
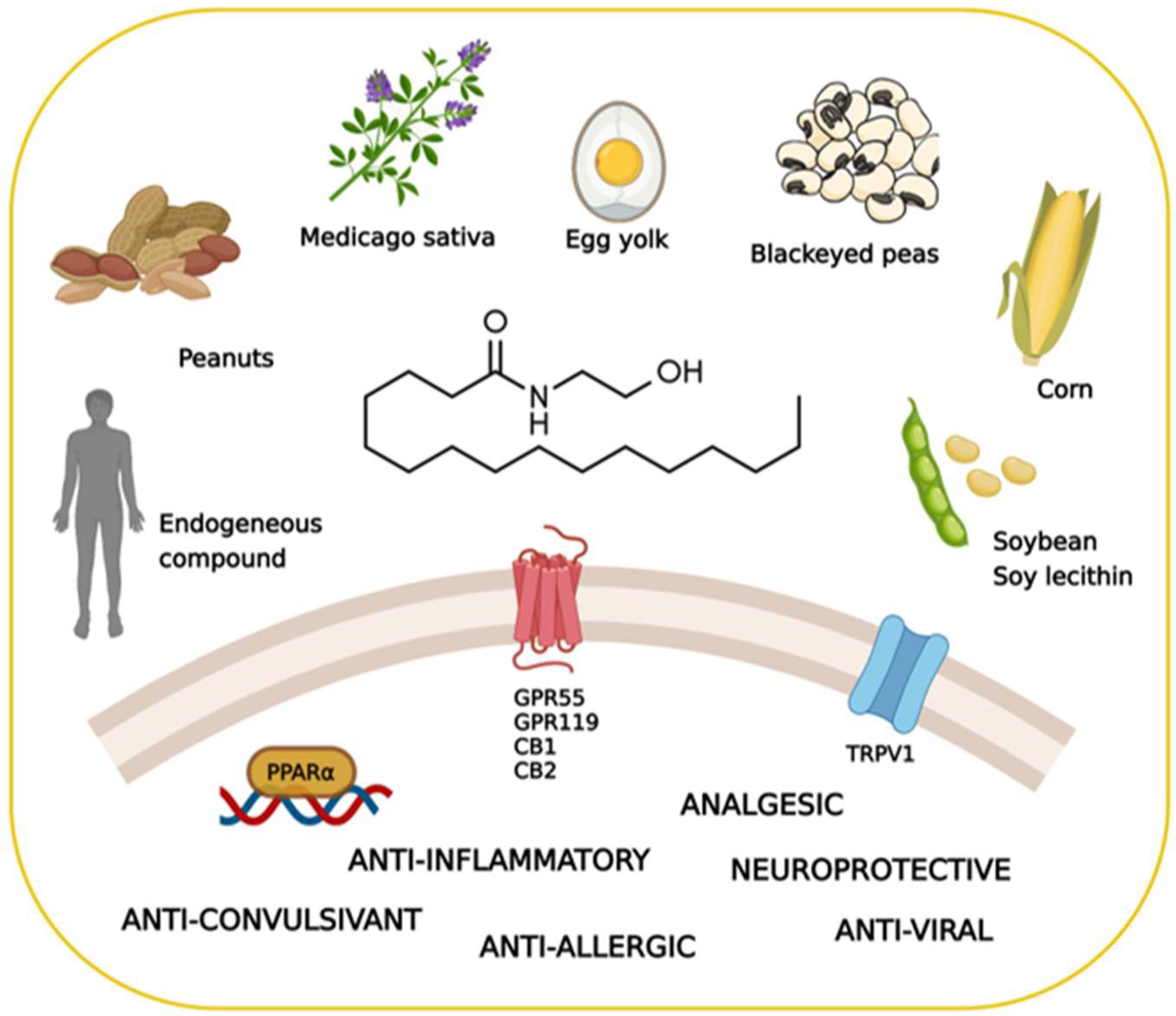
Palmitoylethanolamide and White Matter Lesions: Evidence for Therapeutic Implications


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. How Nature Provides Therapeutic Molecules: The History of Palmitoylethanolamide
2. The Pharmacology of Palmitoylethanolamide
2.1. Biosynthesis, Degradation, and Pharmacokinetics of PEA
2.2. The Search for the Palmitoylethanolamide Receptor
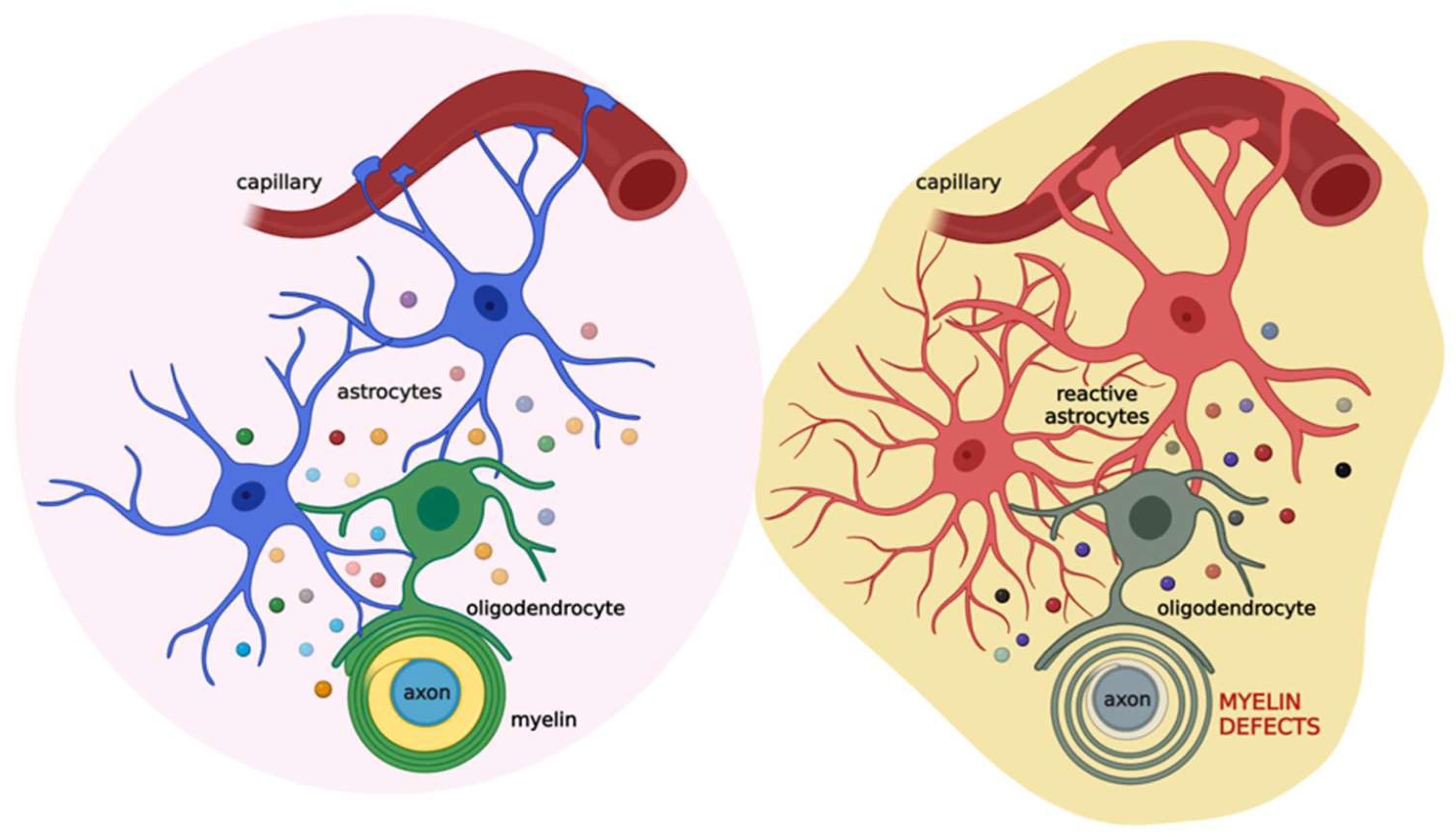
3. Myelin Sheath Organization and Functions
4. The Role of the Astrocyte-Oligodendrocyte Cross-Talk in Myelination
5. White Matter Defects in Demyelinating Diseases and the Therapeutic Potential of Palmitoylethanolamide
6. White Matter Defects in Age-Related Neurodegenerative Diseases and the Therapeutic Potential of Palmitoylethanolamide
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Coburn, A.; Moore, L. American Journal of Diseases of Children. JAMA Pediatrics 1943, 65, 744–756. [Google Scholar]
- Coburn, A.F.; Graham, C.E.; Haninger, J. The effect of egg yolk in diets on anaphylactic arthritis (passive Arthus phenomenon) in the guinea pig. J. Exp. Med. 1954, 100, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Long, D.A.; Martin, A.J. Factor in arachis oil depressing sensitivity to tuberculin in B.C.G.-infected guineapigs. Lancet 1956, 270, 464–466. [Google Scholar] [CrossRef]
- Long, D.; Miles, A. Opposite actions of thyroid and adrenal hormones in allergic hypersensitivity. Lancet 1950, 255, 492–495. [Google Scholar] [CrossRef]
- Bachur, N.R.; Masek, K.; Melmon, K.L.; Udenfriend, S. Fatty Acid Amides of Ethanolamine in Mammalian Tissues. J. Biol. Chem. 1965, 240, 1019–1024. [Google Scholar] [CrossRef]
- Aloe, L.; Leon, A.; Levi-Montalcini, R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions 1993, 39, C145–C147. [Google Scholar] [CrossRef]
- Valenza, M.; Facchinetti, R.; Menegoni, G.; Steardo, L.; Scuderi, C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules 2021, 11, 600. [Google Scholar] [CrossRef]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharm. 2019, 10, 821. [Google Scholar] [CrossRef]
- Romano, A.; Di Bonaventura, M.V.M.; Gallelli, C.A.; Koczwara, J.B.; Smeets, D.; Giusepponi, M.E.; De Ceglia, M.; Friuli, M.; Di Bonaventura, E.M.; Scuderi, C.; et al. Oleoylethanolamide decreases frustration stress-induced binge-like eating in female rats: A novel potential treatment for binge eating disorder. Neuropsychopharmacology 2020, 45, 1931–1941. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharm. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Rankin, L.; Fowler, C.J. The Basal Pharmacology of Palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Izzo, A.A.; Degenhardt, B.; Valenti, M.; Scaglione, G.; Capasso, R.; Sorrentini, I.; Di Marzo, V. Involvement of the cannabimimetic compound, N-palmitoyl-ethanolamine, in inflammatory and neuropathic conditions: Review of the available pre-clinical data, and first human studies. Neuropharmacology 2005, 48, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Zhukov, O.D. [Distribution of N-([1-14C]-palmitoyl)ethanolamine in rat tissues]. Ukr. Biokhimichnyi Zhurnal 1999, 71, 124–125. [Google Scholar]
- Artamonov, M.; Zhukov, O.; Shuba, I.; Storozhuk, L.; Khmel, T.; Klimashevsky, V.; Mikosha, A.; Gula, N. Incorporation of labelled N-acylethanolamine (NAE) into rat brain regions in vivo and adaptive properties of saturated NAE under X-ray irradiation. Ukr. Biokhimichnyi Zhurnal 2005, 77, 51–62. [Google Scholar]
- Gabrielsson, L.; Mattsson, S.; Fowler, C.J. Palmitoylethanolamide for the treatment of pain: Pharmacokinetics, safety and efficacy. Br. J. Clin. Pharm. 2016, 82, 932–942. [Google Scholar] [CrossRef]
- Balvers, M.G.; Verhoeckx, K.C.; Meijerink, J.; Wortelboer, H.M.; Witkamp, R.F. Measurement of palmitoylethanolamide and other N-acylethanolamines during physiological and pathological conditions. CNS Neurol. Disord. Drug Targets 2013, 12, 23–33. [Google Scholar] [CrossRef]
- Straus, D.S.; Glass, C.K. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007, 28, 551–558. [Google Scholar] [CrossRef]
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Steardo, L.; Pompili, E.; Fumagalli, L.; Campolongo, P.; et al. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1419. [Google Scholar] [CrossRef]
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratù, M.R.; Steardo, L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J. Neuroinflamm. 2012, 9, 49. [Google Scholar] [CrossRef]
- Paterniti, I.; Impellizzeri, D.; Crupi, R.; Morabito, R.; Campolo, M.; Esposito, E.; Cuzzocrea, S. Molecular evidence for the involvement of PPAR-δ and PPAR-γ in anti-inflammatory and neuroprotective activities of palmitoylethanolamide after spinal cord trauma. J. Neuroinflamm. 2013, 10, 20. [Google Scholar] [CrossRef]
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Meli, R.; Calignano, A.; et al. Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T+tf/J mice: Contribution of central and peripheral mechanisms. Brain Behav. Immun. 2018, 74, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo, L., Jr.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by β-amyloid peptide. J. Cell Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L. Neuroglial roots of neurodegenerative diseases: Therapeutic potential of palmitoylethanolamide in models of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2013, 12, 62–69. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-β25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? Br. J. Pharm. 2007, 152, 984–986. [Google Scholar] [CrossRef] [PubMed]
- Mattace Raso, G.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS health and disease. Pharm. Res. 2014, 86, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharm. 2017, 174, 1349–1365. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Davis, J.B.; Di Marzo, V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001, 506, 253–256. [Google Scholar] [CrossRef]
- Smart, D.; Jonsson, K.O.; Vandevoorde, S.; Lambert, D.M.; Fowler, C.J. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br. J. Pharm. 2002, 136, 452–458. [Google Scholar] [CrossRef]
- Ambrosino, P.; Soldovieri, M.V.; Russo, C.; Taglialatela, M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br. J. Pharm. 2013, 168, 1430–1444. [Google Scholar] [CrossRef]
- Capasso, R.; Orlando, P.; Pagano, E.; Aveta, T.; Buono, L.; Borrelli, F.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide normalizes intestinal motility in a model of post-inflammatory accelerated transit: Involvement of CB₁ receptors and TRPV1 channels. Br. J. Pharm. 2014, 171, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Snaidero, N.; Simons, M. Myelination at a glance. J. Cell Sci. 2014, 127, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef] [PubMed]
- Gensert, J.M.; Goldman, J.E. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 1997, 19, 197–203. [Google Scholar] [CrossRef]
- Fernandez-Castaneda, A.; Gaultier, A. Adult oligodendrocyte progenitor cells—Multifaceted regulators of the CNS in health and disease. Brain Behav. Immun. 2016, 57, 1–7. [Google Scholar] [CrossRef]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef]
- Murtie, J.C.; Zhou, Y.X.; Le, T.Q.; Vana, A.C.; Armstrong, R.C. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol. Dis. 2005, 19, 171–182. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Kitada, M.; Rowitch, D.H. Transcription factor co-expression patterns indicate heterogeneity of oligodendroglial subpopulations in adult spinal cord. Glia 2006, 54, 35–46. [Google Scholar] [CrossRef]
- Hartline, D.K. What is myelin? Neuron Glia Biol. 2008, 4, 153–163. [Google Scholar] [CrossRef]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron-glia interactions. Nat. Rev. NeuroSci. 2006, 7, 423–436. [Google Scholar] [CrossRef]
- Readhead, C.; Popko, B.; Takahashi, N.; Shine, H.D.; Saavedra, R.A.; Sidman, R.L.; Hood, L. Expression of a myelin basic protein gene in transgenic shiverer mice: Correction of the dysmyelinating phenotype. Cell 1987, 48, 703–712. [Google Scholar] [CrossRef]
- Roach, A.; Takahashi, N.; Pravtcheva, D.; Ruddle, F.; Hood, L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell 1985, 42, 149–155. [Google Scholar] [CrossRef]
- Snaidero, N.; Möbius, W.; Czopka, T.; Hekking, L.H.; Mathisen, C.; Verkleij, D.; Goebbels, S.; Edgar, J.; Merkler, D.; Lyons, D.A.; et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 2014, 156, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Krämer, E.M.; Thiele, C.; Stoffel, W.; Trotter, J. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J. Cell Biol. 2000, 151, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Snaidero, N.; Velte, C.; Myllykoski, M.; Raasakka, A.; Ignatev, A.; Werner, H.B.; Erwig, M.S.; Möbius, W.; Kursula, P.; Nave, K.A.; et al. Antagonistic Functions of MBP and CNP Establish Cytosolic Channels in CNS Myelin. Cell Rep. 2017, 18, 314–323. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and Microglia as Major Players of Myelin Production in Normal and Pathological Conditions. Front. Cell NeuroSci. 2020, 14, 79. [Google Scholar] [CrossRef]
- Müller, E. Die multiple Sklerose des Gehirns und Rückenmarks: Ihre Pathologie und Behandlung. Pathol. Anat. Und. Pathog. 1904, 300–344. [Google Scholar]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Barnett, S.C.; Linington, C. Myelination: Do astrocytes play a role? Neuroscientist 2013, 19, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Lundgaard, I.; Osório, M.J.; Kress, B.T.; Sanggaard, S.; Nedergaard, M. White matter astrocytes in health and disease. Neuroscience 2014, 276, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are astrocytes important? NeuroChem. Res. 2015, 40, 389–401. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Li, B.; Scuderi, C. Astrocytes: The Housekeepers and Guardians of the CNS. Adv. Neurobiol. 2021, 26, 21–53. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef]
- Orthmann-Murphy, J.L.; Abrams, C.K.; Scherer, S.S. Gap junctions couple astrocytes and oligodendrocytes. J. Mol. NeuroSci. 2008, 35, 101–116. [Google Scholar] [CrossRef]
- Nutma, E.; van Gent, D.; Amor, S.; Peferoen, L.A.N. Astrocyte and Oligodendrocyte Cross-Talk in the Central Nervous System. Cells 2020, 9, 600. [Google Scholar] [CrossRef]
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Connexin29 and connexin32 at oligodendrocyte and astrocyte gap junctions and in myelin of the mouse central nervous system. J. Comp. Neurol. 2003, 464, 356–370. [Google Scholar] [CrossRef]
- Markoullis, K.; Sargiannidou, I.; Gardner, C.; Hadjisavvas, A.; Reynolds, R.; Kleopa, K.A. Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 2012, 60, 1053–1066. [Google Scholar] [CrossRef]
- Richardson, W.D.; Pringle, N.; Mosley, M.J.; Westermark, B.; Dubois-Dalcq, M. A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell 1988, 53, 309–319. [Google Scholar] [CrossRef]
- McKinnon, R.D.; Waldron, S.; Kiel, M.E. PDGF alpha-receptor signal strength controls an RTK rheostat that integrates phosphoinositol 3’-kinase and phospholipase Cgamma pathways during oligodendrocyte maturation. J. NeuroSci. 2005, 25, 3499–3508. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.S.; Milner, R.; Nishiyama, A.; Frausto, R.F.; Serwanski, D.R.; Pagarigan, R.R.; Whitton, J.L.; Miller, R.H.; Crocker, S.J. Astrocytic tissue inhibitor of metalloproteinase-1 (TIMP-1) promotes oligodendrocyte differentiation and enhances CNS myelination. J. NeuroSci. 2011, 31, 6247–6254. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.J.; Agranoff, B.W.; Albers, R.W.; Fisher, S.K.; Uhler, M.D. Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Lippincott-Raven: New York, NY, USA, 1999. [Google Scholar]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Bronzuoli, M.R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte Function Is Affected by Aging and Not Alzheimer’s Disease: A Preliminary Investigation in Hippocampi of 3xTg-AD Mice. Front. Pharm. 2019, 10, 644. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Steardo, L., Jr.; Steardo, L.; Verkhratsky, A.; Scuderi, C. Systemic Inflammation and Astrocyte Reactivity in the Neuropsychiatric Sequelae of COVID-19: Focus on Autism Spectrum Disorders. Front. Cell NeuroSci. 2021, 15, 748136. [Google Scholar] [CrossRef]
- Steardo, L., Jr.; Steardo, L.; Verkhratsky, A.; Scuderi, C. Post-COVID-19 neuropsychiatric syndrome: Is maladaptive glial recovery to blame? Acta Physiol. 2021, 233, e13717. [Google Scholar] [CrossRef]
- Scuderi, C.; Verkhratsky, A.; Parpura, V.; Li, B. Neuroglia in Psychiatric Disorders. Adv. Neurobiol. 2021, 26, 3–19. [Google Scholar] [CrossRef]
- Messersmith, D.J.; Murtie, J.C.; Le, T.Q.; Frost, E.E.; Armstrong, R.C. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J. NeuroSci. Res. 2000, 62, 241–256. [Google Scholar] [CrossRef]
- Albrecht, P.J.; Dahl, J.P.; Stoltzfus, O.K.; Levenson, R.; Levison, S.W. Ciliary neurotrophic factor activates spinal cord astrocytes, stimulating their production and release of fibroblast growth factor-2, to increase motor neuron survival. Exp. Neurol. 2002, 173, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Raine, C.S.; Cannella, B.; Brosnan, C.F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Investig. 1991, 87, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Cammer, W.; Zhang, H. Maturation of oligodendrocytes is more sensitive to TNF alpha than is survival of precursors and immature oligodendrocytes. J. Neuroimmunol. 1999, 97, 37–42. [Google Scholar] [CrossRef]
- Ding, X.; Yan, Y.; Li, X.; Li, K.; Ciric, B.; Yang, J.; Zhang, Y.; Wu, S.; Xu, H.; Chen, W.; et al. Silencing IFN-γ binding/signaling in astrocytes versus microglia leads to opposite effects on central nervous system autoimmunity. J. Immunol. 2015, 194, 4251–4264. [Google Scholar] [CrossRef]
- De Waard, D.M.; Bugiani, M. Astrocyte-Oligodendrocyte-Microglia Crosstalk in Astrocytopathies. Front. Cell NeuroSci. 2020, 14, 608073. [Google Scholar] [CrossRef]
- Dooves, S.; Bugiani, M.; Postma, N.L.; Polder, E.; Land, N.; Horan, S.T.; van Deijk, A.L.; van de Kreeke, A.; Jacobs, G.; Vuong, C.; et al. Astrocytes are central in the pathomechanisms of vanishing white matter. J. Clin. Investig. 2016, 126, 1512–1524. [Google Scholar] [CrossRef]
- Li, L.; Tian, E.; Chen, X.; Chao, J.; Klein, J.; Qu, Q.; Sun, G.; Sun, G.; Huang, Y.; Warden, C.D.; et al. GFAP Mutations in Astrocytes Impair Oligodendrocyte Progenitor Proliferation and Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell 2018, 23, 239–251.e6. [Google Scholar] [CrossRef]
- Love, S. Demyelinating diseases. J. Clin. Pathol. 2006, 59, 1151–1159. [Google Scholar] [CrossRef]
- Touma, L.; Muccilli, A. Diagnosis and Management of Central Nervous System Demyelinating Disorders. Neurol. Clin. 2022, 40, 113–131. [Google Scholar] [CrossRef]
- Henríquez, K.; Molt, F.; Gajardo, J.; Cortés, B.; Ramirez-Santana, M. Sociodemographic and clinical characteristics of people with multiple sclerosis and neuro-myelitis optica spectrum disorder in a central northern region of Chile: A prevalence study. Mult. Scler. Relat. Disord. 2022, 61, 103750. [Google Scholar] [CrossRef]
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.L.M.; Fischer, R. Inflammation and Oxidative Stress in Multiple Sclerosis: Consequences for Therapy Development. Oxid. Med. Cell Longev. 2020, 2020, 7191080. [Google Scholar] [CrossRef] [PubMed]
- Orefice, N.S.; Alhouayek, M.; Carotenuto, A.; Montella, S.; Barbato, F.; Comelli, A.; Calignano, A.; Muccioli, G.G.; Orefice, G. Oral Palmitoylethanolamide Treatment Is Associated with Reduced Cutaneous Adverse Effects of Interferon-β1a and Circulating Proinflammatory Cytokines in Relapsing-Remitting Multiple Sclerosis. Neurotherapeutics 2016, 13, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Barbierato, M.; Borri, M.; Facci, L.; Zusso, M.; Skaper, S.D.; Giusti, P. Expression and Differential Responsiveness of Central Nervous System Glial Cell Populations to the Acute Phase Protein Serum Amyloid A. Sci. Rep. 2017, 7, 12158. [Google Scholar] [CrossRef] [PubMed]
- Ristori, G.; Laurenti, F.; Stacchini, P.; Gasperini, C.; Buttinelli, C.; Pozzilli, C.; Salvetti, M. Serum amyloid A protein is elevated in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 1998, 88, 9–12. [Google Scholar] [CrossRef]
- Chung, T.F.; Sipe, J.D.; McKee, A.; Fine, R.E.; Schreiber, B.M.; Liang, J.S.; Johnson, R.J. Serum amyloid A in Alzheimer’s disease brain is predominantly localized to myelin sheaths and axonal membrane. Amyloid 2000, 7, 105–110. [Google Scholar] [CrossRef]
- Barbierato, M.; Facci, L.; Marinelli, C.; Zusso, M.; Argentini, C.; Skaper, S.D.; Giusti, P. Co-ultramicronized Palmitoylethanolamide/Luteolin Promotes the Maturation of Oligodendrocyte Precursor Cells. Sci. Rep. 2015, 5, 16676. [Google Scholar] [CrossRef]
- Skaper, S.D.; Barbierato, M.; Facci, L.; Borri, M.; Contarini, G.; Zusso, M.; Giusti, P. Co-Ultramicronized Palmitoylethanolamide/Luteolin Facilitates the Development of Differentiating and Undifferentiated Rat Oligodendrocyte Progenitor Cells. Mol. Neurobiol. 2018, 55, 103–114. [Google Scholar] [CrossRef]
- Facci, L.; Barbierato, M.; Fusco, M.; Giusti, P.; Zusso, M. Co-Ultramicronized Palmitoylethanolamide/Luteolin-Induced Oligodendrocyte Precursor Cell Differentiation is Associated With Tyro3 Receptor Upregulation. Front. Pharm. 2021, 12, 698133. [Google Scholar] [CrossRef]
- Cassano, T.; Magini, A.; Giovagnoli, S.; Polchi, A.; Calcagnini, S.; Pace, L.; Lavecchia, M.A.; Scuderi, C.; Bronzuoli, M.R.; Ruggeri, L.; et al. Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer’s disease. Exp. Neurol. 2019, 311, 88–105. [Google Scholar] [CrossRef]
- Contarini, G.; Franceschini, D.; Facci, L.; Barbierato, M.; Giusti, P.; Zusso, M. A co-ultramicronized palmitoylethanolamide/luteolin composite mitigates clinical score and disease-relevant molecular markers in a mouse model of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2019, 16, 126. [Google Scholar] [CrossRef]
- Rahimi, A.; Faizi, M.; Talebi, F.; Noorbakhsh, F.; Kahrizi, F.; Naderi, N. Interaction between the protective effects of cannabidiol and palmitoylethanolamide in experimental model of multiple sclerosis in C57BL/6 mice. Neuroscience 2015, 290, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; D’Agostino, G.; Pacini, A.; Russo, R.; Zanardelli, M.; Ghelardini, C.; Calignano, A. Palmitoylethanolamide is a disease-modifying agent in peripheral neuropathy: Pain relief and neuroprotection share a PPAR-alpha-mediated mechanism. Mediat. Inflamm. 2013, 2013, 328797. [Google Scholar] [CrossRef] [PubMed]
- González-Hernández, A.; Martínez-Lorenzana, G.; Rodríguez-Jiménez, J.; Rojas-Piloni, G.; Condés-Lara, M. Intracisternal injection of palmitoylethanolamide inhibits the peripheral nociceptive evoked responses of dorsal horn wide dynamic range neurons. J. Neural. Transm. 2015, 122, 369–374. [Google Scholar] [CrossRef]
- Truini, A.; Biasiotta, A.; Di Stefano, G.; La Cesa, S.; Leone, C.; Cartoni, C.; Federico, V.; Petrucci, M.T.; Cruccu, G. Palmitoylethanolamide restores myelinated-fibre function in patients with chemotherapy-induced painful neuropathy. CNS Neurol. Disord. Drug Targets 2011, 10, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Peters, A. Structural changes that occur during normal aging of primate cerebral hemispheres. NeuroSci. Biobehav. Rev. 2002, 26, 733–741. [Google Scholar] [CrossRef]
- Peters, A. The structure of myelin sheaths in the central nervous system of Xenopus laevis (Daudin). J. Biophys. BioChem. Cytol. 1960, 7, 121–126. [Google Scholar] [CrossRef]
- Sowell, E.R.; Peterson, B.S.; Thompson, P.M.; Welcome, S.E.; Henkenius, A.L.; Toga, A.W. Mapping cortical change across the human life span. Nat. NeuroSci. 2003, 6, 309–315. [Google Scholar] [CrossRef]
- Feldman, M.L.; Peters, A. Ballooning of myelin sheaths in normally aged macaques. J. Neurocytol. 1998, 27, 605–614. [Google Scholar] [CrossRef]
- Deber, C.M.; Reynolds, S.J. Central nervous system myelin: Structure, function, and pathology. Clin. BioChem. 1991, 24, 113–134. [Google Scholar] [CrossRef]
- Kormas, P.; Moutzouri, A. Current Psychological Approaches in Neurodegenerative Diseases. In Handbook of Computational Neurodegeneration; Vlamos, P., Kotsireas, I.S., Tarnanas, I., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 1–29. [Google Scholar]
- Sohn, Y.H.; Kim, J.S. The influence of white matter hyperintensities on the clinical features of Parkinson’s disease. Yonsei. Med. J. 1998, 39, 50–55. [Google Scholar] [CrossRef]
- Piccini, P.; Pavese, N.; Canapicchi, R.; Paoli, C.; Del Dotto, P.; Puglioli, M.; Rossi, G.; Bonuccelli, U. White matter hyperintensities in Parkinson’s disease. Clinical correlations. Arch. Neurol. 1995, 52, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, J.S.; Lee, K.S.; An, J.Y.; Kim, W.; Kim, Y.I.; Kim, B.S.; Jung, S.L. The severity of leukoaraiosis correlates with the clinical phenotype of Parkinson’s disease. Arch. Gerontol. Geriatr. 2009, 49, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.A.; Evidente, V.G.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; Connor, D.J.; Hentz, J.G.; Adler, C.H.; Beach, T.G. Are there differences in cerebral white matter lesion burdens between Parkinson’s disease patients with or without dementia? Acta Neuropathol. 2010, 119, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.K.; Aarsland, D.; Greve, O.J.; Larsen, J.P. Visual rating of white matter hyperintensities in Parkinson’s disease. Mov. Disord. 2006, 21, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [Google Scholar] [CrossRef]
- Dean, D.C., 3rd; Hurley, S.A.; Kecskemeti, S.R.; O’Grady, J.P.; Canda, C.; Davenport-Sis, N.J.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Asthana, S.; et al. Association of Amyloid Pathology With Myelin Alteration in Preclinical Alzheimer Disease. JAMA Neurol. 2017, 74, 41–49. [Google Scholar] [CrossRef]
- Benitez, A.; Fieremans, E.; Jensen, J.H.; Falangola, M.F.; Tabesh, A.; Ferris, S.H.; Helpern, J.A. White matter tract integrity metrics reflect the vulnerability of late-myelinating tracts in Alzheimer’s disease. Neuroimage Clin. 2014, 4, 64–71. [Google Scholar] [CrossRef]
- Zhan, X.; Jickling, G.C.; Ander, B.P.; Stamova, B.; Liu, D.; Kao, P.F.; Zelin, M.A.; Jin, L.W.; DeCarli, C.; Sharp, F.R. Myelin basic protein associates with AβPP, Aβ1-42, and amyloid plaques in cortex of Alzheimer’s disease brain. J. Alzheimers Dis. 2015, 44, 1213–1229. [Google Scholar] [CrossRef]
- Wang, S.S.; Zhang, Z.; Zhu, T.B.; Chu, S.F.; He, W.B.; Chen, N.H. Myelin injury in the central nervous system and Alzheimer’s disease. Brain Res. Bull. 2018, 140, 162–168. [Google Scholar] [CrossRef]
- Valenza, M.; Scuderi, C. How useful are biomarkers for the diagnosis of Alzheimer’s disease and especially for its therapy? Neural Regen. Res. 2022, 17, 2205–2207. [Google Scholar] [CrossRef]
- Roher, A.E.; Weiss, N.; Kokjohn, T.A.; Kuo, Y.M.; Kalback, W.; Anthony, J.; Watson, D.; Luehrs, D.C.; Sue, L.; Walker, D.; et al. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry 2002, 41, 11080–11090. [Google Scholar] [CrossRef] [PubMed]
- Quintela-López, T.; Ortiz-Sanz, C.; Serrano-Regal, M.P.; Gaminde-Blasco, A.; Valero, J.; Baleriola, J.; Sánchez-Gómez, M.V.; Matute, C.; Alberdi, E. Aβ oligomers promote oligodendrocyte differentiation and maturation via integrin β1 and Fyn kinase signaling. Cell Death Dis. 2019, 10, 445. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, Y.; Liu, Z.; Geng, Q.; Chen, Z.; Zhang, Y. Alterations of myelin morphology and oligodendrocyte development in early stage of Alzheimer’s disease mouse model. NeuroSci. Lett. 2017, 642, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, R.; Valenza, M.; Gomiero, C.; Mancini, G.F.; Steardo, L.; Campolongo, P.; Scuderi, C. Co-Ultramicronized Palmitoylethanolamide/Luteolin Restores Oligodendrocyte Homeostasis via Peroxisome Proliferator-Activated Receptor-α in an In Vitro Model of Alzheimer’s Disease. Biomedicines 2022, 10, 1236. [Google Scholar] [CrossRef] [PubMed]
- Vanzulli, I.; Papanikolaou, M.; De-La-Rocha, I.C.; Pieropan, F.; Rivera, A.D.; Gomez-Nicola, D.; Verkhratsky, A.; Rodríguez, J.J.; Butt, A.M. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 94, 130–139. [Google Scholar] [CrossRef]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, a020453. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—from mechanisms to experimental medicines. Nat. Rev. NeuroSci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Back, S.A.; Tuohy, T.M.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.L.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar] [CrossRef]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-Derived Endothelin-1 Inhibits Remyelination through Notch Activation. Neuron 2014, 81, 1442. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Muramatsu, R.; Fujimura, H.; Mochizuki, H.; Kataoka, H.; Yamashita, T. Circulating transforming growth factor-β1 facilitates remyelination in the adult central nervous system. Elife 2019, 8, 41869. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Rothhammer, V. Protective Functions of Reactive Astrocytes Following Central Nervous System Insult. Front. Immunol. 2020, 11, 573256. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, R.; Valenza, M.; Bronzuoli, M.R.; Menegoni, G.; Ratano, P.; Steardo, L.; Campolongo, P.; Scuderi, C. Looking for a Treatment for the Early Stage of Alzheimer’s Disease: Preclinical Evidence with Co-Ultramicronized Palmitoylethanolamide and Luteolin. Int. J. Mol. Sci. 2020, 21, 3802. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, M.; Esposito, G.; Negro, L.; Capoccia, E.; Sarnelli, G.; Scuderi, C.; De Filippis, D.; Steardo, L.; Iuvone, T. Palmitoylethanolamide Regulates Production of Pro-Angiogenic Mediators in a Model of β Amyloid-Induced Astrogliosis In Vitro. CNS Neurol. Disord. Drug Targets 2015, 14, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L., Jr.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxidative Med. Cell. Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef]
- Manni, B.; Federzoni, L.; Zucchi, P.; Fabbo, A. Co-ultraPEALut Effect on Mild Cognitive Impairment: A Retrospective Observational Study. Acta Sci. Neurol. 2021, 4, 8–14. [Google Scholar] [CrossRef]
- Calabrò, R.S.; Naro, A.; De Luca, R.; Leonardi, S.; Russo, M.; Marra, A.; Bramanti, P. PEALut efficacy in mild cognitive impairment: Evidence from a SPECT case study! Aging Clin. Exp. Res. 2016, 28, 1279–1282. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenza, M.; Facchinetti, R.; Steardo, L.; Scuderi, C. Palmitoylethanolamide and White Matter Lesions: Evidence for Therapeutic Implications. Biomolecules 2022, 12, 1191. https://doi.org/10.3390/biom12091191
Valenza M, Facchinetti R, Steardo L, Scuderi C. Palmitoylethanolamide and White Matter Lesions: Evidence for Therapeutic Implications. Biomolecules. 2022; 12(9):1191. https://doi.org/10.3390/biom12091191
Chicago/Turabian StyleValenza, Marta, Roberta Facchinetti, Luca Steardo, and Caterina Scuderi. 2022. "Palmitoylethanolamide and White Matter Lesions: Evidence for Therapeutic Implications" Biomolecules 12, no. 9: 1191. https://doi.org/10.3390/biom12091191