
AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2

 , ,
, ,
Abstract
:1. Introduction
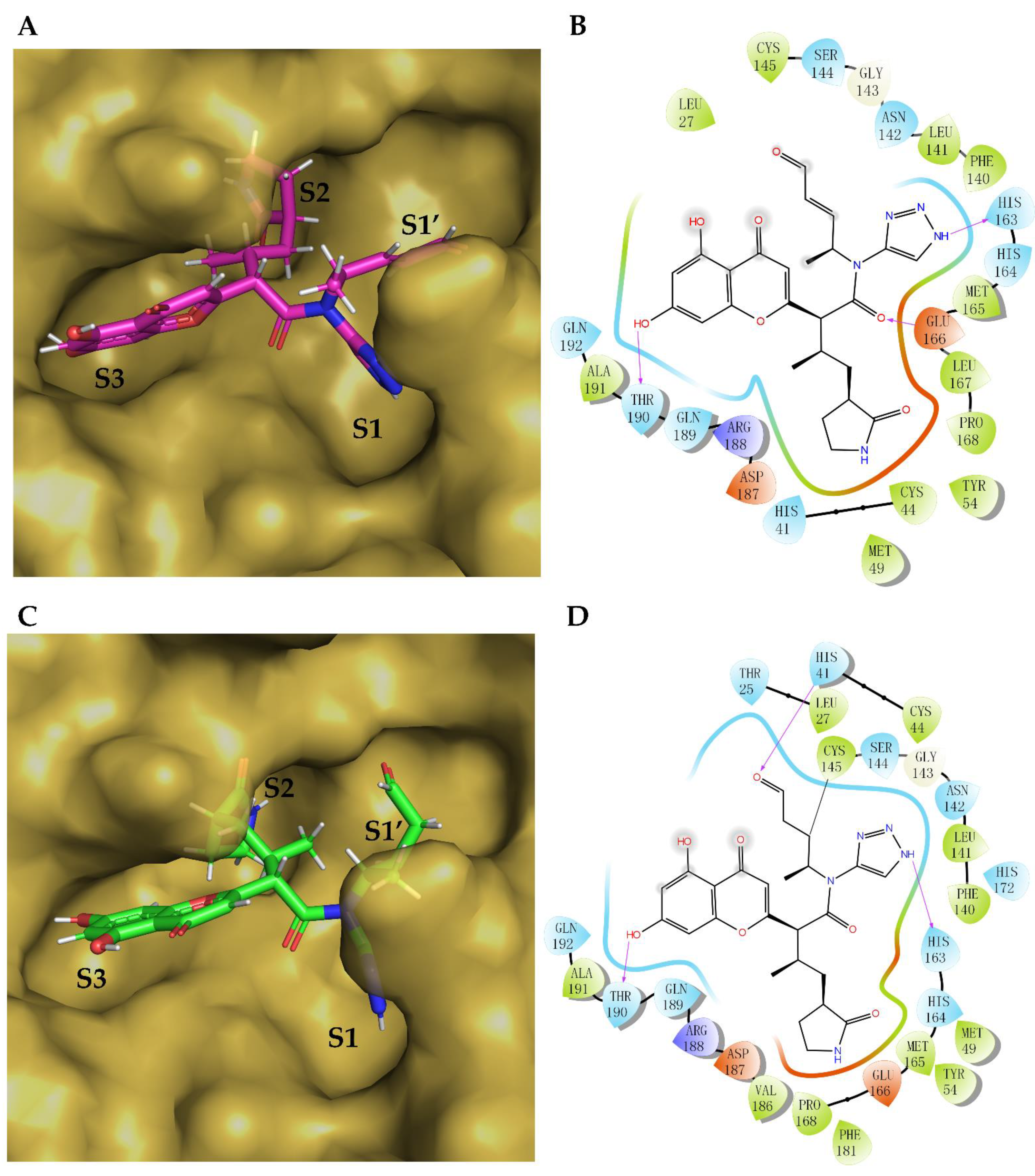
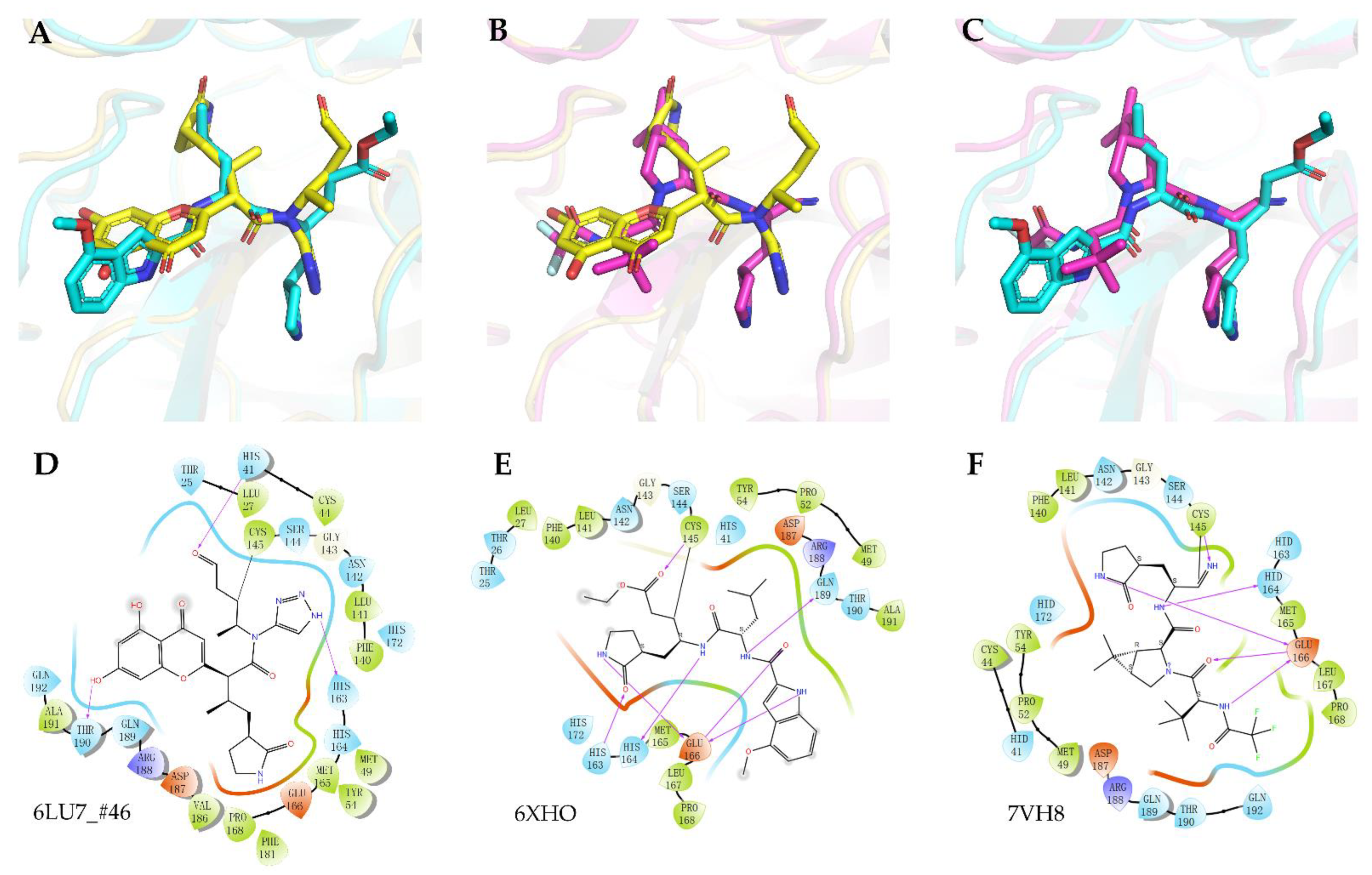
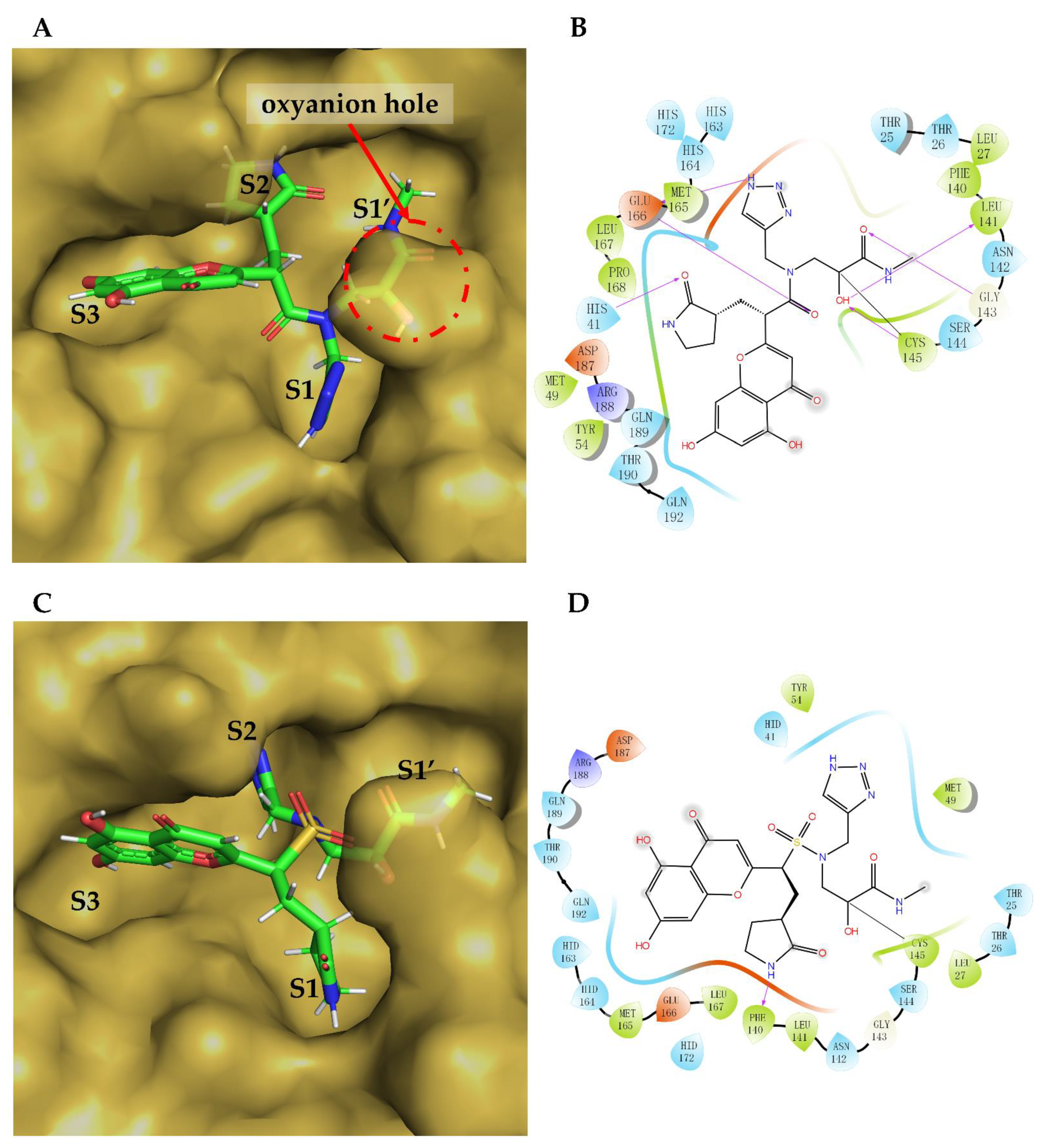
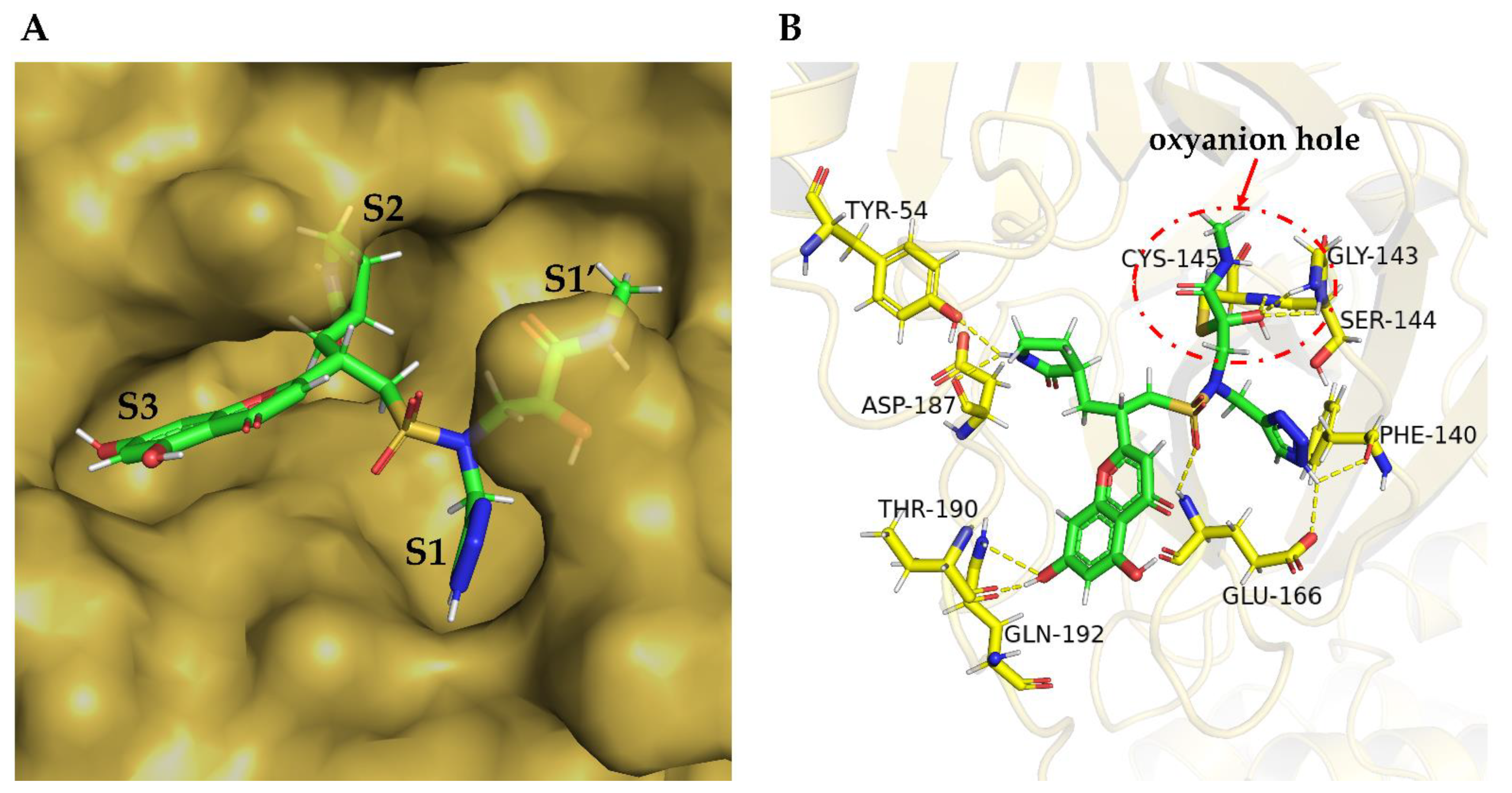
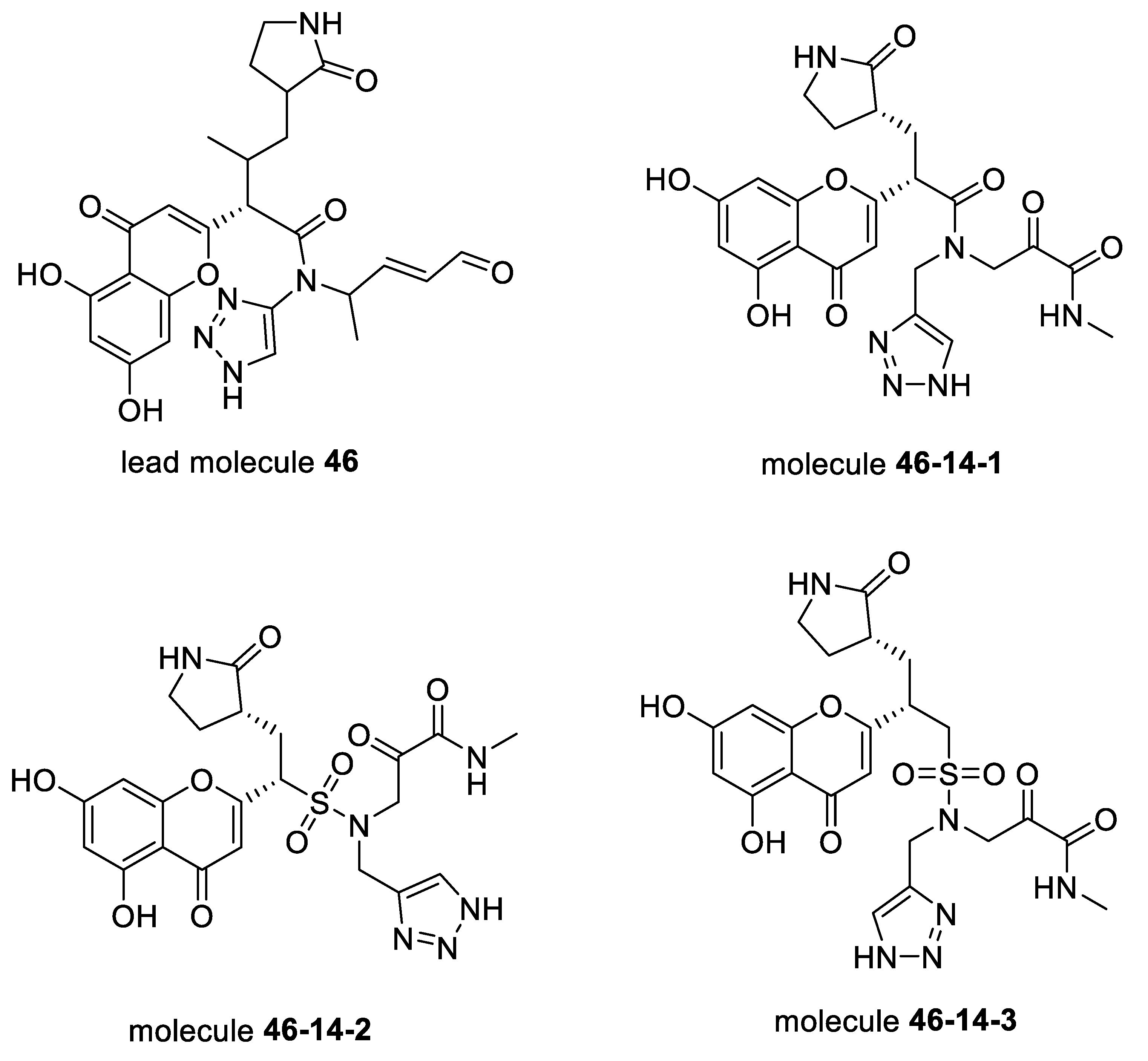
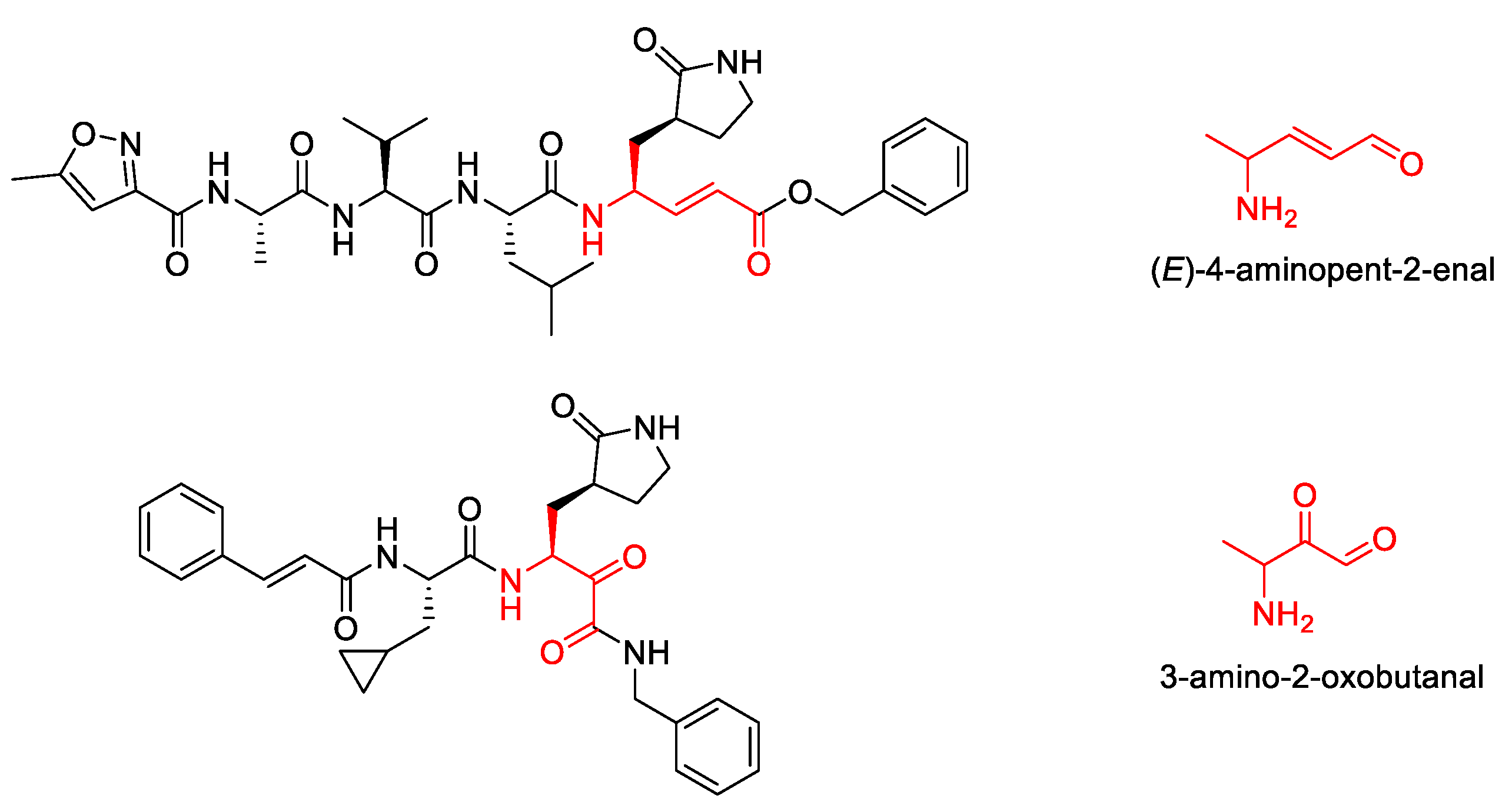
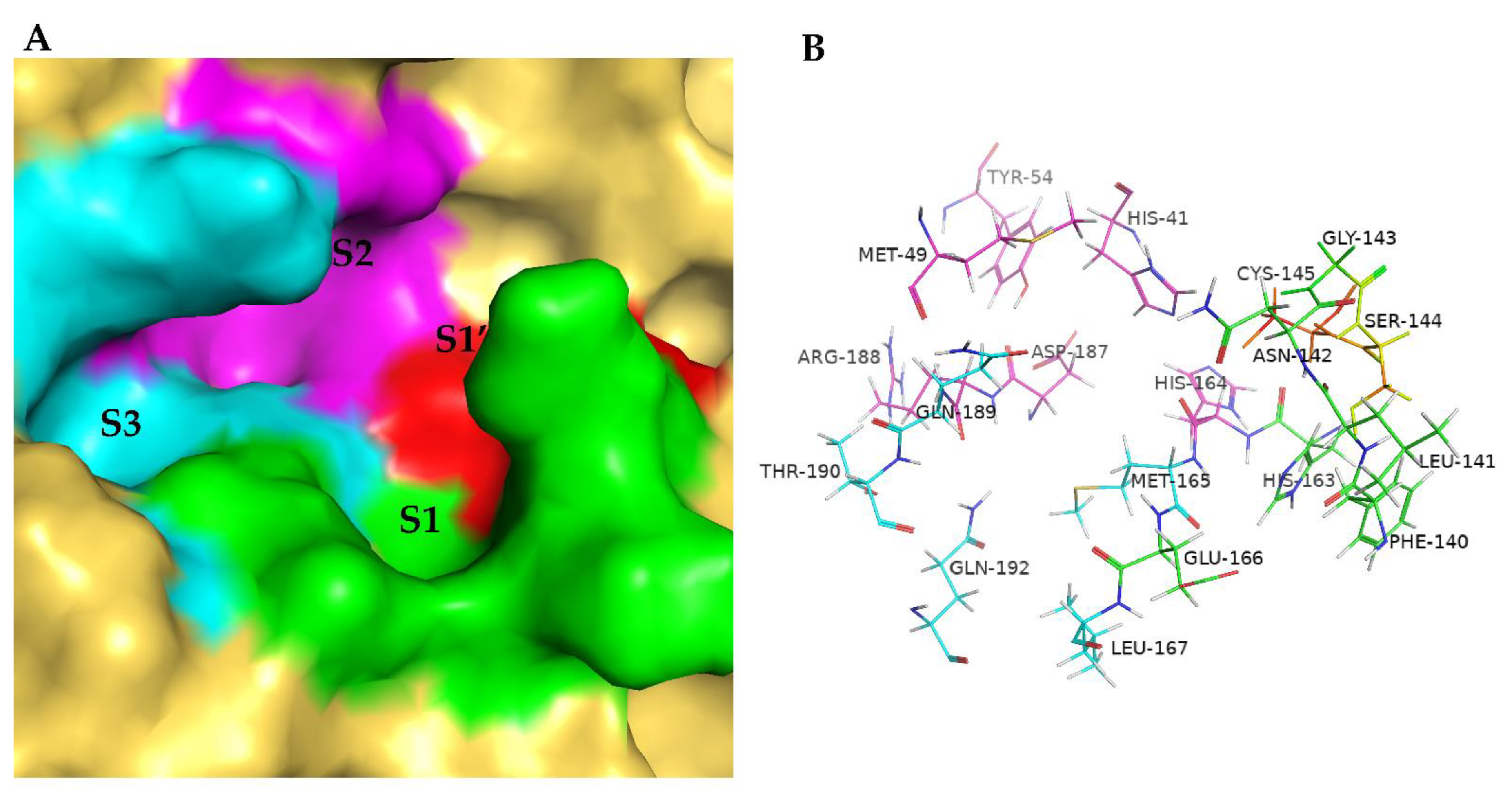
2. Results
3. Discussion
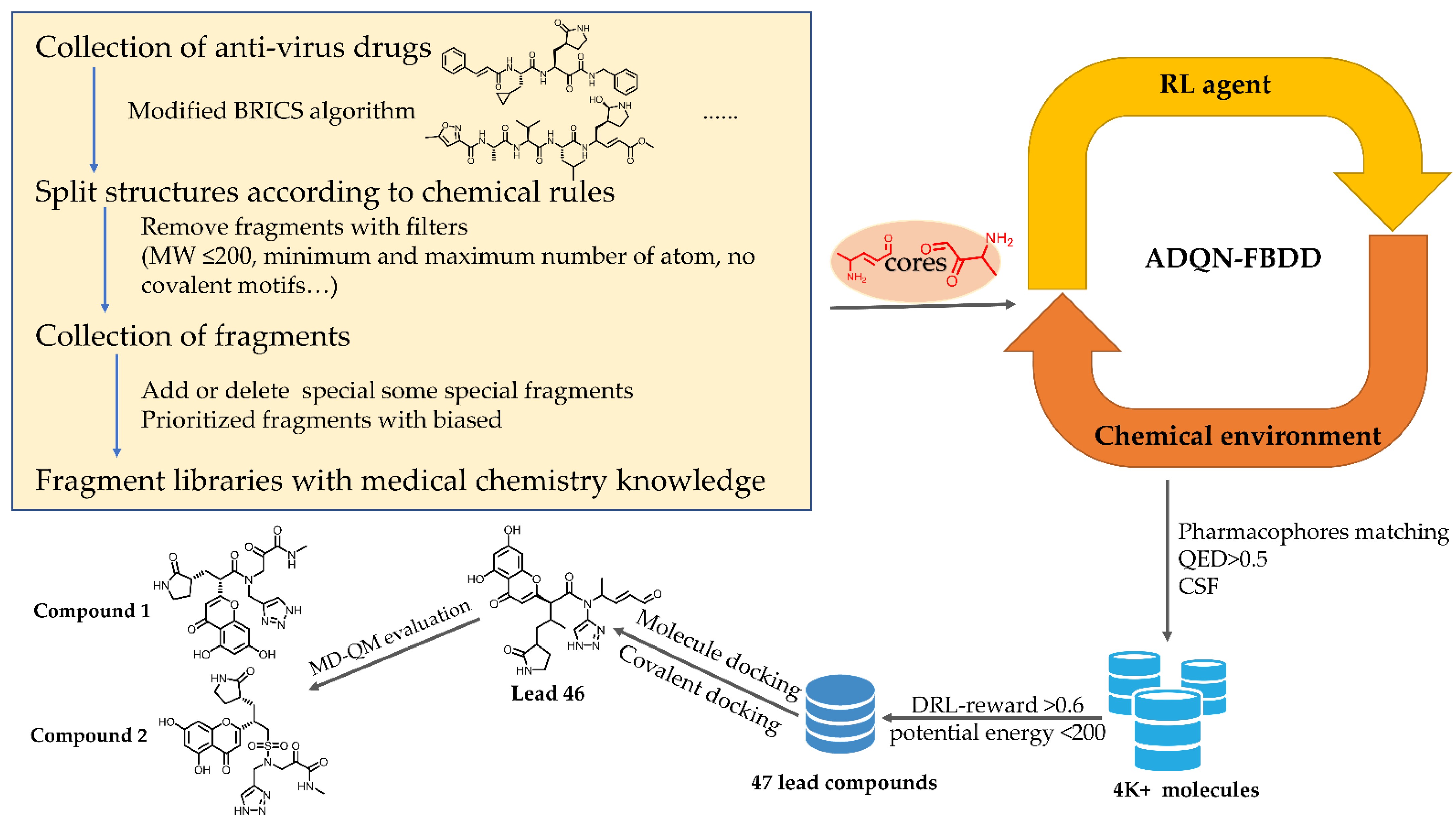
4. Methods
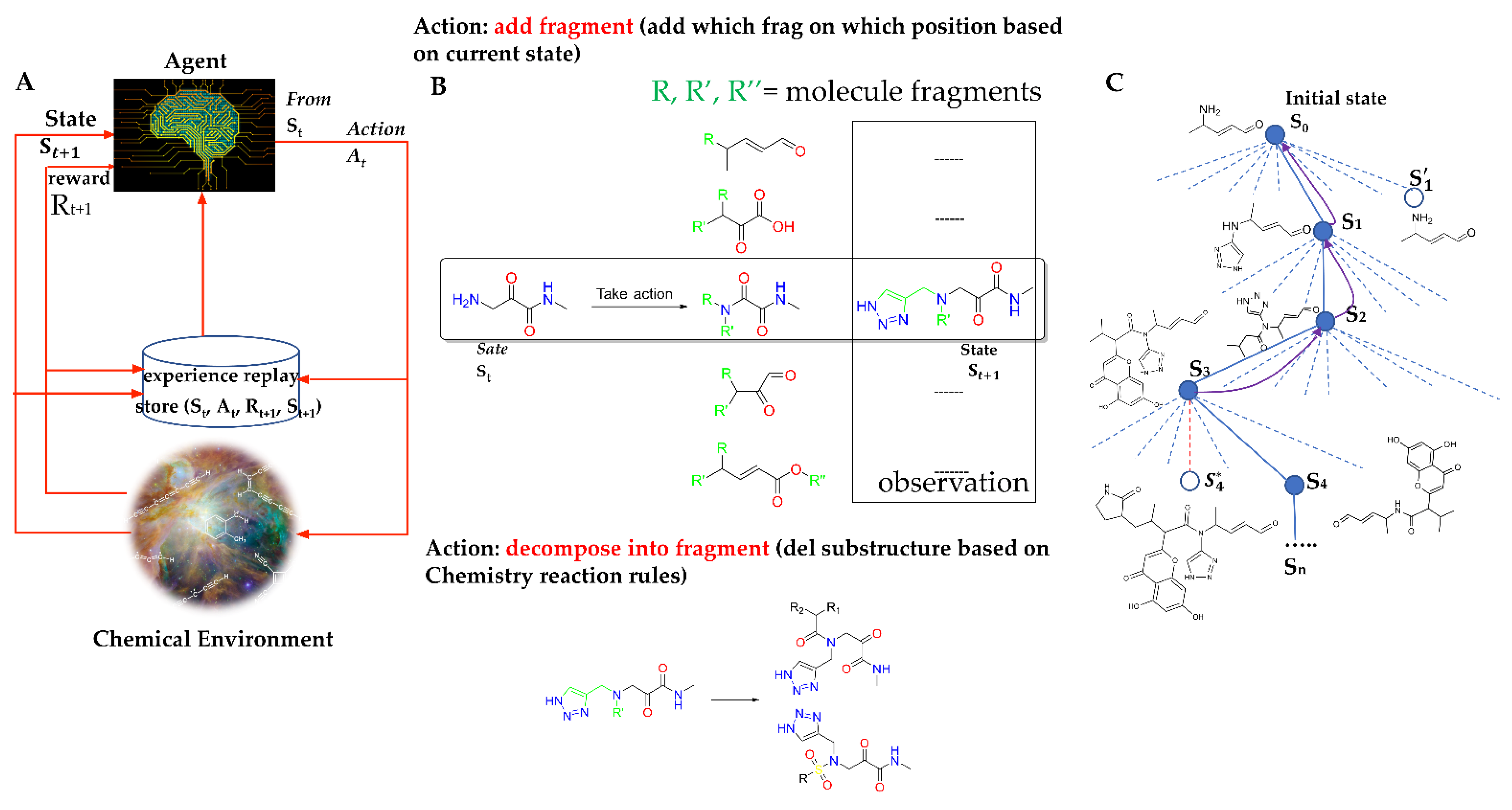
4.1. Markov Decision Process (MDP) for Molecule Generation
4.2. Chemical Environment Design
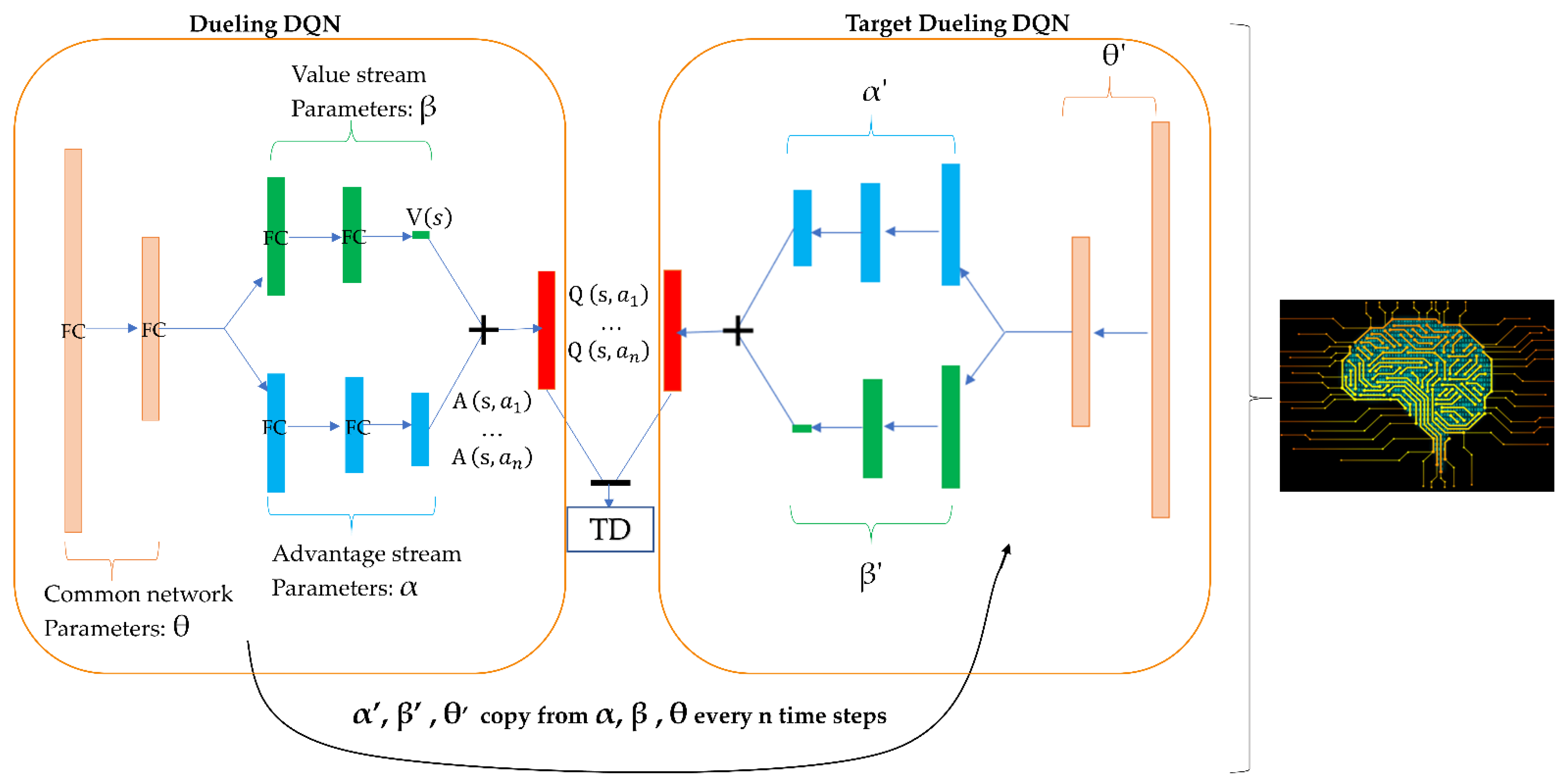
4.3. Agent Design
4.4. Prioritized Experience Replay
4.5. Fragment Library Design
4.6. Core Selection
4.7. Reward Design
4.8. Molecule Generation and Selection
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gorbalenya, A.E. Severe acute respiratory syndrome-related coronavirus–The species and its viruses, a statement of the Coronavirus Study Group. BioRxiv 2020, 937862. [Google Scholar] [CrossRef]
- Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE. Available online: https://coronavirus.jhu.edu/ (accessed on 19 May 2022).
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. Alpha-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef] [PubMed]
- Jun, C.; Yun, L.; Xiuhong, X.; Ping, L.; Feng, L.; Tao, L.; Zhiyin, S.; Mei, W.; Yinzhong, S.; Hongzhou, L. Efficacy study of lopinavir, ritonavir and abirater for the treatment of COVID-19. Chin. J. Anim. Infect. Dis. 2020, 38, 86–89. [Google Scholar]
- Liu, X.; Wang, X.-J. Potential inhibitors for 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom. 2020, 47, 119–121. [Google Scholar] [CrossRef]
- Yang, H.T.; Xie, W.Q.; Xue, X.Y.; Yang, K.L.; Ma, J.; Liang, W.X.; Zhao, Q.; Zhou, Z.; Pei, D.Q.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e428. [Google Scholar]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Tuley, A.; Fast, W. The taxonomy of covalent inhibitors. Biochemistry 2018, 57, 3326–3337. [Google Scholar] [CrossRef]
- Jain, R.P.; Pettersson, H.I.; Zhang, J.; Aull, K.D.; Fortin, P.D.; Huitema, C.; Eltis, L.D.; Parrish, J.C.; James, M.N.G.; Wishart, D.S.; et al. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J. Med. Chem. 2004, 47, 6113–6116. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Jan, J.-T.; Ma, S.-H.; Kuo, C.-J.; Juan, H.-F.; Cheng, Y.-S.E.; Hsu, H.-H.; Huang, H.-C.; Wu, D.; Brik, A.; et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Xi, K.; Ratia, K.; Santarsiero, B.D.; Fu, W.; Harcourt, B.H.; Rota, P.A.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Design and Synthesis of Peptidomimetic Severe Acute Respiratory Syndrome Chymotrypsin-like Protease Inhibitors. J. Med. Chem. 2005, 48, 6767–6771. [Google Scholar] [CrossRef]
- Shie, J.-J.; Fang, J.-M.; Kuo, C.-J.; Kuo, T.-H.; Liang, P.-H.; Huang, H.-J.; Yang, W.-B.; Lin, C.-H.; Chen, J.-L.; Wu, A.Y.-T.; et al. Discovery of Potent Anilide Inhibitors against the Severe Acute Respiratory Syndrome 3CL Protease. J. Med. Chem. 2005, 48, 4469–4473. [Google Scholar] [CrossRef] [PubMed]
- Shie, J.-J.; Fang, J.-M.; Kuo, T.-H.; Kuo, C.-J.; Liang, P.-H.; Huang, H.-J.; Wu, Y.-T.; Jan, J.-T.; Cheng, Y.-S.E.; Wong, C.-H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α, β-unsaturated esters. Bioorganic Med. Chem. 2005, 48, 4469–4473. [Google Scholar] [CrossRef] [PubMed]
- Al-Gharabli, S.I.; Shah, S.T.A.; Weik, S.; Schmidt, M.F.; Mesters, J.R.; Kuhn, D.; Klebe, G.; Hilgenfeld, R.; Rademann, J. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS-CoV Mpro) inhibitors. ChemBioChem 2006, 7, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Lu, I.-L.; Mahindroo, N.; Liang, P.-H.; Peng, Y.-H.; Kuo, C.-J.; Tsai, K.-C.; Hsieh, H.-P.; Chao, Y.-S.; Wu, S.-Y. Structure-Based Drug Design and Structural Biology Study of Novel Nonpeptide Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Main Protease. J. Med. Chem. 2006, 49, 5154–5161. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-C.; Chen, S.-Y.; Liang, P.-H.; Lu, I.-L.; Mahindroo, N.; Hsieh, H.-P.; Chao, Y.-S.; Liu, L.; Liu, D.; Lien, W. Discovery of a novel family of SARS-CoV protease inhibitors by virtual screening and 3D-QSAR studies. J. Med. Chem. 2006, 49, 3485–3495. [Google Scholar] [CrossRef]
- Wu, C.-Y.; King, K.-Y.; Kuo, C.-J.; Fang, J.-M.; Wu, Y.-T.; Ho, M.-Y.; Liao, C.-L.; Shie, J.-J.; Liang, P.-H.; Wong, C.-H. Stable Benzotriazole Esters as Mechanism-Based Inactivators of the Severe Acute Respiratory Syndrome 3CL Protease. Chem. Biol. 2006, 13, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Akaji, K.; Konno, H.; Onozuka, M.; Makino, A.; Saito, H.; Nosaka, K. Evaluation of peptide-aldehyde inhibitors using R188I mutant of SARS 3CL protease as a proteolysis-resistant mutant. Bioorganic Med. Chem. 2008, 16, 9400–9408. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Gong, G.; Grum-Tokars, V.; Mulhearn, D.C.; Baker, S.C.; Coughlin, M.; Prabhakar, B.S.; Sleeman, K.; Johnson, M.E.; Mesecar, A.D. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 5684–5688. [Google Scholar] [CrossRef]
- Shao, Y.-M.; Yang, W.-B.; Kuo, T.-H.; Tsai, K.-C.; Lin, C.-H.; Yang, A.-S.; Liang, P.-H.; Wong, C.-H. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorganic Med. Chem. 2008, 16, 4652–4660. [Google Scholar] [CrossRef]
- Kuo, C.-J.; Liu, H.-G.; Lo, Y.-K.; Seong, C.-M.; Lee, K.-I.; Jung, Y.-S.; Liang, P.-H. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009, 583, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Ramajayam, R.; Tan, K.-P.; Liu, H.-G.; Liang, P.-H. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorganic Med. Chem. 2010, 18, 7849–7854. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.B.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, J.-Y.; Kim, D.; Naguyen, T.T.H.; Park, S.-J.; Chang, J.S.; Park, K.H. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CLpro inhibition. Bioorganic Med. Chem. 2010, 18, 7940–7947. [Google Scholar] [CrossRef] [PubMed]
- Akaji, K.; Konno, H.; Mitsui, H.; Teruya, K.; Shimamoto, Y.; Hattori, Y.; Ozaki, T.; Kusunoki, M.; Sanjoh, A. Structure-Based Design, Synthesis, and Evaluation of Peptide-Mimetic SARS 3CL Protease Inhibitors. J. Med. Chem. 2011, 54, 7962–7973. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013, 56, 534–546. [Google Scholar]
- Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles’ heel”: Current effective inhibitor targeting a 3C-like protease. Protein Cell 2013, 4, 248. [Google Scholar] [CrossRef]
- Thanigaimalai, P.; Konno, S.; Yamamoto, T.; Koiwai, Y.; Taguchi, A.; Takayama, K.; Yakushiji, F.; Akaji, K.; Chen, S.-E. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: Design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013, 68, 372–384. [Google Scholar] [CrossRef]
- Turlington, M.; Chun, A.; Tomar, S.; Eggler, A.; Grum-Tokars, V.; Jacobs, J.; Daniels, J.S.; Dawson, E.; Saldanha, A.; Chase, P. Discovery of N-(benzo [1,2,3] triazol-1-yl)-N-(benzyl) acetamido) phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: Identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorganic Med. Chem. Lett. 2013, 23, 6172–6177. [Google Scholar] [CrossRef]
- Kumar, V.; Shin, J.S.; Shie, J.-J.; Ku, K.B.; Kim, C.; Go, Y.Y.; Huang, K.-F.; Kim, M.; Liang, P.-H. Identification and evaluation of potent Middle East respiratory syndrome coronavirus (MERS-CoV) 3CLPro inhibitors. Antivir. Res. 2017, 141, 101–106. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction Tree Variational Autoencoder for Molecular Graph Generation. In Artificial Intelligence in Drug Discovery; RSC Publishing: London, UK, 2021; Volume 11, pp. 228–249. [Google Scholar] [CrossRef]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2017, 9, 513–530. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Z.; Jiang, M.; Wang, S.; Zhang, S.; Wei, Z. Molecule Property Prediction Based on Spatial Graph Embedding. J. Chem. Inf. Model. 2019, 59, 3817–3828. [Google Scholar] [CrossRef]
- Liu, K.; Sun, X.; Jia, L.; Ma, J.; Xing, H.; Wu, J.; Gao, H.; Sun, Y.; Boulnois, F.; Fan, J. Chemi-Net: A Molecular Graph Convolutional Network for Accurate Drug Property Prediction. Int. J. Mol. Sci. 2019, 20, 3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, J.X.; Liu, B.W.; Ying, R.; Pande, V.; Leskovec, J. Graph Convolutional Policy Network for Goal-Directed Molecular Graph Generation. Adv. Neural Inf. Processing Syst. 2018, 31. [Google Scholar] [CrossRef]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Model. 2020, 60, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, G.L.; Sánchez-Lengeling, B.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Zhou, Z.; Kearnes, S.; Li, L.; Zare, R.N.; Riley, P. Optimization of Molecules via Deep Reinforcement Learning. Sci. Rep. 2019, 9, 10752. [Google Scholar] [CrossRef]
- Tang, B.; Kramer, S.T.; Fang, M.; Qiu, Y.; Wu, Z.; Xu, D. A self-attention based message passing neural network for predicting molecular lipophilicity and aqueous solubility. J. Cheminform. 2020, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- ADQN–FBDD. Available online: https://github.com/tbwxmu/2019-nCov (accessed on 19 May 2022).
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, M. Assessing the impact of generative AI on medicinal chemistry. Nat. Biotechnol. 2020, 38, 143–145. [Google Scholar] [CrossRef]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hasselt, H.; Guez, A.; Silver, D. Deep Reinforcement Learning with Double Q-Learning. In Proceedings of the Thirtieth AAAI Conference on Artificial Intelligence, Phoenix, AZ, USA, 12–17 February 2016; pp. 2094–2100. [Google Scholar]
- Simonini, T. Improvements in Deep Q Learning: Dueling Double DQN, Prioritized Experience Replay, and Fixed Q-Targets. 2018. Available online: https://www.freecodecamp.org/news/improvements-in-deep-q-learning-dueling-double-dqn-prioritized-experience-replay-and-fixed-58b130cc5682/ (accessed on 6 July 2018).
- Schaul, T.; Quan, J.; Antonoglou, I.; Silver, D. Prioritized Experience Replay. arXiv 2015, arXiv:1511.05952. [Google Scholar]
- Brockman, G.; Cheung, V.; Pettersson, L.; Schneider, J.; Schulman, J.; Tang, J.; Zaremba, W. OpenAI Gym. arXiv 2016, arXiv:1606.01540. [Google Scholar]
- Speck-Planche, A. Recent advances in fragment-based computational drug design: Tackling simultaneous targets/biological effects. Futur. Med. Chem. 2018, 10, 2021–2024. [Google Scholar] [CrossRef] [Green Version]
- Varin, T.; Schuffenhauer, A.; Ertl, P.; Renner, S. Mining for Bioactive Scaffolds with Scaffold Networks: Improved Compound Set Enrichment from Primary Screening Data. J. Chem. Inf. Model. 2011, 51, 1528–1538. [Google Scholar] [CrossRef]
- Schuffenhauer, A.; Ertl, P.; Roggo, S.; Wetzel, S.; Koch, M.A.; Waldmann, H. The Scaffold Tree − Visualization of the Scaffold Universe by Hierarchical Scaffold Classification. J. Chem. Inf. Model. 2006, 47, 47–58. [Google Scholar] [CrossRef]
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.; Chung, P.W. Deep learning for molecular design—a review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Duan, Y.; et al. Structure-based drug design, virtual screening and high-throughput screening rapidly identify antiviral leads targeting COVID-19. bioRxiv 2020, 964882. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Representative Tool | Method | Training/Performance |
|---|---|---|
| RNNs [35] | Recurrent neural networks | Extensive training data are required to learn the context of the atom composition; may generate many invalid SMILES |
| GENTRL [36] | Variational-autoencoder-based models | Large training data are required to learn the atom distribution from training SMILES; may still produce some invalid SMILES |
| ORGAN [37],ORGANIC [38] | Generative adversarial networks | Use adversarial attacks to enhance the success rate; however, this still cannot guarantee valid SMILES |
| MolDQN [39] | Reinforcement-learning-based models | Employ self-learning without training data and all generated SMILES are valid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.; He, F.; Liu, D.; He, F.; Wu, T.; Fang, M.; Niu, Z.; Wu, Z.; Xu, D. AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2. Biomolecules 2022, 12, 746. https://doi.org/10.3390/biom12060746
Tang B, He F, Liu D, He F, Wu T, Fang M, Niu Z, Wu Z, Xu D. AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2. Biomolecules. 2022; 12(6):746. https://doi.org/10.3390/biom12060746
Chicago/Turabian StyleTang, Bowen, Fengming He, Dongpeng Liu, Fei He, Tong Wu, Meijuan Fang, Zhangming Niu, Zhen Wu, and Dong Xu. 2022. "AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2" Biomolecules 12, no. 6: 746. https://doi.org/10.3390/biom12060746