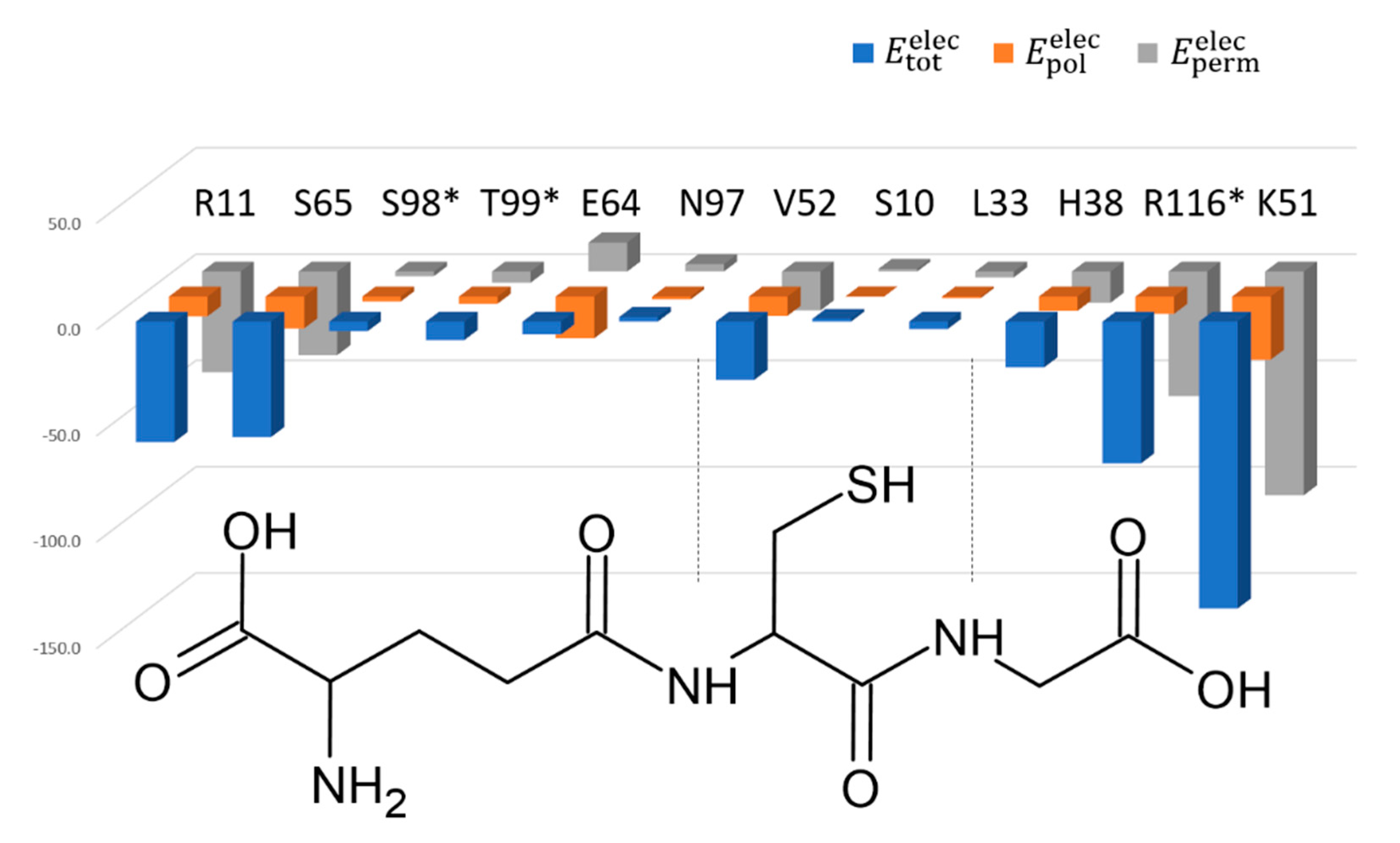
3.3. Active Site Structure and Its Analysis Using Transferred Multipolar Electron-Density
3.4. Biochemical Characterization of SynGSTC1 and Variants
3.5. Comparative Genomic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mashiyama, S.T.; Malabanan, M.M.; Akiva, E.; Bhosle, R.; Branch, M.C.; Hillerich, B.; Jagessar, K.; Kim, J.; Patskovsky, Y.; Seidel, R.D.; et al. Large-scale determination of sequence, structure, and function relationships in cytosolic glutathione transferases across the biosphere. PLoS Biol. 2014, 12, e1001843. [Google Scholar] [CrossRef]
- Allocati, N.; Federici, L.; Masulli, M.; Di Ilio, C. Glutathione transferases in bacteria. FEBS J. 2009, 276, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-X.; Jiang, Y.-L.; He, Y.-X.; Bao, R.; Chen, Y.; Zhou, C.-Z. Structures of yeast glutathione-S-transferase Gtt2 reveal a new catalytic type of GST family. EMBO Rep. 2009, 10, 1320–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, A.J.M.; Tyzack, J.D.; Borkakoti, N.; Holliday, G.L.; Thornton, J.M. A global analysis of function and conservation of catalytic residues in enzymes. J. Biol. Chem. 2020, 295, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Perrot, T.; Schwartz, M.; Saiag, F.; Salzet, G.; Dumarçay, S.; Favier, F.; Gérardin, P.; Girardet, J.-M.; Sormani, R.; Morel-Rouhier, M.; et al. Fungal Glutathione Transferases as Tools to Explore the Chemical Diversity of Amazonian Wood Extractives. ACS Sustain. Chem. Eng. 2018, 6, 13078–13085. [Google Scholar] [CrossRef]
- Schwartz, M.; Perrot, T.; Aubert, E.; Dumarçay, S.; Favier, F.; Gerardin, P.; Morel-Rouhier, M.; Mulliert, G.; Saiag, F.; Didierjean, C.; et al. Molecular recognition of wood polyphenols by phase II detoxification enzymes of the white rot Trametes versicolor. Sci. Rep. 2018, 8, 8472. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nianiou-Obeidat, I.; Madesis, P.; Kissoudis, C.; Voulgari, G.; Chronopoulou, E.; Tsaftaris, A.; Labrou, N.E. Plant glutathione transferase-mediated stress tolerance: Functions and biotechnological applications. Plant Cell Rep. 2017, 36, 791–805. [Google Scholar] [CrossRef]
- Sylvestre-Gonon, E.; Law, S.R.; Schwartz, M.; Robe, K.; Keech, O.; Didierjean, C.; Dubos, C.; Rouhier, N.; Hecker, A. Functional, Structural and Biochemical Features of Plant Serinyl-Glutathione Transferases. Front. Plant Sci. 2019, 10, 608. [Google Scholar] [CrossRef] [Green Version]
- Wiktelius, E.; Stenberg, G. Novel class of glutathione transferases from cyanobacteria exhibit high catalytic activities towards naturally occurring isothiocyanates. Biochem. J. 2007, 406, 115–123. [Google Scholar] [CrossRef]
- Shehu, D.; Abdullahi, N.; Alias, Z. Cytosolic Glutathione S-transferase in Bacteria: A Review. Pol. J. Environ. Stud. 2019, 28, 515–528. [Google Scholar] [CrossRef]
- Meux, E.; Prosper, P.; Ngadin, A.; Didierjean, C.; Morel, M.; Dumarçay, S.; Lamant, T.; Jacquot, J.-P.; Favier, F.; Gelhaye, E. Glutathione Transferases of Phanerochaete chrysosporium: S-Glutathionyl-p-hydroquinone Reductase Belongs to a New Structural Class. J. Biol. Chem. 2011, 286, 9162–9173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masai, E.; Ichimura, A.; Sato, Y.; Miyauchi, K.; Katayama, Y.; Fukuda, M. Roles of the enantioselective glutathione S-transferases in cleavage of beta-aryl ether. J. Bacteriol. 2003, 185, 1768–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meux, E.; Prosper, P.; Masai, E.; Mulliert, G.; Dumarçay, S.; Morel, M.; Didierjean, C.; Gelhaye, E.; Favier, F. Sphingobium sp. SYK-6 LigG involved in lignin degradation is structurally and biochemically related to the glutathione transferase omega class. FEBS Lett. 2012, 586, 3944–3950. [Google Scholar] [CrossRef] [PubMed]
- Kammerscheit, X.; Hecker, A.; Rouhier, N.; Chauvat, F.; Cassier-Chauvat, C. Methylglyoxal Detoxification Revisited: Role of Glutathione Transferase in Model Cyanobacterium Synechocystis sp. Strain PCC 6803. mBio 2020, 11, e00882-20. [Google Scholar] [CrossRef]
- Pandey, T.; Singh, S.K.; Chhetri, G.; Tripathi, T.; Singh, A.K. Characterization of a Highly pH Stable Chi-Class Glutathione S-Transferase from Synechocystis PCC 6803. PLoS ONE 2015, 10, e0126811. [Google Scholar] [CrossRef] [Green Version]
- Feil, S.C.; Tang, J.; Hansen, G.; Gorman, M.A.; Wiktelius, E.; Stenberg, G.; Parker, M.W. Crystallization and preliminary X-ray analysis of glutathione transferases from cyanobacteria. Acta Crystallogr. Sect. F Crystallogr. Commun. 2009, 65, 475–477. [Google Scholar] [CrossRef] [Green Version]
- Allocati, N.; Casalone, E.; Masulli, M.; Ceccarelli, I.; Carletti, E.; Parker, M.W.; Di Ilio, C. Functional analysis of the evolutionarily conserved proline 53 residue in Proteus mirabilis glutathione transferase B1-1. FEBS Lett. 1999, 445, 347–350. [Google Scholar] [CrossRef] [Green Version]
- Dragani, B.; Stenberg, G.; Melino, S.; Petruzzelli, R.; Mannervik, B.; Aceto, A. The Conserved N-capping Box in the Hydrophobic Core of Glutathione S-Transferase P1-1 Is Essential for Refolding. Identification of A buried and Conserved Hydrogen Bond Important for Protein Stability. J. Biol. Chem. 1997, 272, 25518–25523. [Google Scholar] [CrossRef] [Green Version]
- ShylajaNaciyar, M.; Karthick, L.; Prakasam, P.A.; Deviram, G.; Uma, L.; Prabaharan, D.; Saha, S.K. Diversity of Glutathione S-Transferases (GSTs) in Cyanobacteria with Reference to Their Structures, Substrate Recognition and Catalytic Functions. Microorganisms 2020, 8, 712. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagin, A.; Lebedev, A. MoRDa, an automatic molecular replacement pipeline. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, s19. [Google Scholar] [CrossRef] [Green Version]
- Smart, O.S.; Womack, T.O.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Vonrhein, C.; Bricogne, G. Exploiting structure similarity in refinement: Automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. Sect. A Found. Adv. 1978, 34, 909–921. [Google Scholar] [CrossRef]
- Domagala, S.; Fournier, B.; Liebschner, D.; Guillot, B.; Jelsch, C. An improved experimental databank of transferable multipolar atom models—ELMAM2. Construction details and applications. Acta Crystallogr. Sect. A Found. Adv. 2012, 68, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Leduc, T.; Aubert, E.; Espinosa, E.; Jelsch, C.; Iordache, C.; Guillot, B. Polarization of Electron Density Databases of Transferable Multipolar Atoms. J. Phys. Chem. 2019, 123, 7156–7170. [Google Scholar] [CrossRef] [Green Version]
- Guillot, B.; Enrique, E.; Huder, L.; Jelsch, C. MoProViewer: A tool to study proteins from a charge density science perspective. Acta Crystallogr. Sect. A Found. Adv. 2014, 70, C279. [Google Scholar] [CrossRef]
- Vuković, V.; Leduc, T.; Jelić-Matošević, Z.; Didierjean, C.; Favier, F.; Guillot, B.; Jelsch, C. A rush to explore protein–ligand electrostatic interaction energy with Charger. Acta Crystallogr. Sect. D Biol. Crystallogr. 2021, 77, 1292–1304. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jauffrit, F.; Penel, S.; Delmotte, S.; Rey, C.; de Vienne, D.M.; Gouy, M.; Charrier, J.P.; Flandrois, J.P.; Brochier-Armanet, C. RiboDB Database: A Comprehensive Resource for Prokaryotic Systematics. Mol. Biol. Evol. 2016, 33, 2170–2172. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Polekhina, G.; Board, P.G.; Blackburn, A.C.; Parker, M.W. Crystal structure of maleylacetoacetate isomerase/glutathione transferase zeta reveals the molecular basis for its remarkable catalytic promiscuity. Biochemistry 2001, 40, 1567–1576. [Google Scholar] [CrossRef]
- Wilce, M.C.J.; Parker, M.W. Structure and function of glutathione S-transferasese. Biochim. Biophys. Acta Prot. Struct. Mol. Enzymol. 1994, 1205, 1–18. [Google Scholar] [CrossRef]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef]
- Dong, R.; Pan, S.; Peng, Z.; Zhang, Y.; Yang, J. mTM-align: A server for fast protein structure database search and multiple protein structure alignment. Nucleic Acids Res. 2018, 46, W380–W386. [Google Scholar] [CrossRef]
- Jacquot, J.P.; Gelhaye, E.; Rouhier, N.; Corbier, C.; Didierjean, C.; Aubry, A. Thioredoxins and related proteins in photosynthetic organisms: Molecular basis for thiol dependent regulation. Biochem. Pharmacol. 2002, 64, 1065–1069. [Google Scholar] [CrossRef]
- Dixon, D.P.; Edwards, R. Glutathione Transferases. Arabidop. Book 2010, 8, e0131. [Google Scholar] [CrossRef] [Green Version]
- Pegeot, H.; Koh, C.S.; Petre, B.; Mathiot, S.; Duplessis, S.; Hecker, A.; Didierjean, C.; Rouhier, N. The poplar Phi class glutathione transferase: Expression, activity and structure of GSTF1. Front. Plant. Sci. 2014, 5, 712. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef]
- Sylvestre-Gonon, E.; Morette, L.; Viloria, M.; Mathiot, S.; Boutilliat, A.; Favier, F.; Rouhier, N.; Didierjean, C.; Hecker, A. Biochemical and structural insights on the poplar tau glutathione transferase GSTU19 and 20 paralogs binding flavonoids. Front. Mol. Biosci. 2022, 9, 9585866. [Google Scholar] [CrossRef] [PubMed]
- Dourado, D.F.; Fernandes, P.A.; Mannervik, B.; Ramos, M.J. Glutathione transferase: New model for glutathione activation. Chemistry 2008, 14, 9591–9598. [Google Scholar] [CrossRef]
- Skopelitou, K.; Dhavala, P.; Papageorgiou, A.C.; Labrou, N.E. A glutathione transferase from Agrobacterium tumefaciens reveals a novel class of bacterial GST superfamily. PLoS ONE 2012, 7, e34263. [Google Scholar] [CrossRef] [Green Version]
- Moreira, D.; Tavera, R.; Benzerara, K.; Skouri-Panet, F.; Couradeau, E.; Gerard, E.; Fonta, C.L.; Novelo, E.; Zivanovic, Y.; Lopez-Garcia, P. Description of Gloeomargarita lithophora gen. nov., sp. nov., a thylakoid-bearing, basal-branching cyanobacterium with intracellular carbonates, and proposal for Gloeomargaritales ord. nov. Int. J. Syst. Evol. Microbiol. 2017, 67, 653–658. [Google Scholar] [CrossRef]
- Dvornyk, V.; Mei, Q. Evolution of kaiA, a key circadian gene of cyanobacteria. Sci. Rep. 2021, 11, 9995. [Google Scholar] [CrossRef]
- Halabi, N.; Rivoire, O.; Leibler, S.; Ranganathan, R. Protein sectors: Evolutionary units of three-dimensional structure. Cell 2009, 138, 774–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Roret, T.; Thuillier, A.; Favier, F.; Gelhaye, E.; Didierjean, C.; Morel-Rouhier, M. Evolutionary divergence of Ure2pA glutathione transferases in wood degrading fungi. Fungal Genet. Biol. 2015, 83, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Aceto, A.; Dragani, B.; Melino, S.; Allocati, N.; Masulli, M.; Ilio, C.D.; Petruzzelli, R. Identification of an N-capping box that affects the α6-helix propensity in glutathione S-transferase superfamily proteins: A role for an invariant aspartic residue. Biochem. J. 1997, 322, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Dirr, H.; Reinemer, P.; Huber, R. X-ray crystal structures of cytosolic glutathione Stransferases: Implications for protein architecture, substrate recognition and catalytic function. Eur. J. Biochem. 1994, 220, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild-Type | S10T | R11A | |
|---|---|---|---|
| Data Collection | |||
| Diffraction source | ESRF-BM07 | ESRF-BM07 | ESRF-BM07 |
| Detector | Pilatus 6M | Pilatus 6M | Pilatus 6M |
| Wavelength (Å) | 0.97951 | 0.97951 | 0.97951 |
| Space Group | P43212 | P43212 | P43212 |
| Unit-cell a; c (Å) | 92.5; 193.6 | 92.9; 193.6 | 92.2; 193.1 |
| Resolution Range (Å) | 48.4 1.7 (1.73 1.70) | 48.4 1.7 (1.73 1.70) | 46.1 2.2 (2.27 2.20) |
| Tot. no. of meas. int. | 1,200,217 (41158) | 1,244,521 (62,785) | 503,802 (22,268) |
| Unique reflections | 92,986 (4529) | 93,931 (4590) | 39,298 (2006) |
| Average redundancy | 13 (9) | 13.2 (14) | 13 (11) |
| Mean I/σ (I) | 24.8 (1.8) | 17.0 (2.0) | 18.4 (2.4) |
| Completeness (%) | 100.0 (99.6) | 100.0 (100.0) | 91.3 (55.5) |
| Rmerge | 0.056 (1.097) | 0.084 (1.52) | 0.097 (1.039) |
| Rmeas | 0.061 (1.168) | 0.087 (1.59) | 0.100 (1.142) |
| CC1/2 | 1.00 (0.83) | 1.00 (0.84) | 1.00 (0.85) |
| Wilson B-factor (Å2) | 29.6 | 28.6 | 41.5 |
| Refinement | |||
| Resolution Range (Å) | 24.8 1.7 | 24.2 1.7 | 31.3 2.2 |
| No. of reflections | 92839 | 93783 | 39248 |
| Rwork/Rfree | 0.204/0.221 | 0.206/0.221 | 0.215/0.242 |
| Corr Fo-Fc/Fo-Fcfree | 0.938/0.936 | 0.940/0.939 | 0.907/0.888 |
| Total number of atoms | 3469 | 3500 | 3253 |
| Average B-factor (Å2) | 34.0 | 32.5 | 44.0 |
| Model quality | |||
| RMSZ Bond lengths | 0.41 | 0.42 | 0.42 |
| RMSZ Bond angles | 0.54 | 0.56 | 0.56 |
| Ramachandran fav. (%) | 98 | 98 | 98 |
| Ramachandran all. (%) | 2 | 2 | 2 |
| Rotamer outliers (%) | 0 | 0 | 1 |
| Clashscore | 1 | 1 | 1 |
| PDB entry | 8AI8 | 8AI9 | 8AIB |
| PITC | GSH | HED | |
|---|---|---|---|
| kcat (s−1) | |||
| WT | 12.6 ± 0.2 | ND | |
| S10T | 7.2 ± 0.1 | ND | |
| S10A | 2.60 ± 0.05 | ND | |
| S10C | 1.19 ± 0.02 | 0.0111 ± 0.0002 | |
| R11A | 0.170 ± 0.003 | ND | |
| Km (µM) | |||
| WT | 33.8 ± 2.5 | 135.2 ± 7.9 | ND |
| S10T | 33.6 ± 2.6 | 142,8 ± 14.6 | ND |
| S10A | 89.7 ± 6.4 | 528.4 ± 33.0 | ND |
| S10C | 33.0 ± 3.0 | 2149 ± 123 | 3.3 ± 0.4 |
| R11A | 108.4 ± 6.0 | 719.2 ± 58.2 | ND |
| kcat/Km (M−1·s−1) | |||
| WT | 3.73 × 105 ± 0.06 × 105 | ND | |
| S10T | 2.14 × 105 ± 0.04 × 105 | ND | |
| S10A | 2.89 × 104 ± 0.06 × 104 | ND | |
| S10C | 3.60 × 104 ± 0.06 × 104 | 3.36 × 103 ± 0.08 × 103 | |
| R11A | 1.53 × 103 ± 0.02 × 103 | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mocchetti, E.; Morette, L.; Mulliert, G.; Mathiot, S.; Guillot, B.; Dehez, F.; Chauvat, F.; Cassier-Chauvat, C.; Brochier-Armanet, C.; Didierjean, C.; et al. Biochemical and Structural Characterization of Chi-Class Glutathione Transferases: A Snapshot on the Glutathione Transferase Encoded by sll0067 Gene in the Cyanobacterium Synechocystis sp. Strain PCC 6803. Biomolecules 2022, 12, 1466. https://doi.org/10.3390/biom12101466
Mocchetti E, Morette L, Mulliert G, Mathiot S, Guillot B, Dehez F, Chauvat F, Cassier-Chauvat C, Brochier-Armanet C, Didierjean C, et al. Biochemical and Structural Characterization of Chi-Class Glutathione Transferases: A Snapshot on the Glutathione Transferase Encoded by sll0067 Gene in the Cyanobacterium Synechocystis sp. Strain PCC 6803. Biomolecules. 2022; 12(10):1466. https://doi.org/10.3390/biom12101466
Chicago/Turabian StyleMocchetti, Eva, Laura Morette, Guillermo Mulliert, Sandrine Mathiot, Benoît Guillot, François Dehez, Franck Chauvat, Corinne Cassier-Chauvat, Céline Brochier-Armanet, Claude Didierjean, and et al. 2022. "Biochemical and Structural Characterization of Chi-Class Glutathione Transferases: A Snapshot on the Glutathione Transferase Encoded by sll0067 Gene in the Cyanobacterium Synechocystis sp. Strain PCC 6803" Biomolecules 12, no. 10: 1466. https://doi.org/10.3390/biom12101466