
Synaptic Effects of Palmitoylethanolamide in Neurodegenerative Disorders


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
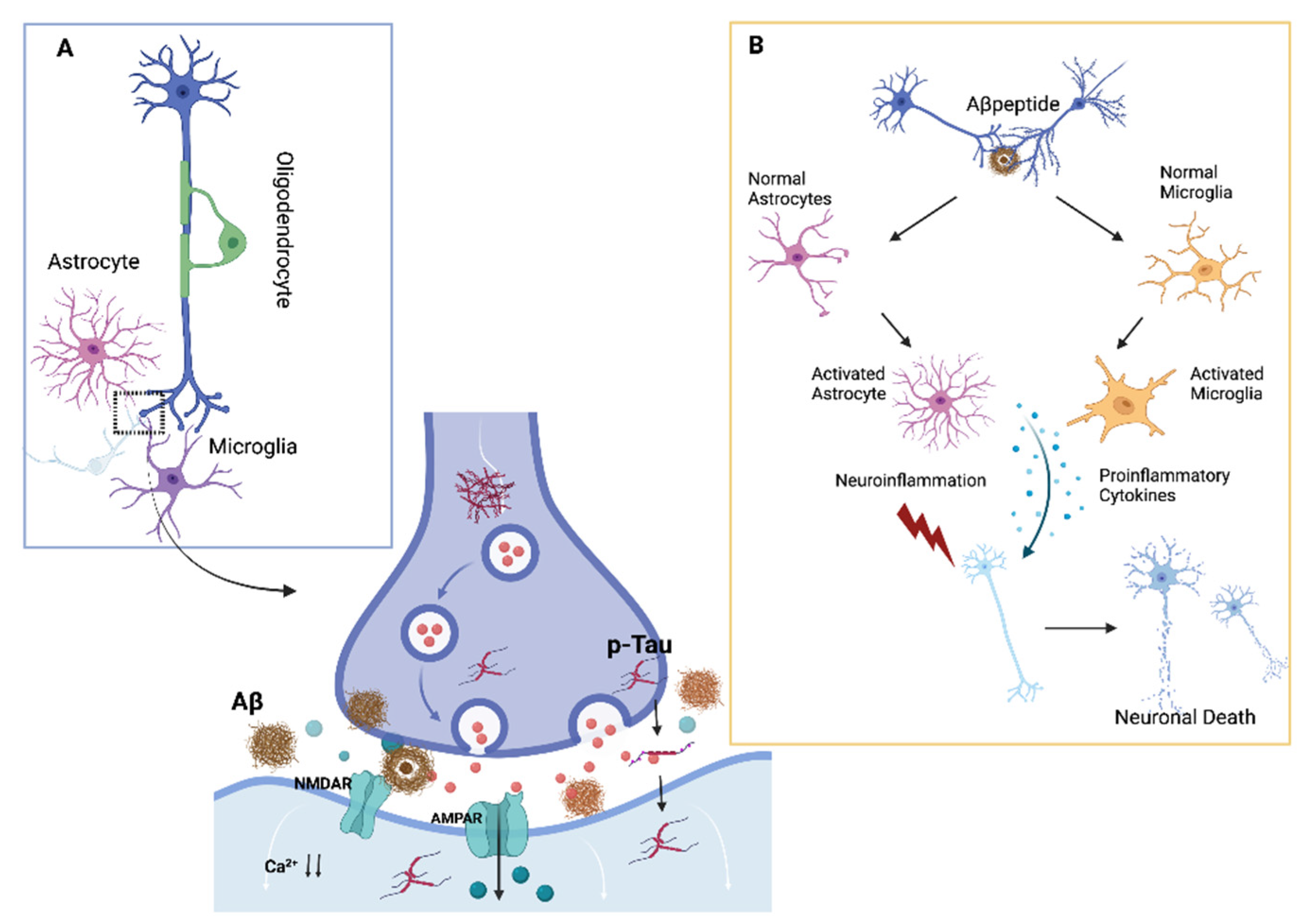
1.1. Synaptic Impairment and Neuroinflammation in Neurodegenerative Disorders
1.1.1. Alzheimer’s Disease
Synaptic Impairment
Neuroinflammation in AD
1.1.2. Frontotemporal Lobar Degeneration
Synaptic Impairment in FTLD
Neuroinflammation in FTLD
1.1.3. Amyotrophic Lateral Sclerosis
Synaptic Impairment in ALS
Neuroinflammation ALS
2. PEA and PEA Combined with Luteolin Mechanisms of Action
2.1. PEA Synaptic Mechanisms of Action
2.2. PEA Combined with Luteolin Effects
3. Synaptic Effects of PEA and PEALut in Neurodegenerative Disorders
3.1. Preclinical Models
3.2. Clinical Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cova, I.; Markova, A.; Campini, I.; Grande, G.; Mariani, C.; Pomati, S. Worldwide Trends in the Prevalence of Dementia. J. Neurol. Sci. 2017, 379, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Basic Mechanisms of Neurodegeneration: A Critical Update. J. Cell Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhou, X.-W.; Wang, J.-Z. The Dual Roles of Cytokines in Alzheimer’s Disease: Update on Interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheff, S.W.; Price, D.A. Synaptic Pathology in Alzheimer’s Disease: A Review of Ultrastructural Studies. Neurobiol. Aging 2003, 24, 1029–1046. [Google Scholar] [CrossRef]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley-Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Abeta Accumulation and Progressive Synaptic Loss, Gliosis, and Tangle Formation in AD Brain. Neurology 2004, 62, 925–931. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Martorana, A.; Di Lorenzo, F.; Belli, L.; Sancesario, G.; Toniolo, S.; Sallustio, F.; Sancesario, G.M.; Koch, G. Cerebrospinal Fluid Aβ 42 Levels: When Physiological Become Pathological State. CNS Neurosci. Ther. 2015, 21, 921–925. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau Oligomers Impair Memory and Induce Synaptic and Mitochondrial Dysfunction in Wild-Type Mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Llorens, F.; Schmitz, M.; Karch, A.; Cramm, M.; Lange, P.; Gherib, K.; Varges, D.; Schmidt, C.; Zerr, I.; Stoeck, K. Comparative Analysis of Cerebrospinal Fluid Biomarkers in the Differential Diagnosis of Neurodegenerative Dementia. Alzheimers Dement. 2016, 12, 577–589. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
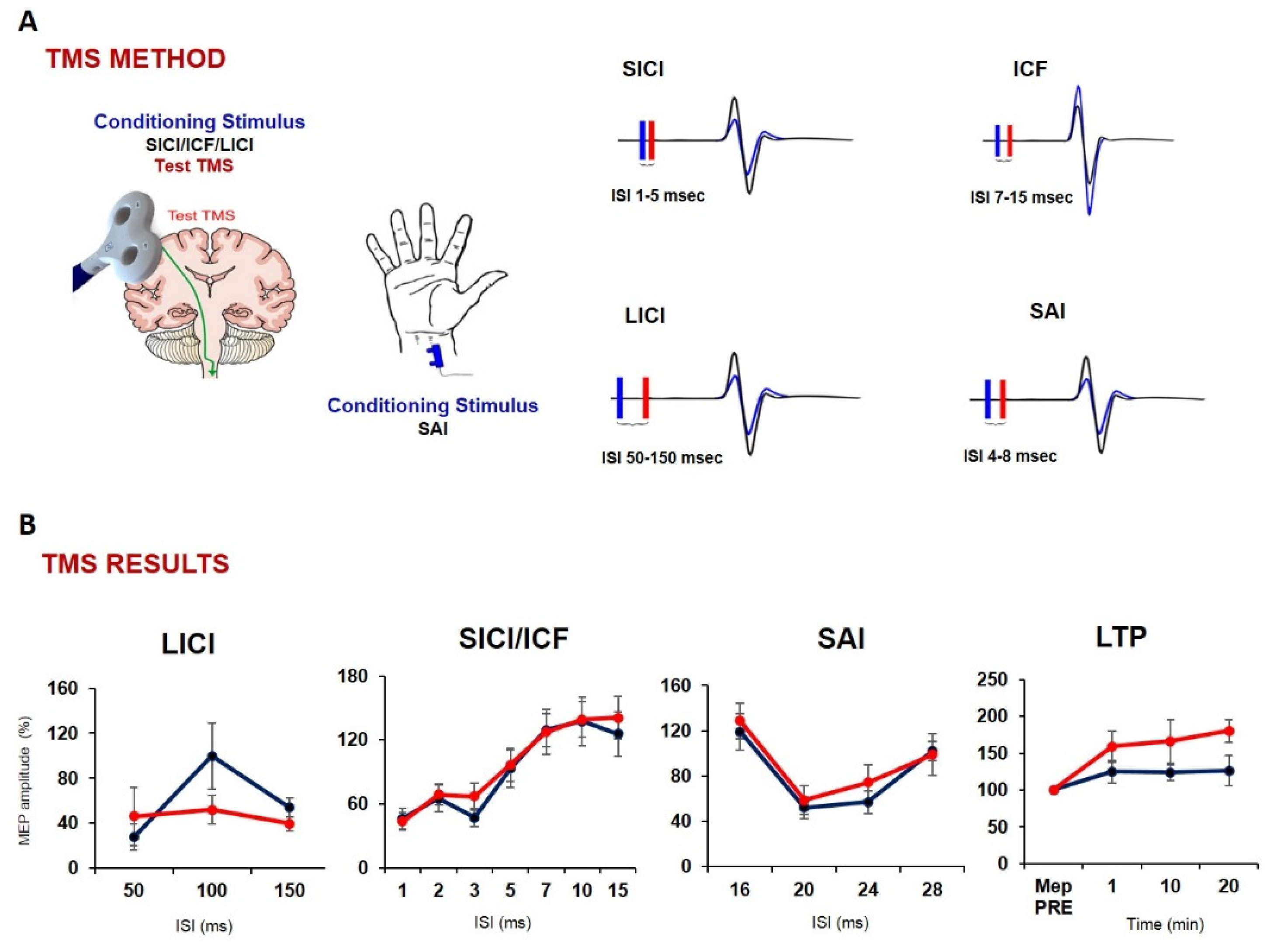
- Benussi, A.; Grassi, M.; Palluzzi, F.; Koch, G.; Di Lazzaro, V.; Nardone, R.; Cantoni, V.; Dell’Era, V.; Premi, E.; Martorana, A.; et al. Classification Accuracy of Transcranial Magnetic Stimulation for the Diagnosis of Neurodegenerative Dementias. Ann. Neurol. 2020, 87, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, F.; Motta, C.; Casula, E.P.; Bonnì, S.; Assogna, M.; Caltagirone, C.; Martorana, A.; Koch, G. LTP-like Cortical Plasticity Predicts Conversion to Dementia in Patients with Memory Impairment. Brain Stimul. 2020, 13, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A Synaptic Model of Memory: Long-Term Potentiation in the Hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Koch, G.; Esposito, Z.; Kusayanagi, H.; Monteleone, F.; Codecá, C.; Di Lorenzo, F.; Caltagirone, C.; Bernardi, G.; Martorana, A. CSF Tau Levels Influence Cortical Plasticity in Alzheimer’s Disease Patients. JAD 2011, 26, 181–186. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Ponzo, V.; Motta, C.; Bonnì, S.; Picazio, S.; Caltagirone, C.; Bozzali, M.; Martorana, A.; Koch, G. Impaired Spike Timing Dependent Cortico-Cortical Plasticity in Alzheimer’s Disease Patients. J. Alzheimers Dis. 2018, 66, 983–991. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Martorana, A.; Ponzo, V.; Bonnì, S.; D’Angelo, E.; Caltagirone, C.; Koch, G. Cerebellar Theta Burst Stimulation Modulates Short Latency Afferent Inhibition in Alzheimer’s Disease Patients. Front. Aging Neurosci. 2013, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Martorana, A.; Di Lorenzo, F.; Manenti, G.; Semprini, R.; Koch, G. Homotaurine Induces Measurable Changes of Short Latency Afferent Inhibition in a Group of Mild Cognitive Impairment Individuals. Front. Aging Neurosci. 2014, 6, 254. [Google Scholar] [CrossRef]
- Arroyo-García, L.E.; Isla, A.G.; Andrade-Talavera, Y.; Balleza-Tapia, H.; Loera-Valencia, R.; Alvarez-Jimenez, L.; Pizzirusso, G.; Tambaro, S.; Nilsson, P.; Fisahn, A. Impaired Spike-Gamma Coupling of Area CA3 Fast-Spiking Interneurons as the Earliest Functional Impairment in the AppNL-G-F Mouse Model of Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5557–5567. [Google Scholar] [CrossRef]
- Nakazono, T.; Lam, T.N.; Patel, A.Y.; Kitazawa, M.; Saito, T.; Saido, T.C.; Igarashi, K.M. Impaired In Vivo Gamma Oscillations in the Medial Entorhinal Cortex of Knock-in Alzheimer Model. Front. Syst. Neurosci. 2017, 11, 48. [Google Scholar] [CrossRef]
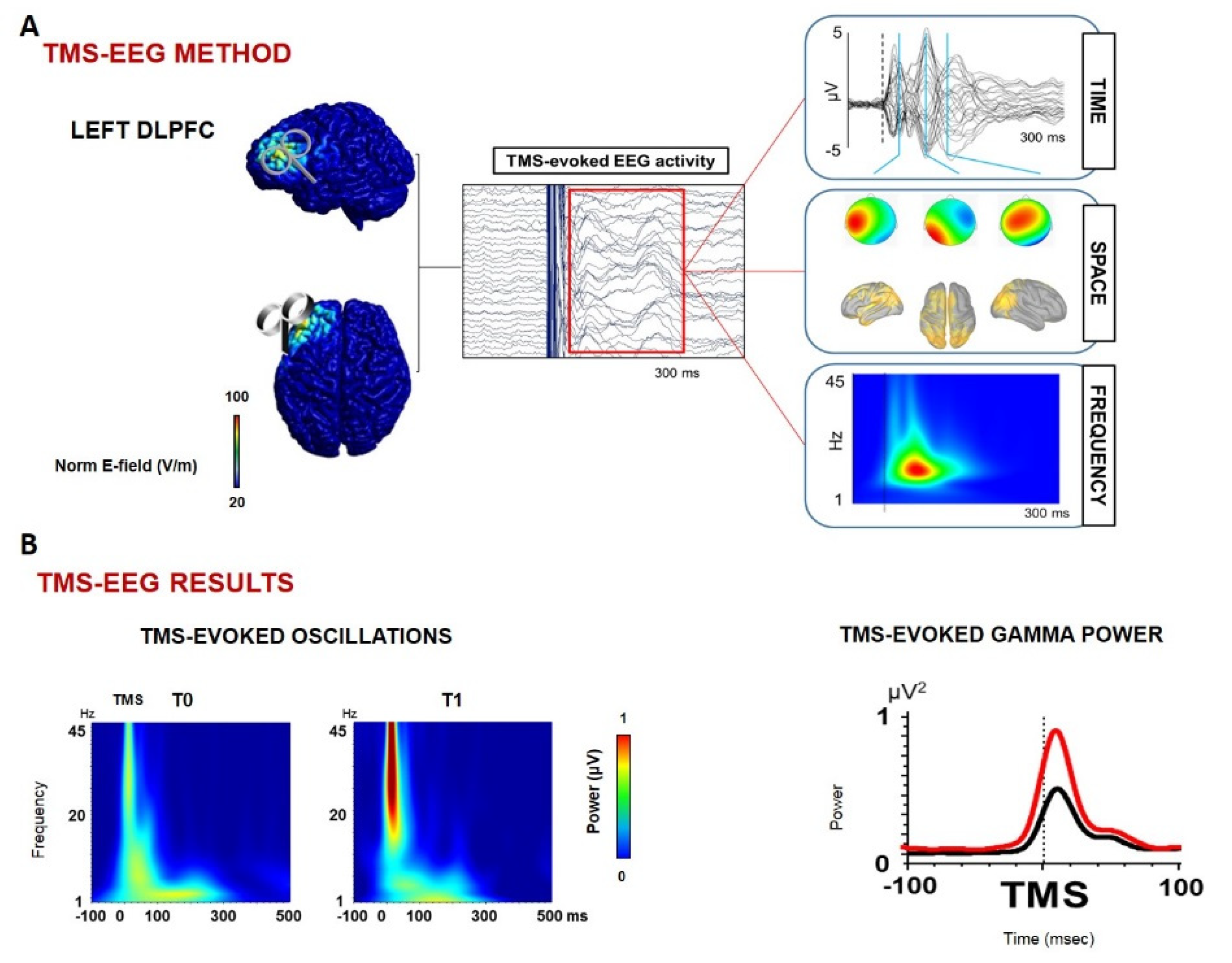
- Casula, E.P.; Pellicciari, M.C.; Bonnì, S.; Borghi, I.; Maiella, M.; Assogna, M.; Minei, M.; Motta, C.; D’Acunto, A.; Porrazzini, F.; et al. Decreased Frontal Gamma Activity in Alzheimer Disease Patients. Ann. Neurol. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma Frequency Entrainment Attenuates Amyloid Load and Modifies Microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Emre, C.; Arroyo-García, L.E.; Do, K.V.; Jun, B.; Ohshima, M.; Alcalde, S.G.; Cothern, M.L.; Maioli, S.; Nilsson, P.; Hjorth, E.; et al. Intranasal Delivery of Pro-Resolving Lipid Mediators Rescues Memory and Gamma Oscillation Impairment in AppNL-G-F/NL-G-F Mice. Commun. Biol. 2022, 5, 245. [Google Scholar] [CrossRef] [PubMed]
- Udeochu, J.C.; Shea, J.M.; Villeda, S.A. Microglia Communication: Parallels between Aging and Alzheimer’s Disease. Clin. Exp. Neuroimmunol. 2016, 7, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory Cytokines Induce Amyloid Beta Neurotoxicity through Modulating Amyloid Precursor Protein Levels/Metabolism. Biomed. Res. Int. 2018, 2018, 3087475. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Di Lorenzo, F.; Loizzo, S.; Motta, C.; Travaglione, S.; Baiula, M.; Rimondini, R.; Ponzo, V.; Bonnì, S.; Toniolo, S.; et al. CSF Tau Is Associated with Impaired Cortical Plasticity, Cognitive Decline and Astrocyte Survival Only in APOE4-Positive Alzheimer’s Disease. Sci. Rep. 2017, 7, 13728. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-Beta-Induced Neuronal Dysfunction in Alzheimer’s Disease: From Synapses toward Neural Networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef]
- Klyubin, I.; Cullen, W.K.; Hu, N.-W.; Rowan, M.J. Alzheimer’s Disease Aβ Assemblies Mediating Rapid Disruption of Synaptic Plasticity and Memory. Mol. Brain 2012, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Motta, C.; Finardi, A.; Toniolo, S.; Di Lorenzo, F.; Scaricamazza, E.; Loizzo, S.; Mercuri, N.B.; Furlan, R.; Koch, G.; Martorana, A. Protective Role of Cerebrospinal Fluid Inflammatory Cytokines in Patients with Amnestic Mild Cognitive Impairment and Early Alzheimer’s Disease Carrying Apolipoprotein E4 Genotype. J. Alzheimers Dis. 2020, 76, 681–689. [Google Scholar] [CrossRef]
- Clare, R.; King, V.G.; Wirenfeldt, M.; Vinters, H.V. Synapse Loss in Dementias. J. Neurosci. Res. 2010, 88, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- van der Ende, E.L.; Xiao, M.; Xu, D.; Poos, J.M.; Panman, J.L.; Jiskoot, L.C.; Meeter, L.H.; Dopper, E.G.; Papma, J.M.; Heller, C.; et al. Neuronal Pentraxin 2: A Synapse-Derived CSF Biomarker in Genetic Frontotemporal Dementia. J. Neurol. Neurosurg. Psychiatry 2020, 91, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, A.M.; Cullum, C.M.; Satumtira, S.; Sontag, E.; Hynan, L.S.; White, C.L.; Bigio, E.H. Contribution of Asymmetric Synapse Loss to Lateralizing Clinical Deficits in Frontotemporal Dementias. Arch. Neurol. 2001, 58, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I. Neurons and Their Dendrites in Frontotemporal Dementia. Dement. Geriatr. Cogn. Disord. 1999, 10, 55–60. [Google Scholar] [CrossRef]
- Liu, X.; Erikson, C.; Brun, A. Cortical Synaptic Changes and Gliosis in Normal Aging, Alzheimer’s Disease and Frontal Lobe Degeneration. Dementia 1996, 7, 128–134. [Google Scholar] [CrossRef]
- Connelly, S.J.; Mukaetova-Ladinska, E.B.; Abdul-All, Z.; Alves da Silva, J.; Brayne, C.; Honer, W.G.; Mann, D.M.A. Synaptic Changes in Frontotemporal Lobar Degeneration: Correlation with MAPT Haplotype and APOE Genotype. Neuropathol. Appl. Neurobiol. 2011, 37, 366–380. [Google Scholar] [CrossRef]
- Lippa, C.F. Synaptophysin Immunoreactivity in Pick’s Disease: Comparison with Alzheimer’s Disease and Dementia with Lewy Bodies. Am. J. Alzheimers Dis. Other Dement. 2004, 19, 341–344. [Google Scholar] [CrossRef]
- Salmon, E.; Bahri, M.A.; Plenevaux, A.; Becker, G.; Seret, A.; Delhaye, E.; Degueldre, C.; Balteau, E.; Lemaire, C.; Luxen, A.; et al. In Vivo Exploration of Synaptic Projecti.ions in Frontotemporal Dementia. Sci. Rep. 2021, 11, 16092. [Google Scholar] [CrossRef]
- Benussi, A.; Cosseddu, M.; Filareto, I.; Dell’Era, V.; Archetti, S.; Sofia Cotelli, M.; Micheli, A.; Padovani, A.; Borroni, B. Impaired Long-Term Potentiation-like Cortical Plasticity in Presymptomatic Genetic Frontotemporal Dementia. Ann. Neurol. 2016, 80, 472–476. [Google Scholar] [CrossRef]
- Broce, I.; Karch, C.M.; Wen, N.; Fan, C.C.; Wang, Y.; Tan, C.H.; Kouri, N.; Ross, O.A.; Höglinger, G.U.; Muller, U.; et al. Immune-Related Genetic Enrichment in Frontotemporal Dementia: An Analysis of Genome-Wide Association Studies. PLoS Med. 2018, 15, e1002487. [Google Scholar] [CrossRef]
- Miller, Z.A.; Rankin, K.P.; Graff-Radford, N.R.; Takada, L.T.; Sturm, V.E.; Cleveland, C.M.; Criswell, L.A.; Jaeger, P.A.; Stan, T.; Heggeli, K.A.; et al. TDP-43 Frontotemporal Lobar Degeneration and Autoimmune Disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 956–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, Z.A.; Sturm, V.E.; Camsari, G.B.; Karydas, A.; Yokoyama, J.S.; Grinberg, L.T.; Boxer, A.L.; Rosen, H.J.; Rankin, K.P.; Gorno-Tempini, M.L.; et al. Increased Prevalence of Autoimmune Disease within C9 and FTD/MND Cohorts. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualetti, G.; Brooks, D.J.; Edison, P. The Role of Neuroinflammation in Dementias. Curr. Neurol. Neurosci. Rep. 2015, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S.T. Common Inflammatory Me.echanisms in Lewy Body Disease and Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2007, 66, 683–686. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Ono, K.; Suzuki, H.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Cytokine Production of Activated Microglia and Decrease in Neurotrophic Factors of Neurons in the Hippocampus of Lewy Body Disease Brains. Acta Neuropathol. 2005, 109, 141–150. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in Neurodegenerative Disease—A Double-Edged Sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate Immune Activation in Neurodegenerative Disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.L.; Arias, N. Synaptopathy Mechanisms in ALS Caused by C9orf72 Repeat Expansion. Front. Cell. Neurosci. 2021, 15, 660693. [Google Scholar] [CrossRef]
- Fogarty, M.J. Amyotrophic Lateral Sclerosis as a Synaptopathy. Neural Regen Res. 2019, 14, 189–192. [Google Scholar] [CrossRef]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic Lateral Sclerosis--a Model of Corticofugal Axonal Spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of PTDP-43 Pathology in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in Treating Amyotrophic Lateral Sclerosis: Insights from Pathophysiological Studies. Trends Neurosci. 2014, 37, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Mu, E.W.H.; Lavidis, N.A.; Noakes, P.G.; Bellingham, M.C. Motor Areas Show Altered Dendritic Structure in an Amyotrophic Lateral Sclerosis Mouse Model. Front. Neurosci. 2017, 11, 609. [Google Scholar] [CrossRef]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic Dysfunction in Neurological Disorders—A Review from Students to Students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef]
- Wishart, T.M.; Parson, S.H.; Gillingwater, T.H. Synaptic Vulnerability in Neurodegenerative Disease. J. Neuropathol. Exp. Neurol. 2006, 65, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Genç, B.; Jara, J.H.; Lagrimas, A.K.B.; Pytel, P.; Roos, R.P.; Mesulam, M.M.; Geula, C.; Bigio, E.H.; Özdinler, P.H. Apical Dendrite Degeneration, a Novel Cellular Pathology for Betz Cells in ALS. Sci. Rep. 2017, 7, 41765. [Google Scholar] [CrossRef] [Green Version]
- Jara, J.H.; Villa, S.R.; Khan, N.A.; Bohn, M.C.; Özdinler, P.H. AAV2 Mediated Retrograde Transduction of Corticospinal Motor Neurons Reveals Initial and Selective Apical Dendrite Degeneration in ALS. Neurobiol. Dis. 2012, 47, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hämäläinen, R.H.; Koistinaho, J. Astrocyte Alterations in Neurodegenerative Pathologies and Their Modeling in Human Induced Pluripotent Stem Cell Platforms. Cell. Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar] [CrossRef] [Green Version]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of Dendritic Cells, MCP-1, and Activated Microglia/Macrophages in Amyotrophic Lateral Sclerosis Spinal Cord Tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic Reactions in Amyotrophic Lateral Sclerosis Brain and Spinal Cord Tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of Widespread Cerebral Microglial Activation in Amyotrophic Lateral Sclerosis: An [11C](R)-PK11195 Positron Emission Tomography Study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Engler, H.; Blomquist, G.; Scott, B.; Wall, A.; Aquilonius, S.-M.; Långström, B.; Askmark, H. Evidence for Astrocytosis in ALS Demonstrated by [11C](L)-Deprenyl-D2 PET. J. Neurol. Sci. 2007, 255, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Green, C.; Altschuler, G.; Wei, W.; Bury, J.J.; Heath, P.R.; Wyles, M.; Gelsthorpe, C.; Highley, J.R.; Lorente-Pons, A.; et al. A Data-Driven Approach Links Microglia to Pathology and Prognosis in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2017, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Mattace Raso, G.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS Health and Disease. Pharmacol. Res. 2014, 86, 32–41. [Google Scholar] [CrossRef]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and Related N-Acylethanolamines: Biological Activities and Metabolism. Inflamm. Regener. 2018, 38, 28. [Google Scholar] [CrossRef]
- Petrosino, S.; Di Marzo, V. The Pharmacology of Palmitoylethanolamide and First Data on the Therapeutic Efficacy of Some of Its New Formulations: Palmitoylethanolamide and Its New Formulations. Br. J. Pharmacol. 2017, 174, 1349–1365. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and Endocannabinoid-Related Mediators: Targets, Metabolism and Role in Neurological Disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Pertwee, R.G. GPR55: A New Member of the Cannabinoid Receptor Clan? Commentary. Br. J. Pharmacol. 2009, 152, 984–986. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, G.; La Rana, G.; Russo, R.; Sasso, O.; Iacono, A.; Esposito, E.; Mattace Raso, G.; Cuzzocrea, S.; Loverme, J.; Piomelli, D.; et al. Central Administration of Palmitoylethanolamide Reduces Hyperalgesia in Mice via Inhibition of NF-KappaB Nuclear Signalling in Dorsal Root Ganglia. Eur. J. Pharmacol. 2009, 613, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Jones, D.C. Emerging Roles of PPARs in Inflammation and Immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo Jr, L.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide Counteracts Reactive Astrogliosis Induced by β-Amyloid Peptide. J. Cell Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide Protects Against the Amyloid-Β25-35-Induced Learning and Memory Impairment in Mice, an Experimental Model of Alzheimer Disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective Activities of Palmitoylethanolamide in an Animal Model of Parkinson’s Disease. PLoS ONE 2012, 7, e41880. [Google Scholar] [CrossRef] [Green Version]
- Paterniti, I.; Cordaro, M.; Campolo, M.; Siracusa, R.; Cornelius, C.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Neuroprotection by Association of Palmitoylethanolamide with Luteolin in Experimental Alzheimer’s Disease Models: The Control of Neuroinflammation. CNS Neurol. Disord. Drug Targets 2014, 13, 1530–1541. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 Is a Cannabinoid Receptor That Increases Intracellular Calcium and Inhibits M Current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a Naturally Occurring Lipid, Is an Orally Effective Intestinal Anti-Inflammatory Agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [Green Version]
- Celorrio, M.; Rojo-Bustamante, E.; Fernández-Suárez, D.; Sáez, E.; Estella-Hermoso de Mendoza, A.; Müller, C.E.; Ramírez, M.J.; Oyarzábal, J.; Franco, R.; Aymerich, M.S. GPR55: A Therapeutic Target for Parkinson’s Disease? Neuropharmacology 2017, 125, 319–332. [Google Scholar] [CrossRef]
- Rinne, P.; Guillamat-Prats, R.; Rami, M.; Bindila, L.; Ring, L.; Lyytikäinen, L.-P.; Raitoharju, E.; Oksala, N.; Lehtimäki, T.; Weber, C.; et al. Palmitoylethanolamide Promotes a Proresolving Macrophage Phenotype and Attenuates Atherosclerotic Plaque Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2562–2575. [Google Scholar] [CrossRef]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Mendez-Díaz, M.; Prospero-García, O. Advances in the Physiology of GPR55 in the Central Nervous System. Curr. Neuropharmacol. 2017, 15, 771–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musella, A.; Fresegna, D.; Rizzo, F.R.; Gentile, A.; Bullitta, S.; De Vito, F.; Guadalupi, L.; Centonze, D.; Mandolesi, G. A Novel Crosstalk within the Endocannabinoid System Controls GABA Transmission in the Striatum. Sci. Rep. 2017, 7, 7363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.E.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R.; et al. Palmitoylethanolamide Induces Microglia Changes Associated with Increased Migration and Phagocytic Activity: Involvement of the CB2 Receptor. Sci. Rep. 2017, 7, 375. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, A.; Molteni, L.; Gullo, F.; Bresciani, E.; Artusa, V.; Rizzi, L.; Ceriani, M.; Meanti, R.; Lecchi, M.; Coco, S.; et al. Palmitoylethanolamide Modulation of Microglia Activation: Characterization of Mechanisms of Action and Implication for Its Neuroprotective Effects. Int. J. Mol. Sci. 2021, 22, 3054. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Ermund, A.; Movahed, P.; Andersson, D.A.; Simonsen, C.; Jönsson, B.A.G.; Blomgren, A.; Birnir, B.; Bevan, S.; Eschalier, A.; et al. Monoacylglycerols Activate TRPV1—A Link between Phospholipase C and TRPV1. PLoS ONE 2013, 8, e81618. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.A. Anandamide and Vanilloid TRPV1 Receptors. Br. J. Pharmacol. 2003, 140, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.H.; Park, E.S.; Won, S.-Y.; Lee, Y.A.; Kim, K.I.; Jeong, J.Y.; Baek, J.Y.; Cho, E.J.; Jin, M.; Chung, Y.C.; et al. TRPV1 on Astrocytes Rescues Nigral Dopamine Neurons in Parkinson’s Disease via CNTF. Brain 2015, 138, 3610–3622. [Google Scholar] [CrossRef] [Green Version]
- Jayant, S.; Sharma, B.M.; Sharma, B. Protective Effect of Transient Receptor Potential Vanilloid Subtype 1 (TRPV1) Modulator, against Behavioral, Biochemical and Structural Damage in Experimental Models of Alzheimer’s Disease. Brain Res. 2016, 1642, 397–408. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef] [Green Version]
- Balleza-Tapia, H.; Crux, S.; Andrade-Talavera, Y.; Dolz-Gaiton, P.; Papadia, D.; Chen, G.; Johansson, J.; Fisahn, A. TrpV1 Receptor Activation Rescues Neuronal Function and Network Gamma Oscillations from Aβ-Induced Impairment in Mouse Hippocampus In Vitro. eLife 2018, 7, e37703. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network Abnormalities and Interneuron Dysfunction in Alzheimer Disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Lu, C.-W.; Wu, C.-C.; Huang, S.-K.; Wang, S.-J. Palmitoylethanolamide Inhibits Glutamate Release in Rat Cerebrocortical Nerve Terminals. Int. J. Mol. Sci. 2015, 16, 5555–5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, C.; Valentão, P.; Ferreres, F.; Andrade, P.B. The Use of Flavonoids in Central Nervous System Disorders. Curr. Med. Chem. 2013, 20, 4694–4719. [Google Scholar] [CrossRef]
- Daily, J.W.; Kang, S.; Park, S. Protection against Alzheimer’s Disease by Luteolin: Role of Brain Glucose Regulation, Anti-Inflammatory Activity, and the Gut Microbiota-Liver-Brain Axis. Biofactors 2021, 47, 218–231. [Google Scholar] [CrossRef]
- Crupi, R.; Paterniti, I.; Ahmad, A.; Campolo, M.; Esposito, E.; Cuzzocrea, S. Effects of Palmitoylethanolamide and Luteolin in an Animal Model of Anxiety/Depression. CNS Neurol. Disord. Drug Targets 2013, 12, 989–1001. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Esposito, E.; Di Paola, R.; Ahmad, A.; Campolo, M.; Peli, A.; Morittu, V.M.; Britti, D.; Cuzzocrea, S. Palmitoylethanolamide and Luteolin Ameliorate Development of Arthritis Caused by Injection of Collagen Type II in Mice. Arthritis Res. Ther. 2013, 15, R192. [Google Scholar] [CrossRef] [Green Version]
- Bertolino, B.; Crupi, R.; Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Beneficial Effects of Co-Ultramicronized Palmitoylethanolamide/Luteolin in a Mouse Model of Autism and in a Case Report of Autism. CNS Neurosci. Ther. 2017, 23, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siracusa, R.; Paterniti, I.; Impellizzeri, D.; Cordaro, M.; Crupi, R.; Navarra, M.; Cuzzocrea, S.; Esposito, E. The Association of Palmitoylethanolamide with Luteolin Decreases Neuroinflammation and Stimulates Autophagy in Parkinson’s Disease Model. CNS Neurol. Disord. Drug Targets 2015, 14, 1350–1365. [Google Scholar] [CrossRef]
- Paterniti, I.; Impellizzeri, D.; Di Paola, R.; Navarra, M.; Cuzzocrea, S.; Esposito, E. A New Co-Ultramicronized Composite Including Palmitoylethanolamide and Luteolin to Prevent Neuroinflammation in Spinal Cord Injury. J. Neuroinflamm. 2013, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 821. [Google Scholar] [CrossRef] [Green Version]
- Landolfo, E.; Cutuli, D.; Petrosini, L.; Caltagirone, C. Effects of Palmitoylethanolamide on Neurodegenerative Diseases: A Review from Rodents to Humans. Biomolecules 2022, 12, 667. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratù, M.R.; Steardo, L. Palmitoylethanolamide Exerts Neuroprotective Effects in Mixed Neuroglial Cultures and Organotypic Hippocampal Slices via Peroxisome Proliferator-Activated Receptor-α. J. Neuroinflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuderi, C.; Steardo, L. Neuroglial Roots of Neurodegenerative Diseases: Therapeutic Potential of Palmitoylethanolamide in Models of Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2013, 12, 62–69. [Google Scholar] [CrossRef]
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Steardo, L.; Pompili, E.; Fumagalli, L.; Campolongo, P.; et al. Palmitoylethanolamide Controls Reactive Gliosis and Exerts Neuroprotective Functions in a Rat Model of Alzheimer’s Disease. Cell Death Dis. 2014, 5, e1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbierato, M.; Borri, M.; Facci, L.; Zusso, M.; Skaper, S.D.; Giusti, P. Expression and Differential Responsiveness of Central Nervous System Glial Cell Populations to the Acute Phase Protein Serum Amyloid A. Sci. Rep. 2017, 7, 12158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchinetti, R.; Valenza, M.; Gomiero, C.; Mancini, G.F.; Steardo, L.; Campolongo, P.; Scuderi, C. Co-Ultramicronized Palmitoylethanolamide/Luteolin Restores Oligodendrocyte Homeostasis via Peroxisome Proliferator-Activated Receptor-α in an In Vitro Model of Alzheimer’s Disease. Biomedicines 2022, 10, 1236. [Google Scholar] [CrossRef]
- Cipriano, M.; Esposito, G.; Negro, L.; Capoccia, E.; Sarnelli, G.; Scuderi, C.; De Filippis, D.; Steardo, L.; Iuvone, T. Palmitoylethanolamide Regulates Production of Pro-Angiogenic Mediators in a Model of β Amyloid-Induced Astrogliosis In Vitro. CNS Neurol. Disord. Drug Targets 2015, 14, 828–837. [Google Scholar] [CrossRef]
- Tomasini, M.C.; Borelli, A.C.; Beggiato, S.; Ferraro, L.; Cassano, T.; Tanganelli, S.; Antonelli, T. Differential Effects of Palmitoylethanolamide against Amyloid-β Induced Toxicity in Cortical Neuronal and Astrocytic Primary Cultures from Wild-Type and 3×Tg-AD Mice. J. Alzheimers Dis. 2015, 46, 407–421. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxid. Med. Cell Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef] [Green Version]
- Guida, F.; Luongo, L.; Marmo, F.; Romano, R.; Iannotta, M.; Napolitano, F.; Belardo, C.; Marabese, I.; D’Aniello, A.; De Gregorio, D.; et al. Palmitoylethanolamide Reduces Pain-Related Behaviors and Restores Glutamatergic Synapses Homeostasis in the Medial Prefrontal Cortex of Neuropathic Mice. Mol. Brain 2015, 8, 47. [Google Scholar] [CrossRef]
- Boccella, S.; Marabese, I.; Iannotta, M.; Belardo, C.; Neugebauer, V.; Mazzitelli, M.; Pieretti, G.; Maione, S.; Palazzo, E. Metabotropic Glutamate Receptor 5 and 8 Modulate the Ameliorative Effect of Ultramicronized Palmitoylethanolamide on Cognitive Decline Associated with Neuropathic Pain. Int. J. Mol. Sci. 2019, 20, 1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- al-Ghoul, W.M.; Li Volsi, G.; Weinberg, R.J.; Rustioni, A. Glutamate Immunocytochemistry in the Dorsal Horn after Injury or Stimulation of the Sciatic Nerve of Rats. Brain Res. Bull. 1993, 30, 453–459. [Google Scholar] [CrossRef]
- Boccella, S.; Cristiano, C.; Romano, R.; Iannotta, M.; Belardo, C.; Farina, A.; Guida, F.; Piscitelli, F.; Palazzo, E.; Mazzitelli, M.; et al. Ultra-Micronized Palmitoylethanolamide Rescues the Cognitive Decline-Associated Loss of Neural Plasticity in the Neuropathic Mouse Entorhinal Cortex-Dentate Gyrus Pathway. Neurobiol. Dis. 2019, 121, 106–119. [Google Scholar] [CrossRef]
- Beggiato, S.; Tomasini, M.C.; Cassano, T.; Ferraro, L. Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease. JCM 2020, 9, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellanti, F.; Bukke, V.N.; Moola, A.; Villani, R.; Scuderi, C.; Steardo, L.; Palombelli, G.; Canese, R.; Beggiato, S.; Altamura, M.; et al. Effects of Ultramicronized Palmitoylethanolamide on Mitochondrial Bioenergetics, Cerebral Metabolism, and Glutamatergic Transmission: An Integrated Approach in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 890855. [Google Scholar] [CrossRef] [PubMed]
- Kramar, C.; Loureiro, M.; Renard, J.; Laviolette, S.R. Palmitoylethanolamide Modulates GPR55 Receptor Signaling in the Ventral Hippocampus to Regulate Mesolimbic Dopamine Activity, Social Interaction, and Memory Processing. Cannabis Cannabinoid Res. 2017, 2, 8–20. [Google Scholar] [CrossRef]
- Facchinetti, R.; Valenza, M.; Bronzuoli, M.R.; Menegoni, G.; Ratano, P.; Steardo, L.; Campolongo, P.; Scuderi, C. Looking for a Treatment for the Early Stage of Alzheimer’s Disease: Preclinical Evidence with Co-Ultramicronized Palmitoylethanolamide and Luteolin. Int. J. Mol. Sci. 2020, 21, 3802. [Google Scholar] [CrossRef]
- Clemente, S. Amyotrophic Lateral Sclerosis Treatment with Ultramicronized Palmitoylethanolamide: A Case Report. CNS Neurol. Disord. Drug Targets 2012, 11, 933–936. [Google Scholar] [CrossRef]
- Palma, E.; Reyes-Ruiz, J.M.; Lopergolo, D.; Roseti, C.; Bertollini, C.; Ruffolo, G.; Cifelli, P.; Onesti, E.; Limatola, C.; Miledi, R.; et al. Acetylcholine Receptors from Human Muscle as Pharmacological Targets for ALS Therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 3060–3065. [Google Scholar] [CrossRef] [Green Version]
- Calabrò, R.S.; Naro, A.; De Luca, R.; Leonardi, S.; Russo, M.; Marra, A.; Bramanti, P. PEALut Efficacy in Mild Cognitive Impairment: Evidence from a SPECT Case Study! Aging Clin. Exp. Res. 2016, 28, 1279–1282. [Google Scholar] [CrossRef]
- Assogna, M.; Casula, E.P.; Borghi, I.; Bonnì, S.; Samà, D.; Motta, C.; Di Lorenzo, F.; D’Acunto, A.; Porrazzini, F.; Minei, M.; et al. Effects of Palmitoylethanolamide Combined with Luteoline on Frontal Lobe Functions, High Frequency Oscillations, and GABAergic Transmission in Patients with Frontotemporal Dementia. J. Alzheimers Dis. 2020, 76, 1297–1308, The publication is available at IOS Press through https://doi.org/10.3233/JAD-200426. [Google Scholar] [PubMed]
- Cummings, J. New Approaches to Symptomatic Treatments for Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.-H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toniolo, S.; Sen, A.; Husain, M. Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9318. [Google Scholar] [CrossRef]
- Francesco, D.L.; Koch, G. Synaptic Impairment: The New Battlefield of Alzheimer’s Disease. Alzheimers Dement. 2021, 17, 314–315. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assogna, M.; Di Lorenzo, F.; Martorana, A.; Koch, G. Synaptic Effects of Palmitoylethanolamide in Neurodegenerative Disorders. Biomolecules 2022, 12, 1161. https://doi.org/10.3390/biom12081161
Assogna M, Di Lorenzo F, Martorana A, Koch G. Synaptic Effects of Palmitoylethanolamide in Neurodegenerative Disorders. Biomolecules. 2022; 12(8):1161. https://doi.org/10.3390/biom12081161
Chicago/Turabian StyleAssogna, Martina, Francesco Di Lorenzo, Alessandro Martorana, and Giacomo Koch. 2022. "Synaptic Effects of Palmitoylethanolamide in Neurodegenerative Disorders" Biomolecules 12, no. 8: 1161. https://doi.org/10.3390/biom12081161