
The Beneficial Effects of Ultramicronized Palmitoylethanolamide in the Management of Neuropathic Pain and Associated Mood Disorders Induced by Paclitaxel in Mice

 , ,
, , 
Abstract
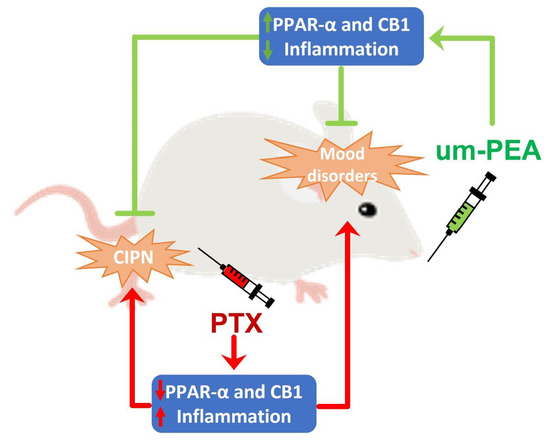
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drug Treatment
2.3. Experimental Groups and Procedures
- Vehicle: mice receiving saline intraperitoneally (IP).
- PTX: mice receiving PTX (8 mg/kg, 100 µL/mouse) IP at day 1, 3, 5, and 7.
- PTX + um - PEA: mice receiving PTX and then um-PEA (30 mg/kg for 7 days) by oral gavage.
- PTX + um - PEA+AM281: mice receiving PTX, then um-PEA, and on the last day AM281 (1 mg/kg) IP.
- PTX+um-PEA+GW6471: mice receiving PTX, then um-PEA, and on the last day GW6471 (2 mg/kg) IP.
2.4. Behavioral Tests
2.4.1. Depressive-like Behavior
2.4.2. Anxiety-like Behavior
2.4.3. Pain Behavior
2.5. Ex Vivo Experiments
2.5.1. Determination of Brain and Spinal Cord Markers of Inflammation
2.5.2. Western Blotting
2.6. Statistical Analysis
3. Results
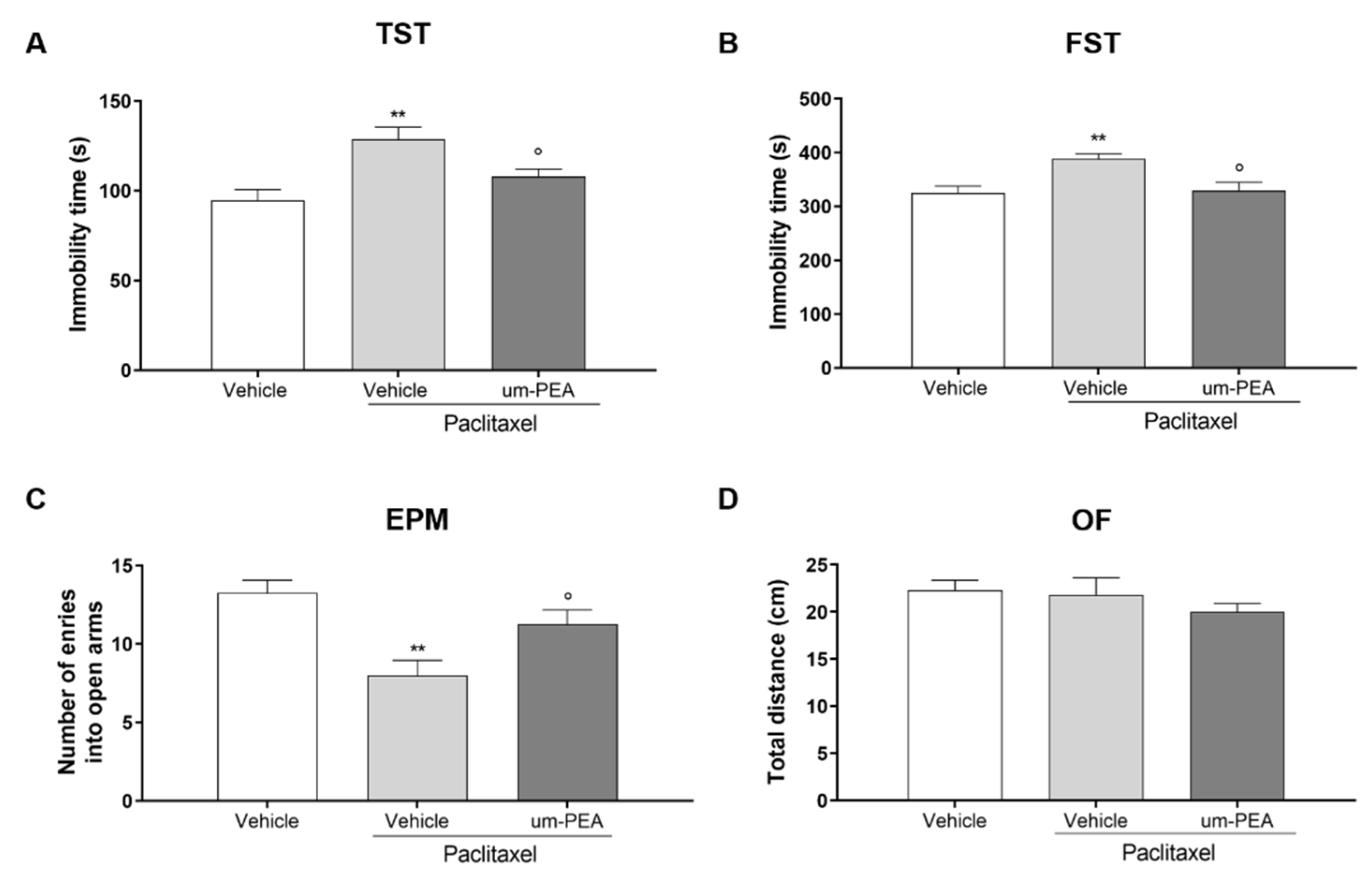
3.1. Effect of um-PEA on PTX-Induced Depressive- and Anxiety-like Behaviors
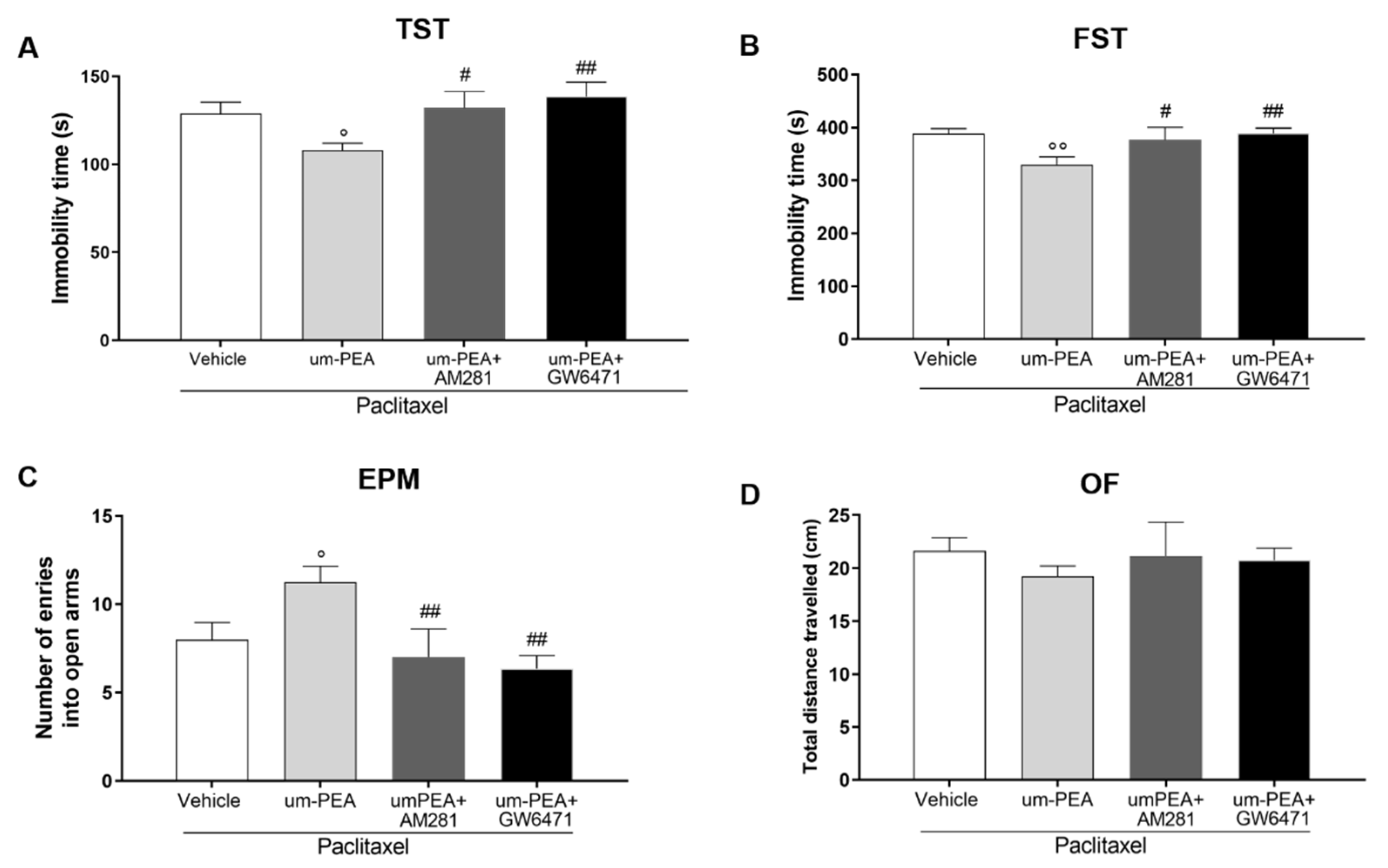
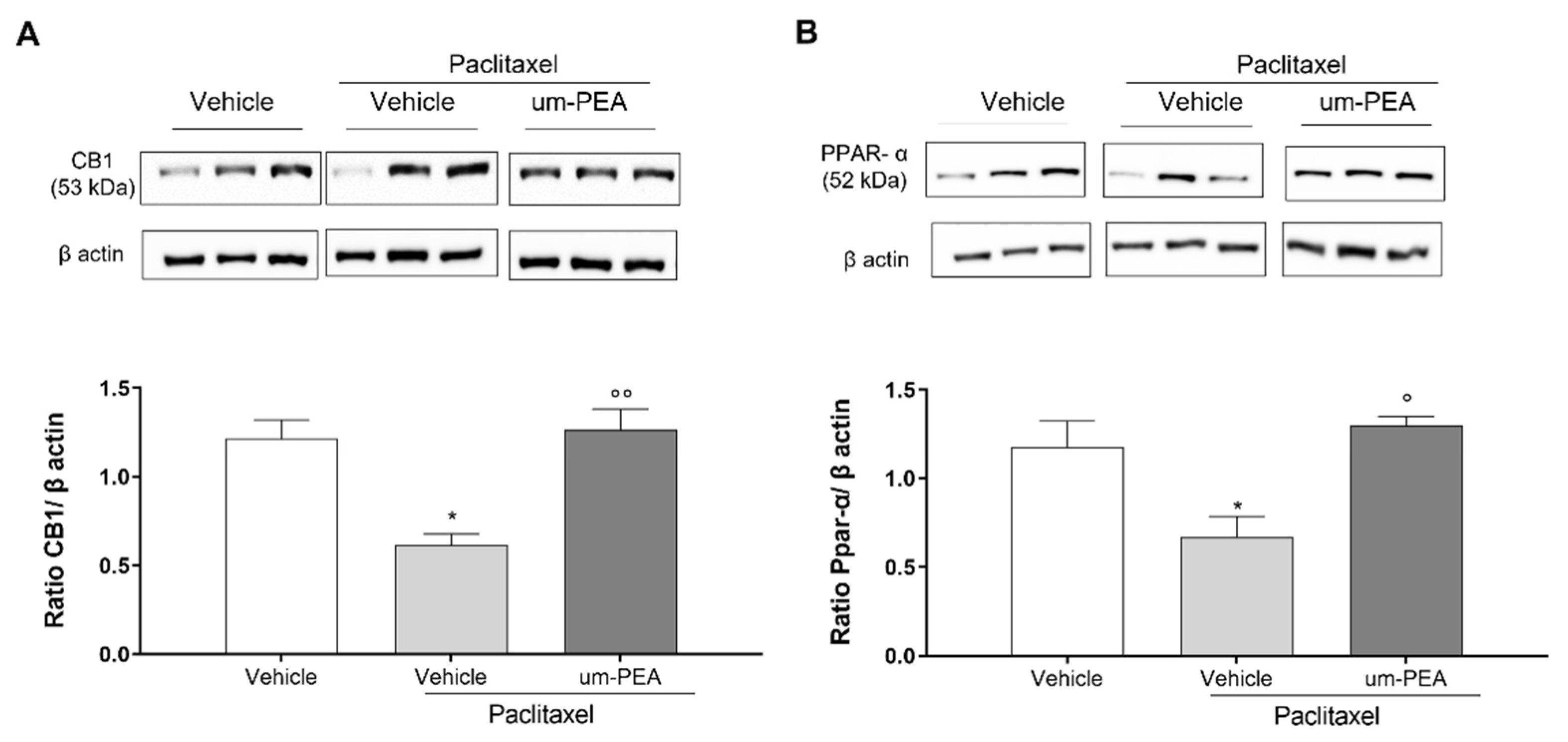
3.2. CB1 and PPAR-α Are Involved in um-PEA Central Activity
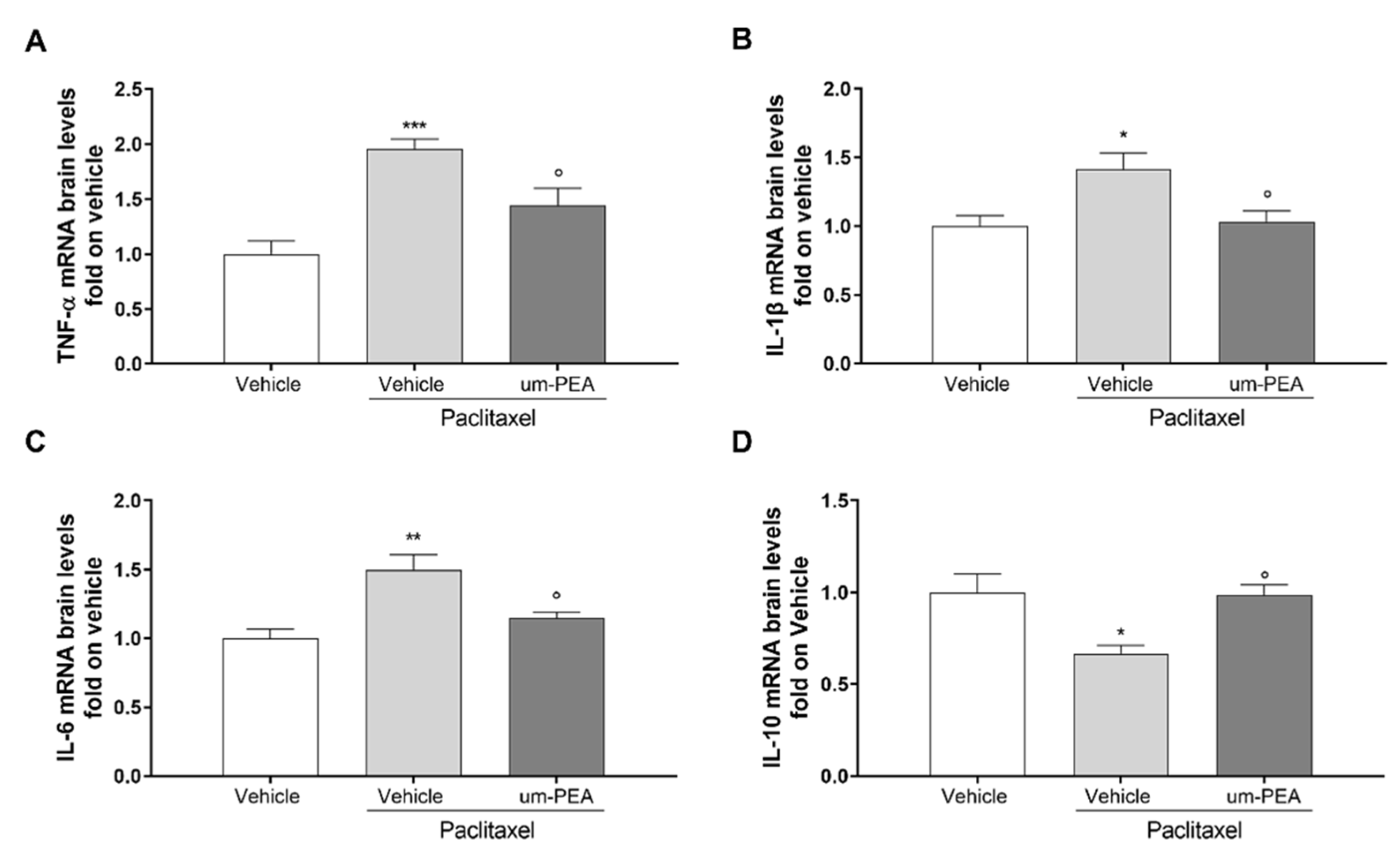
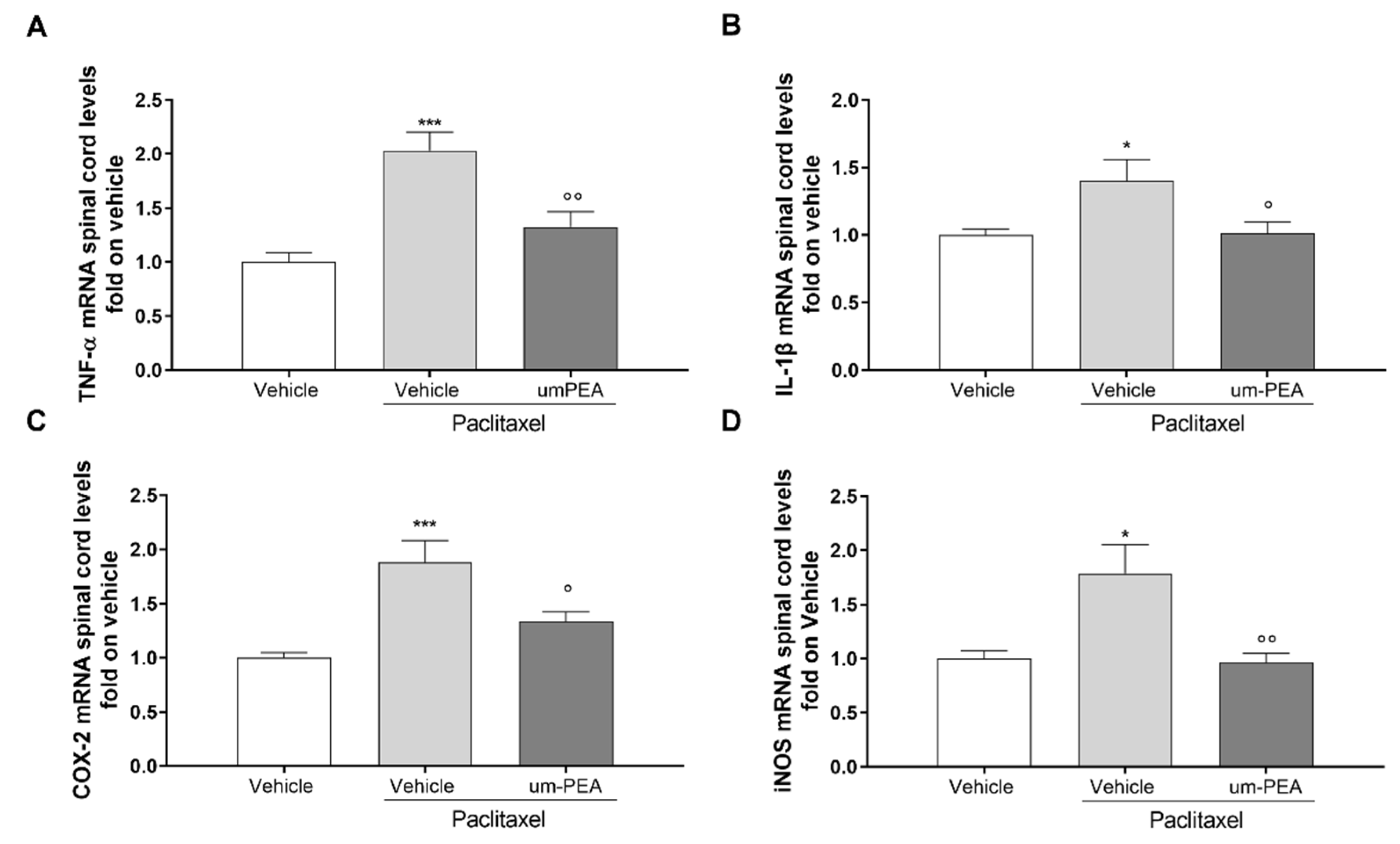
3.3. Effect of um-PEA on Spinal and Sovraspinal Inflammatory Mediators in PTX Mice
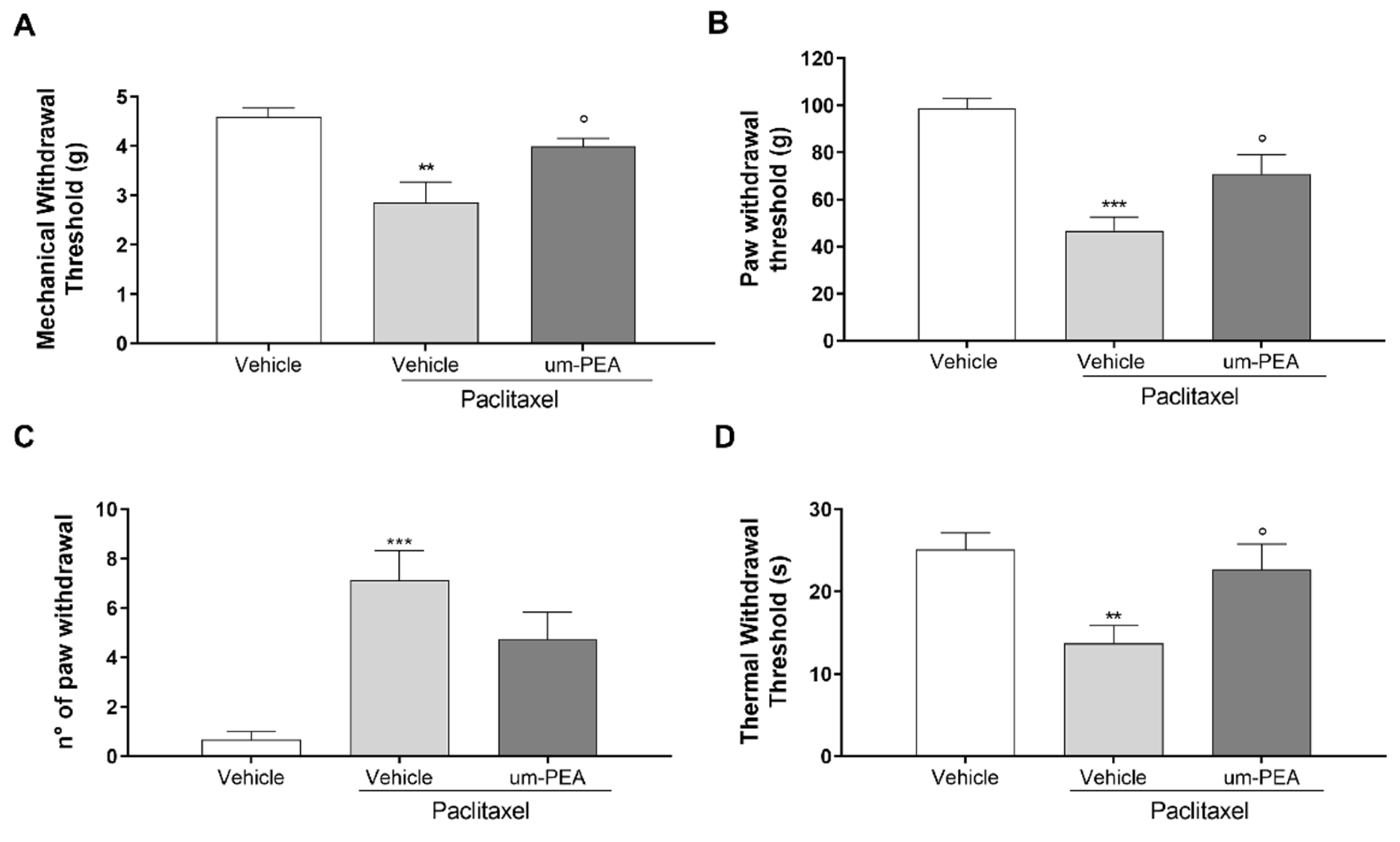
3.4. Effect of um-PEA on PTX-Induced Peripheral Neuropathy
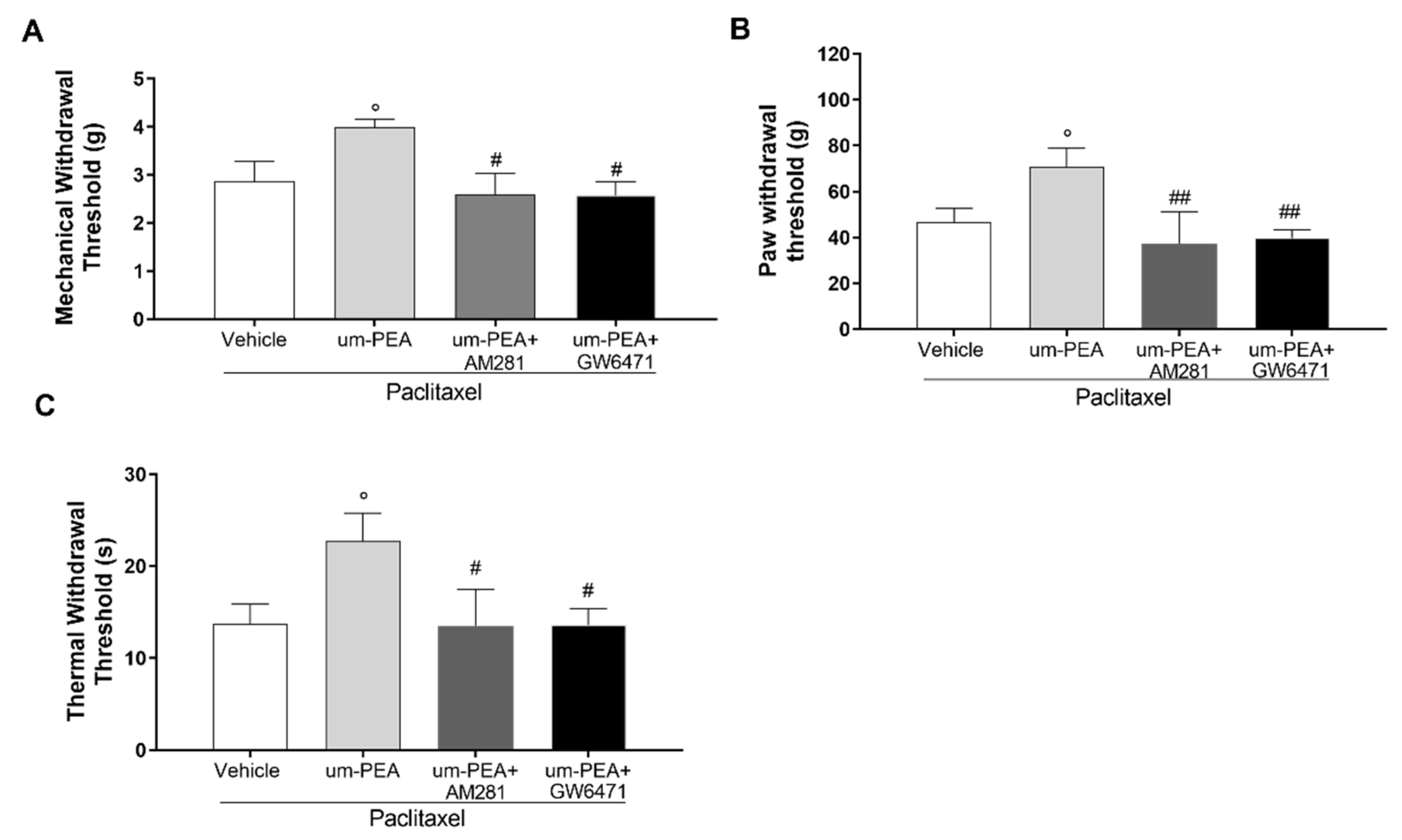
3.5. Effect of um-PEA in PTX-Treated Mice Is PPAR-α and CB1 Mediated
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flatters, S.J.L.; Dougherty, P.M.; Colvin, L.A. Clinical and Preclinical Perspectives on Chemotherapy-Induced Peripheral Neuropathy (CIPN): A Narrative Review. Br. J. Anaesth. 2017, 119, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dranitsaris, G.; King, J.; Kaura, S.; Yu, B.; Zhang, A. Nab-Paclitaxel, Docetaxel, or Solvent-Based Paclitaxel in Metastatic Breast Cancer: A Cost-Utility Analysis from a Chinese Health Care Perspective. Clin. Outcomes Res. 2015, 7, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, Prevalence, and Predictors of Chemotherapy-Induced Peripheral Neuropathy: A Systematic Review and Meta-Analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Juranek, J.K.; Zygulska, A.L. Chemotherapy-Induced Neuropathies-a Growing Problem for Patients and Health Care Providers. Brain Behav. 2017, 7, e00558. [Google Scholar] [CrossRef] [PubMed]
- Staff, N.P.; Grisold, A.; Grisold, W.; Windebank, A.J. Chemotherapy-Induced Peripheral Neuropathy: A Current Review: CIPN. Ann. Neurol. 2017, 81, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Bonhof, C.S.; Poll-Franse, L.V.; Vissers, P.A.J.; Wasowicz, D.K.; Wegdam, J.A.; Révész, D.; Vreugdenhil, G.; Mols, F. Anxiety and Depression Mediate the Association between Chemotherapy-induced Peripheral Neuropathy and Fatigue: Results from the Population-based PROFILES Registry. Psycho-Oncology 2019, 28, 1926–1933. [Google Scholar] [CrossRef] [Green Version]
- Massie, M.J. Prevalence of Depression in Patients With Cancer. J. Natl. Cancer Inst. Monogr. 2004, 2004, 57–71. [Google Scholar] [CrossRef]
- Mehnert, A.; Brähler, E.; Faller, H.; Härter, M.; Keller, M.; Schulz, H.; Wegscheider, K.; Weis, J.; Boehncke, A.; Hund, B.; et al. Four-Week Prevalence of Mental Disorders in Patients With Cancer Across Major Tumor Entities. J. Clin. Oncol. 2014, 32, 3540–3546. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Duggett, N.A.; Griffiths, L.A.; McKenna, O.E.; de Santis, V.; Yongsanguanchai, N.; Mokori, E.B.; Flatters, S.J.L. Oxidative Stress in the Development, Maintenance and Resolution of Paclitaxel-Induced Painful Neuropathy. Neuroscience 2016, 333, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Kidd, J.F.; Pilkington, M.F.; Schell, M.J.; Fogarty, K.E.; Skepper, J.N.; Taylor, C.W.; Thorn, P. Paclitaxel Affects Cytosolic Calcium Signals by Opening the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 2002, 277, 6504–6510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carozzi, V.A.; Canta, A.; Chiorazzi, A. Chemotherapy-Induced Peripheral Neuropathy: What Do We Know about Mechanisms? Neurosci. Lett. 2015, 596, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, I.; Macedo, D.; Alho, I.; Dionísio, M.R.; Costa, L. Treatment of Cancer Pain by Targeting Cytokines. Mediat. Inflamm. 2015, 2015, 984570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluthé, R.M.; Layé, S.; Michaud, B.; Combe, C.; Dantzer, R.; Parnet, P. Role of Interleukin-1beta and Tumour Necrosis Factor-Alpha in Lipopolysaccharide-Induced Sickness Behaviour: A Study with Interleukin-1 Type I Receptor-Deficient Mice. Eur. J. Neurosci. 2000, 12, 4447–4456. [Google Scholar] [CrossRef] [PubMed]
- Bluthé, R. Role of IL-6 in Cytokine-Induced Sickness Behavior a Study with IL-6 Deficient Mice. Physiol. Behav. 2000, 70, 367–373. [Google Scholar] [CrossRef]
- Szajnik, M.; Szczepanski, M.J.; Czystowska, M.; Elishaev, E.; Mandapathil, M.; Nowak-Markwitz, E.; Spaczynski, M.; Whiteside, T.L. TLR4 Signaling Induced by Lipopolysaccharide or Paclitaxel Regulates Tumor Survival and Chemoresistance in Ovarian Cancer. Oncogene 2009, 28, 4353–4363. [Google Scholar] [CrossRef] [Green Version]
- Rajput, S.; Volk-Draper, L.D.; Ran, S. TLR4 is a Novel Determinant of the Response to Paclitaxel in Breast Cancer. Mol. Cancer Ther. 2013, 12, 1676–1687. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.-C.; Ma, Y.-B.; Wu, F.-X.; Ma, Z.-F.; Liu, N.-F.; Gao, R.; Gao, Y.-S.; Sheng, X.-G. TLR4 Induces Tumor Growth and Inhibits Paclitaxel Activity in MyD88-Positive Human Ovarian Carcinoma in Vitro. Oncol. Lett. 2014, 7, 871–877. [Google Scholar] [CrossRef] [Green Version]
- Varrassi, G.; Alon, E.; Bagnasco, M.; Lanata, L.; Mayoral-Rojals, V.; Paladini, A.; Pergolizzi, J.V.; Perrot, S.; Scarpignato, C.; Tölle, T. Towards an Effective and Safe Treatment of Inflammatory Pain: A Delphi-Guided Expert Consensus. Adv. Ther. 2019, 36, 2618–2637. [Google Scholar] [CrossRef] [Green Version]
- Ghazizadeh-Hashemi, M.; Ghajar, A.; Shalbafan, M.-R.; Ghazizadeh-Hashemi, F.; Afarideh, M.; Malekpour, F.; Ghaleiha, A.; Ardebili, M.E.; Akhondzadeh, S. Palmitoylethanolamide as Adjunctive Therapy in Major Depressive Disorder: A Double-Blind, Randomized and Placebo-Controlled Trial. J. Affect. Disord. 2018, 232, 127–133. [Google Scholar] [CrossRef]
- Nau, R.; Ribes, S.; Djukic, M.; Eiffert, H. Strategies to Increase the Activity of Microglia as Efficient Protectors of the Brain against Infections. Front. Cell. Neurosci. 2014, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxidative Med. Cell. Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, L.; Fowler, C.J. The Basal Pharmacology of Palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Crupi, R.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Micronized/Ultramicronized Palmitoylethanolamide Displays Superior Oral Efficacy Compared to Nonmicronized Palmitoylethanolamide in a Rat Model of Inflammatory Pain. J. Neuroinflammation 2014, 11, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, B.; Comelli, F.; Bettoni, I.; Colleoni, M.; Giagnoni, G. The Endogenous Fatty Acid Amide, Palmitoylethanolamide, Has Anti-Allodynic and Anti-Hyperalgesic Effects in a Murine Model of Neuropathic Pain: Involvement of CB1, TRPV1 and PPARγ Receptors and Neurotrophic Factors. Pain 2008, 139, 541–550. [Google Scholar] [CrossRef]
- Bettoni, I.; Comelli, F.; Colombo, A.; Bonfanti, P.; Costa, B. Non-Neuronal Cell Modulation Relieves Neuropathic Pain: Efficacy of the Endogenous Lipid Palmitoylethanolamide. CNS Neurol. Disord. -Drug Targets 2013, 12, 34–44. [Google Scholar] [CrossRef]
- D’Amico, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. ALIAmides Update: Palmitoylethanolamide and Its Formulations on Management of Peripheral Neuropathic Pain. Int. J. Mol. Sci. 2020, 21, 5330. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Fusco, M.; della Valle, M.F.; Zusso, M.; Costa, B.; Giusti, P. Palmitoylethanolamide, a Naturally Occurring Disease-Modifying Agent in Neuropathic Pain. Inflammopharmacol 2014, 22, 79–94. [Google Scholar] [CrossRef]
- Peritore, A.F.; Siracusa, R.; Fusco, R.; Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Crupi, R.; Genovese, T.; Impellizzeri, D.; Cuzzocrea, S.; et al. Ultramicronized Palmitoylethanolamide and Paracetamol, a New Association to Relieve Hyperalgesia and Pain in a Sciatic Nerve Injury Model in Rat. Int. J. Mol. Sci. 2020, 21, 3509. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Peritore, A.F.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Crupi, R.; D’Amico, R.; Fusco, R.; Evangelista, M.; Cuzzocrea, S.; et al. The Neuroprotective Effects of Micronized PEA (PEA-m) Formulation on Diabetic Peripheral Neuropathy in Mice. FASEB J. 2019, 33, 11364–11380. [Google Scholar] [CrossRef] [PubMed]
- Steels, E.; Venkatesh, R.; Steels, E.; Vitetta, G.; Vitetta, L. A Double-Blind Randomized Placebo Controlled Study Assessing Safety, Tolerability and Efficacy of Palmitoylethanolamide for Symptoms of Knee Osteoarthritis. Inflammopharmacol 2019, 27, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Marini, I.; Bartolucci, M.L.; Bortolotti, F.; Gatto, M.R.; Bonetti, G.A. Palmitoylethanolamide versus a Nonsteroidal Anti-Inflammatory Drug in the Treatment of Temporomandibular Joint Inflammatory Pain. J. Orofac. Pain 2012, 26, 99–104. [Google Scholar] [PubMed]
- Keppel Hesselink, J.; Kopsky, D. Palmitoylethanolamide, a Neutraceutical, in Nerve Compression Syndromes: Efficacy and Safety in Sciatic Pain and Carpal Tunnel Syndrome. J. Pain Res. 2015, 8, 729–734. [Google Scholar] [CrossRef] [Green Version]
- Schifilliti, C.; Cucinotta, L.; Fedele, V.; Ingegnosi, C.; Luca, S.; Leotta, C. Micronized Palmitoylethanolamide Reduces the Symptoms of Neuropathic Pain in Diabetic Patients. Pain Res. Treat. 2014, 2014, 849623. [Google Scholar] [CrossRef]
- Bacci, C.; Cassetta, G.; Emanuele, B.; Berengo, M. Randomized Split-Mouth Study on Postoperative Effects of Palmitoylethanolamide for Impacted Lower Third Molar Surgery. ISRN Surg. 2011, 2011, 917350. [Google Scholar] [CrossRef] [Green Version]
- Del Giorno, R.; Skaper, S.; Paladini, A.; Varrassi, G.; Coaccioli, S. Palmitoylethanolamide in Fibromyalgia: Results from Prospective and Retrospective Observational Studies. Pain Ther. 2015, 4, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Stochino-Loi, E.; Pontis, A.; Cofelice, V.; Pirarba, S.; Fais, M.F.; Daniilidis, A.; Melis, I.; Paoletti, A.M.; Angioni, S. Effect of Ultramicronized-Palmitoylethanolamide and Co-Micronized Palmitoylethanolamide/Polydatin on Chronic Pelvic Pain and Quality of Life in Endometriosis Patients: An Open-Label Pilot Study. Int. J. Womens Health 2019, 11, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.M.; Okorokov, A.L.; Hill, M.N.; Bras, J.T.; Lee, M.-C.; Li, S.; Gossage, S.J.; van Drimmelen, M.; Morena, M.; Houlden, H.; et al. Microdeletion in a FAAH Pseudogene Identified in a Patient with High Anandamide Concentrations and Pain Insensitivity. Br. J. Anaesth. 2019, 123, e249–e253. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare Mannelli, L.; Pacini, A.; Corti, F.; Boccella, S.; Luongo, L.; Esposito, E.; Cuzzocrea, S.; Maione, S.; Calignano, A.; Ghelardini, C. Antineuropathic Profile of N-Palmitoylethanolamine in a Rat Model of Oxaliplatin-Induced Neurotoxicity. PLoS ONE 2015, 10, e0128080. [Google Scholar] [CrossRef] [Green Version]
- Donvito, G.; Wilkerson, J.L.; Damaj, M.I.; Lichtman, A.H. Palmitoylethanolamide Reverses Paclitaxel-Induced Allodynia in Mice. J. Pharmacol. Exp. Ther. 2016, 359, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caillaud, M.; Patel, N.H.; Toma, W.; White, A.; Thompson, D.; Mann, J.; Tran, T.H.; Roberts, J.L.; Poklis, J.L.; Bigbee, J.W.; et al. A Fenofibrate Diet Prevents Paclitaxel-Induced Peripheral Neuropathy in Mice. Cancers 2020, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide Protects Against the Amyloid-Β25-35-Induced Learning and Memory Impairment in Mice, an Experimental Model of Alzheimer Disease. Neuropsychopharmacol 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [Green Version]
- Esposito, E.; Cuzzocrea, S. Palmitoylethanolamide in Homeostatic and Traumatic Central Nervous System Injuries. CNS Neurol. Disord. -Drug Targets 2013, 12, 55–61. [Google Scholar] [CrossRef]
- Herrera, M.I.; Udovin, L.D.; Toro-Urrego, N.; Kusnier, C.F.; Luaces, J.P.; Capani, F. Palmitoylethanolamide Ameliorates Hippocampal Damage and Behavioral Dysfunction After Perinatal Asphyxia in the Immature Rat Brain. Front. Neurosci. 2018, 12, 145. [Google Scholar] [CrossRef] [Green Version]
- Brotini, S.; Schievano, C.; Guidi, L. Ultra-Micronized Palmitoylethanolamide: An Efficacious Adjuvant Therapy for Parkinson’s Disease. CNS Neurol. Disord. -Drug Targets 2017, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kruk-Slomka, M.; Dzik, A.; Budzynska, B.; Biala, G. Endocannabinoid System: The Direct and Indirect Involvement in the Memory and Learning Processes—A Short Review. Mol. Neurobiol. 2017, 54, 8332–8347. [Google Scholar] [CrossRef] [Green Version]
- Crupi, R.; Paterniti, I.; Ahmad, A.; Campolo, M.; Esposito, E.; Cuzzocrea, S. Effects of Palmitoylethanolamide and Luteolin in an Animal Model of Anxiety/Depression. CNS Neurol. Disord. -Drug Targets 2013, 12, 989–1001. [Google Scholar] [CrossRef]
- Karabatsiakis, A.; Hamuni, G.; Wilker, S.; Kolassa, S.; Renu, D.; Kadereit, S.; Schauer, M.; Hennessy, T.; Kolassa, I.-T. Metabolite Profiling in Posttraumatic Stress Disorder. J. Mol. Psychiatry 2015, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Meli, R.; Calignano, A.; et al. Palmitoylethanolamide Counteracts Autistic-like Behaviours in BTBR T+tf/J Mice: Contribution of Central and Peripheral Mechanisms. Brain Behav. Immun. 2018, 74, 166–175. [Google Scholar] [CrossRef]
- Fusco, R.; Gugliandolo, E.; Campolo, M.; Evangelista, M.; Di Paola, R.; Cuzzocrea, S. Effect of a New Formulation of Micronized and Ultramicronized N-Palmitoylethanolamine in a Tibia Fracture Mouse Model of Complex Regional Pain Syndrome. PLoS ONE 2017, 12, e0178553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toma, W.; Caillaud, M.; Patel, N.H.; Tran, T.H.; Donvito, G.; Roberts, J.; Bagdas, D.; Jackson, A.; Lichtman, A.; Gewirtz, D.A.; et al. N-acylethanolamine-hydrolysing Acid Amidase: A New Potential Target to Treat Paclitaxel-induced Neuropathy. Eur. J. Pain 2021, 25, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR Isotypes in the Adult Mouse and Human Brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef] [PubMed]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef] [Green Version]
- Roch, G.; Batallé, G.; Bai, X.; Pouso-Vázquez, E.; Rodríguez, L.; Pol, O. The Beneficial Effects of Heme Oxygenase 1 and Hydrogen Sulfide Activation in the Management of Neuropathic Pain, Anxiety- and Depressive-like Effects of Paclitaxel in Mice. Antioxidants 2022, 11, 122. [Google Scholar] [CrossRef]
- Fornasari, D. Pain Mechanisms in Patients with Chronic Pain. Clin. Drug Investig. 2012, 32, 45–52. [Google Scholar] [CrossRef]
- Clayton, P.; Subah, S.; Venkatesh, R.; Hill, M.; Bogoda, N. Palmitoylethanolamide: A Potential Alternative to Cannabidiol. J. Diet. Suppl. 2021, 1–26. [Google Scholar] [CrossRef]
- Domi, E.; Uhrig, S.; Soverchia, L.; Spanagel, R.; Hansson, A.C.; Barbier, E.; Heilig, M.; Ciccocioppo, R.; Ubaldi, M. Genetic Deletion of Neuronal PPARγ Enhances the Emotional Response to Acute Stress and Exacerbates Anxiety: An Effect Reversed by Rescue of Amygdala PPARγ Function. J. Neurosci. 2016, 36, 12611–12623. [Google Scholar] [CrossRef]
- Locci, A.; Pinna, G. Stimulation of Peroxisome Proliferator-Activated Receptor-α by N-Palmitoylethanolamine Engages Allopregnanolone Biosynthesis to Modulate Emotional Behavior. Biol. Psychiatry 2019, 85, 1036–1045. [Google Scholar] [CrossRef]
- Yu, H.-L.; Deng, X.-Q.; Li, Y.-J.; Li, Y.-C.; Quan, Z.-S.; Sun, X.-Y. Short Communication—N-Palmitoylethanolamide, an Endocannabinoid, Exhibits Antidepressant Effects in the Forced Swim Test and the Tail Suspension Test in Mice. Pharmacol. Rep. 2011, 63, 834–839. [Google Scholar] [CrossRef]
- Guida, F.; Luongo, L.; Marmo, F.; Romano, R.; Iannotta, M.; Napolitano, F.; Belardo, C.; Marabese, I.; D’Aniello, A.; De Gregorio, D.; et al. Palmitoylethanolamide Reduces Pain-Related Behaviors and Restores Glutamatergic Synapses Homeostasis in the Medial Prefrontal Cortex of Neuropathic Mice. Mol. Brain 2015, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, P.; Gołda, A.; McCreary, A.C.; Filip, M.; Przegaliński, E. Activation of Endocannabinoid Transmission Induces Antidepressant-like Effects in Rats. J. Physiol. Pharmacol. 2008, 59, 217–228. [Google Scholar] [PubMed]
- Melis, M.; Carta, G.; Pistis, M.; Banni, S. Physiological Role of Peroxisome Proliferator-Activated Receptors Type Alpha on Dopamine Systems. CNS Neurol. Disord. -Drug Targets 2013, 12, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Mattace Raso, G.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS Health and Disease. Pharmacol. Res. 2014, 86, 32–41. [Google Scholar] [CrossRef]
- Matrisciano, F.; Pinna, G. PPAR-α Hypermethylation in the Hippocampus of Mice Exposed to Social Isolation Stress Is Associated with Enhanced Neuroinflammation and Aggressive Behavior. Int. J. Mol. Sci. 2021, 22, 10678. [Google Scholar] [CrossRef]
- Maione, S.; Costa, B.; Di Marzo, V. Endocannabinoids: A Unique Opportunity to Develop Multitarget Analgesics. Pain 2013, 154, S87–S93. [Google Scholar] [CrossRef]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.E.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R.; et al. Palmitoylethanolamide Induces Microglia Changes Associated with Increased Migration and Phagocytic Activity: Involvement of the CB2 Receptor. Sci. Rep. 2017, 7, 375. [Google Scholar] [CrossRef]
- Ambrosino, P.; Soldovieri, M.V.; De Maria, M.; Russo, C.; Taglialatela, M. Functional and Biochemical Interaction between PPARα Receptors and TRPV1 Channels: Potential Role in PPARα Agonists-Mediated Analgesia. Pharmacol. Res. 2014, 87, 113–122. [Google Scholar] [CrossRef]
- Hillard, C.J. Stress Regulates Endocannabinoid-CB1 Receptor Signaling. Semin. Immunol. 2014, 26, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Pinna, G. Endocannabinoids and Precision Medicine for Mood Disorders and Suicide. Front. Psychiatry 2021, 12, 658433. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Hillard, C.J. Pharmacological Evaluation of Cannabinoid Receptor Ligands in a Mouse Model of Anxiety: Further Evidence for an Anxiolytic Role for Endogenous Cannabinoid Signaling. J. Pharmacol. Exp. Ther. 2006, 318, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherma, M.; Medalie, J.; Fratta, W.; Vadivel, S.K.; Makriyannis, A.; Piomelli, D.; Mikics, E.; Haller, J.; Yasar, S.; Tanda, G.; et al. The Endogenous Cannabinoid Anandamide Has Effects on Motivation and Anxiety That Are Revealed by Fatty Acid Amide Hydrolase (FAAH) Inhibition. Neuropharmacology 2008, 54, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Esmaeili, M.A.; Yadav, S.; Gupta, R.K.; Waggoner, G.R.; Deloach, A.; Calingasan, N.Y.; Beal, M.F.; Kiaei, M. Preferential PPAR-α Activation Reduces Neuroinflammation, and Blocks Neurodegeneration in Vivo. Hum. Mol. Genet. 2016, 25, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Rolland, B.; Deguil, J.; Jardri, R.; Cottencin, O.; Thomas, P.; Bordet, R. Therapeutic Prospects of PPARs in Psychiatric Disorders: A Comprehensive Review. Curr. Drug Targets 2013, 14, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Racke, M.K.; Drew, P.D. PPARs in Neuroinflammation. PPAR Res. 2008, 2008, 638356. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, A. Stress, Emotion, and Human Immune Function. Psychol. Bull. 1990, 108, 363–382. [Google Scholar] [CrossRef]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo Jr, L.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide Counteracts Reactive Astrogliosis Induced by β-Amyloid Peptide. J. Cell. Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [Green Version]
- Bergandi, L.; Apprato, G.; Silvagno, F. Antioxidant and Anti-Inflammatory Activity of Combined Phycocyanin and Palmitoylethanolamide in Human Lung and Prostate Epithelial Cells. Antioxidants 2022, 11, 201. [Google Scholar] [CrossRef]
- D’Aloia, A.; Molteni, L.; Gullo, F.; Bresciani, E.; Artusa, V.; Rizzi, L.; Ceriani, M.; Meanti, R.; Lecchi, M.; Coco, S.; et al. Palmitoylethanolamide Modulation of Microglia Activation: Characterization of Mechanisms of Action and Implication for Its Neuroprotective Effects. Int. J. Mol. Sci. 2021, 22, 3054. [Google Scholar] [CrossRef]
- Antonucci, N.; Cirillo, A.; Siniscalco, D. Beneficial Effects of Palmitoylethanolamide on Expressive Language, Cognition, and Behaviors in Autism: A Report of Two Cases. Case Rep. Psychiatry 2015, 2015, 325061. [Google Scholar] [CrossRef] [Green Version]
- Altamura, C.; Ventriglia, M.; Martini, M.G.; Montesano, D.; Errante, Y.; Piscitelli, F.; Scrascia, F.; Quattrocchi, C.; Palazzo, P.; Seccia, S.; et al. Elevation of Plasma 2-Arachidonoylglycerol Levels in Alzheimer’s Disease Patients as a Potential Protective Mechanism against Neurodegenerative Decline. JAD 2015, 46, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Xu, Z.-Z.; Gao, Y.-J. Emerging Targets in Neuroinflammation-Driven Chronic Pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Xanthos, D.N.; Sandkühler, J. Neurogenic Neuroinflammation: Inflammatory CNS Reactions in Response to Neuronal Activity. Nat. Rev. Neurosci. 2014, 15, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-Mediated Neuroinflammation in Neurodegenerative Diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Landolfo, E.; Cutuli, D.; Petrosini, L.; Caltagirone, C. Effects of Palmitoylethanolamide on Neurodegenerative Diseases: A Review from Rodents to Humans. Biomolecules 2022, 12, 667. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsson, L.; Mattsson, S.; Fowler, C.J. Palmitoylethanolamide for the Treatment of Pain: Pharmacokinetics, Safety and Efficacy. Br. J. Clin. Pharmacol. 2016, 82, 932–942. [Google Scholar] [CrossRef] [Green Version]
- Seol, T.-K.; Lee, W.; Park, S.; Kim, K.N.; Kim, T.Y.; Oh, Y.N.; Jun, J.H. Effect of Palmitoylethanolamide on Inflammatory and Neuropathic Pain in Rats. Korean J. Anesthesiol. 2017, 70, 561. [Google Scholar] [CrossRef] [Green Version]
- Re, G.; Barbero, R.; Miolo, A.; Di Marzo, V. Palmitoylethanolamide, Endocannabinoids and Related Cannabimimetic Compounds in Protection against Tissue Inflammation and Pain: Potential Use in Companion Animals. Vet. J. 2007, 173, 21–30. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Chamessian, A.; Zhang, Y.-Q. Pain Regulation by Non-Neuronal Cells and Inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mazidi, S.; Alotaibi, M.; Nedjadi, T.; Chaudhary, A.; Alzoghaibi, M.; Djouhri, L. Blocking of Cytokines Signalling Attenuates Evoked and Spontaneous Neuropathic Pain Behaviours in the Paclitaxel Rat Model of Chemotherapy-Induced Neuropathy. Eur. J. Pain 2018, 22, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Agostino, G.; La Rana, G.; Russo, R.; Sasso, O.; Iacono, A.; Esposito, E.; Mattace Raso, G.; Cuzzocrea, S.; LoVerme, J.; Piomelli, D.; et al. Central Administration of Palmitoylethanolamide Reduces Hyperalgesia in Mice via Inhibition of NF-ΚB Nuclear Signalling in Dorsal Root Ganglia. Eur. J. Pharmacol. 2009, 613, 54–59. [Google Scholar] [CrossRef]
- Di Cesare Mannelli, L.; D’Agostino, G.; Pacini, A.; Russo, R.; Zanardelli, M.; Ghelardini, C.; Calignano, A. Palmitoylethanolamide Is a Disease-Modifying Agent in Peripheral Neuropathy: Pain Relief and Neuroprotection Share a PPAR-Alpha-Mediated Mechanism. Mediat. Inflamm. 2013, 2013, 328797. [Google Scholar] [CrossRef]
- Khasabova, I.A.; Seybold, V.S.; Simone, D.A. The Role of PPARγ in Chemotherapy-Evoked Pain. Neurosci. Lett. 2021, 753, 135845. [Google Scholar] [CrossRef]
- Roa-Coria, J.E.; Navarrete-Vázquez, G.; Fowler, C.J.; Flores-Murrieta, F.J.; Déciga-Campos, M.; Granados-Soto, V. N-(4-Methoxy-2-Nitrophenyl)Hexadecanamide, a Palmitoylethanolamide Analogue, Reduces Formalin-Induced Nociception. Life Sci. 2012, 91, 1288–1294. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cristiano, C.; Avagliano, C.; Cuozzo, M.; Liguori, F.M.; Calignano, A.; Russo, R. The Beneficial Effects of Ultramicronized Palmitoylethanolamide in the Management of Neuropathic Pain and Associated Mood Disorders Induced by Paclitaxel in Mice. Biomolecules 2022, 12, 1155. https://doi.org/10.3390/biom12081155
Cristiano C, Avagliano C, Cuozzo M, Liguori FM, Calignano A, Russo R. The Beneficial Effects of Ultramicronized Palmitoylethanolamide in the Management of Neuropathic Pain and Associated Mood Disorders Induced by Paclitaxel in Mice. Biomolecules. 2022; 12(8):1155. https://doi.org/10.3390/biom12081155
Chicago/Turabian StyleCristiano, Claudia, Carmen Avagliano, Mariarosaria Cuozzo, Fabrizio Maria Liguori, Antonio Calignano, and Roberto Russo. 2022. "The Beneficial Effects of Ultramicronized Palmitoylethanolamide in the Management of Neuropathic Pain and Associated Mood Disorders Induced by Paclitaxel in Mice" Biomolecules 12, no. 8: 1155. https://doi.org/10.3390/biom12081155