
Extracellular Vesicles from Steatotic Hepatocytes Provoke Pro-Fibrotic Responses in Cultured Stellate Cells

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples, Reagents and Cell Lines
2.2. Isolation of EVs
2.3. Quantification of EVs
2.4. Cryo-Transmission Electron Microscopy (Cryo-TEM)
2.5. Cell Migration in a Boyden Chamber
2.6. Qualitative and Quantitative DNA Analysis by qRT-PCR
2.7. Protein Concentration Determination
2.8. SDS-PAGE and Western Blot
2.9. Cell Viability
2.10. Oil-Red-O and Phalloidin Staining
2.11. Cell Proliferation Measurement
2.12. Flow Cytometry Analysis of CCR5 Surface Expression
2.13. Statistics
3. Results
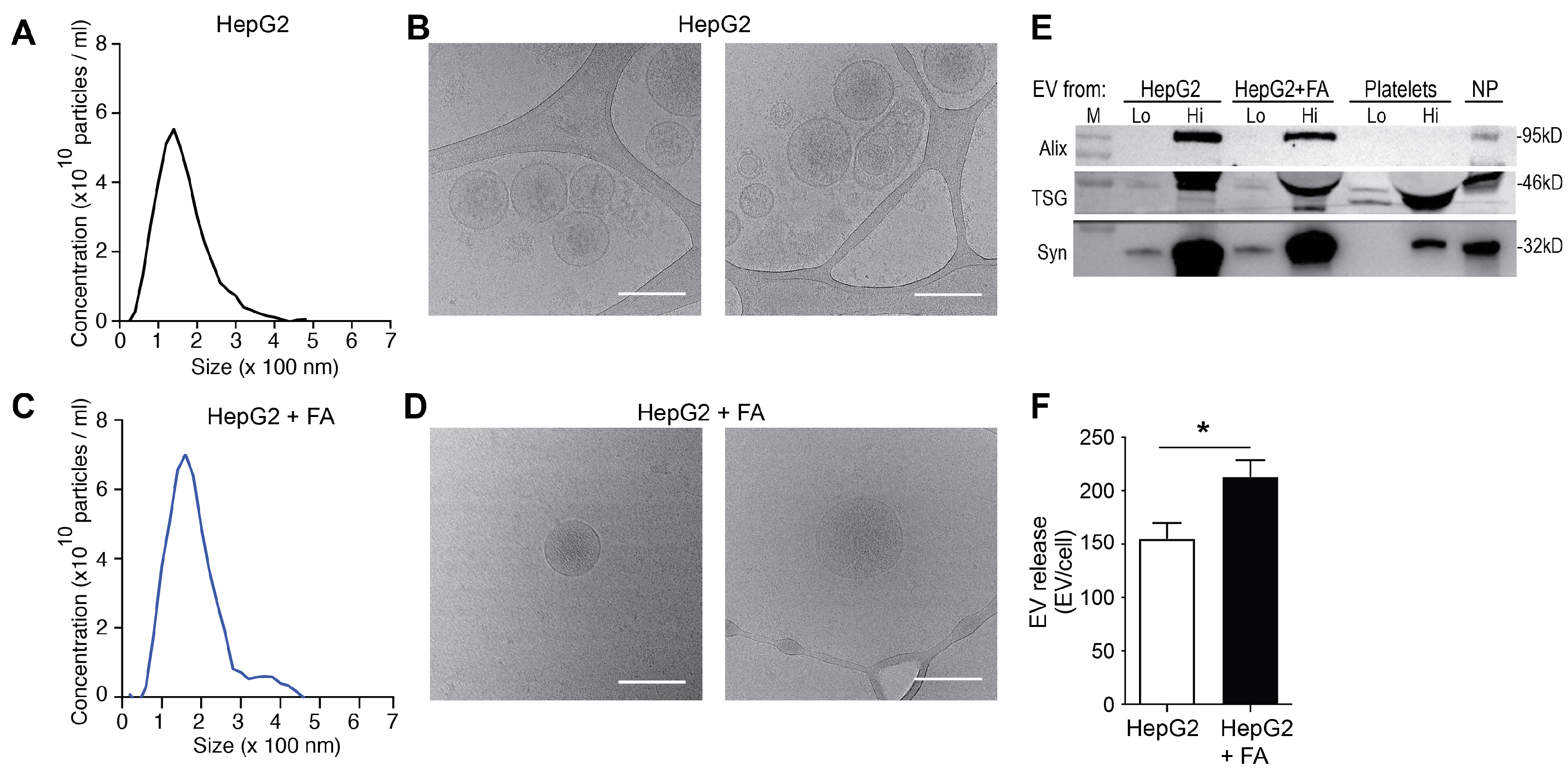
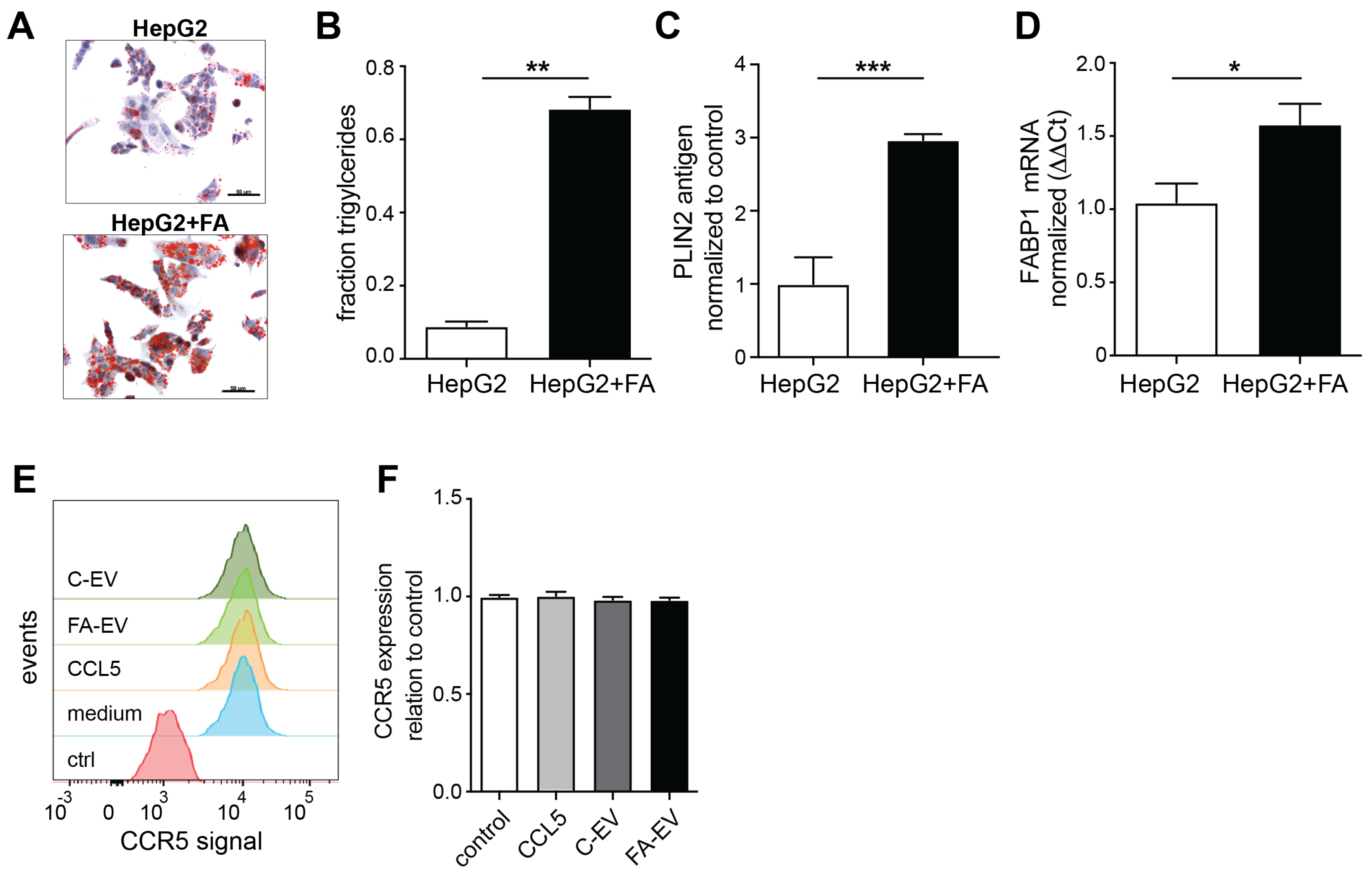
3.1. HepG2 Cells Release Increased Amounts of EVs under Steatogenic Conditions
3.2. EVs from HepG2 Cells Modulate Migration of Stellate Cells
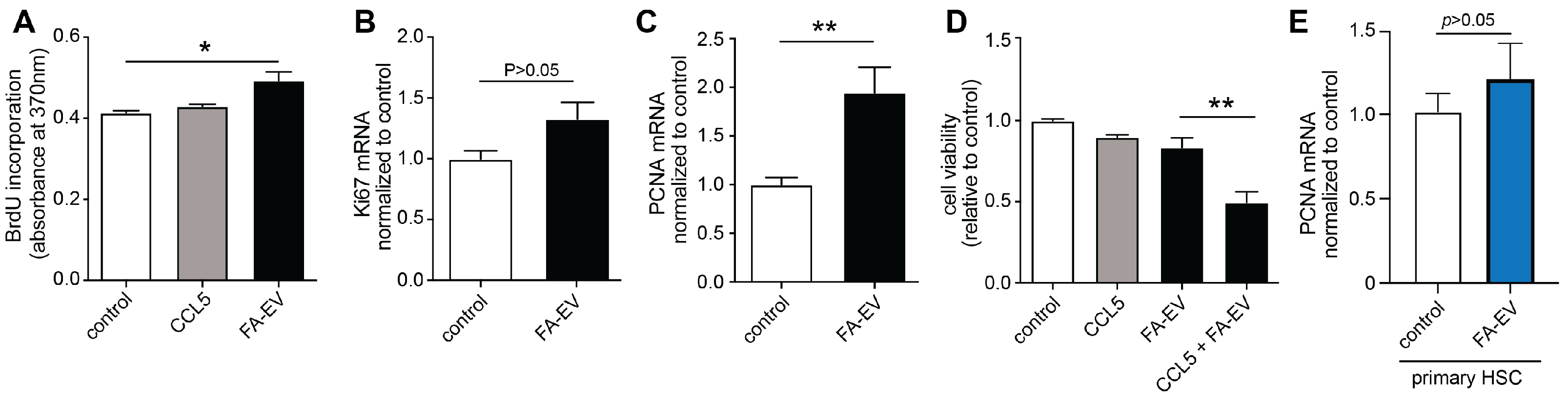
3.3. EVs from HepG2 Cells Increase the Proliferation of TWNT-4 Stellate Cells
3.4. EVs from Steatotic HepG2 Modulate the Expression of Matrix Myofibroblast Markers and Remodeling Markers in Stellate Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Walenbergh, S.M.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol. 2013, 58, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A.; Luedde, T. The transition from inflammation to cancer in the liver. Clin. Liver Dis. 2016, 8, 89–93. [Google Scholar] [CrossRef]
- Cordeiro, A.; Costa, R.; Andrade, N.; Silva, C.; Canabrava, N.; Pena, M.J.; Rodrigues, I.; Andrade, S.; Ramalho, A. Does adipose tissue inflammation drive the development of non-alcoholic fatty liver disease in obesity? Clin. Res. Hepatol. Gastroenterol. 2020, 44, 394–402. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clement, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Hellerbrand, C.; Stefanovic, B.; Giordano, F.; Burchardt, E.R.; Brenner, D.A. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J. Hepatol. 1999, 30, 77–87. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, F.; Hammerich, L.; Storch, D.; Bartneck, M.; Huss, S.; Russeler, V.; Gassler, N.; Lira, S.A.; Luedde, T.; Trautwein, C.; et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology 2012, 55, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Berres, M.L.; Koenen, R.R.; Rueland, A.; Zaldivar, M.M.; Heinrichs, D.; Sahin, H.; Schmitz, P.; Streetz, K.L.; Berg, T.; Gassler, N.; et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Investig. 2010, 120, 4129–4140. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell. Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Aranda, E.; Hayakawa, Y.; Bhanja, P.; Atay, S.; Brodin, N.P.; Li, J.; Asfaha, S.; Liu, L.; Tailor, Y.; et al. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat. Commun. 2016, 7, 13096. [Google Scholar] [CrossRef]
- Vajen, T.; Benedikter, B.J.; Heinzmann, A.C.A.; Vasina, E.M.; Henskens, Y.; Parsons, M.; Maguire, P.B.; Stassen, F.R.; Heemskerk, J.W.M.; Schurgers, L.J.; et al. Platelet extracellular vesicles induce a pro-inflammatory smooth muscle cell phenotype. J. Extracell. Vesicles 2017, 6, 1322454. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.S.; Lee, Y.S.; Park, S.H.; Jang, M.J.; Jo, E.; Kim, S.C.; Han, Y.M.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Watanabe, T.; Okitsu, T.; Sakaguchi, M.; Takesue, M.; Kunieda, T.; Omoto, K.; Yamamoto, S.; Tanaka, N.; Kobayashi, N. Establishment of an immortalized human hepatic stellate cell line to develop antifibrotic therapies. Cell Transpl. 2003, 12, 499–507. [Google Scholar] [CrossRef]
- Koenen, R.R.; Christella, M.; Thomassen, L.G.; Tans, G.; Rosing, J.; Hackeng, T.M. Effect of oral contraceptives on the anticoagulant activity of protein S in plasma. Thromb. Haemost. 2005, 93, 853–859. [Google Scholar] [CrossRef]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrichs, D.; Berres, M.L.; Coeuru, M.; Knauel, M.; Nellen, A.; Fischer, P.; Philippeit, C.; Bucala, R.; Trautwein, C.; Wasmuth, H.E.; et al. Protective role of macrophage migration inhibitory factor in nonalcoholic steatohepatitis. FASEB J. 2014, 28, 5136–5147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugeratski, F.G.; Hodge, K.; Lilla, S.; McAndrews, K.M.; Zhou, X.; Hwang, R.F.; Zanivan, S.; Kalluri, R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat. Cell. Biol. 2021, 23, 631–641. [Google Scholar] [CrossRef]
- Guo, Q.; Furuta, K.; Lucien, F.; Gutierrez Sanchez, L.H.; Hirsova, P.; Krishnan, A.; Kabashima, A.; Pavelko, K.D.; Madden, B.; Alhuwaish, H.; et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J. Hepatol. 2019, 71, 1193–1205. [Google Scholar] [CrossRef]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.H.; Hirsova, P.; Tomita, K.; Bronk, S.F.; Werneburg, N.W.; Harrison, S.A.; Goodfellow, V.S.; Malhi, H.; Gores, G.J. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016, 63, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, S.Y.; Ko, E.; Lee, J.H.; Yi, H.S.; Yoo, Y.J.; Je, J.; Suh, S.J.; Jung, Y.K.; Kim, J.H.; et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 2017, 7, 3710. [Google Scholar] [CrossRef]
- Luo, X.; Luo, S.Z.; Xu, Z.X.; Zhou, C.; Li, Z.H.; Zhou, X.Y.; Xu, M.Y. Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J. Gastroenterol. 2021, 27, 1419–1434. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef] [PubMed]
- Lepreux, S.; Desmouliere, A. Human liver myofibroblasts during development and diseases with a focus on portal (myo)fibroblasts. Front. Physiol. 2015, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, E.; Perumal, E.; Subramaniyan, R.; Mustapha, N. Liver fibrosis: Extracellular vesicles mediated intercellular communication in perisinusoidal space. Hepatology, 2021; 1–11, ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Chiabotto, G.; Ceccotti, E.; Tapparo, M.; Camussi, G.; Bruno, S. Human Liver Stem Cell-Derived Extracellular Vesicles Target Hepatic Stellate Cells and Attenuate Their Pro-fibrotic Phenotype. Front. Cell Dev. Biol. 2021, 9, 777462. [Google Scholar] [CrossRef] [PubMed]
- Diehl, P.; Nienaber, F.; Zaldivia, M.T.K.; Stamm, J.; Siegel, P.M.; Mellett, N.A.; Wessinger, M.; Wang, X.; McFadyen, J.D.; Bassler, N.; et al. Lysophosphatidylcholine is a Major Component of Platelet Microvesicles Promoting Platelet Activation and Reporting Atherosclerotic Plaque Instability. Thromb. Haemost. 2019, 119, 1295–1310. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell. 2019, 75, 644–660.e5. [Google Scholar] [CrossRef]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Species, Type | Supplier |
|---|---|---|
| Alix | mouse monoclonal | Bio-Rad, OTI1A4 |
| -actin | rabbit monoclonal | Cell Signal. Tech., D6A8 |
| CCR5 | rat monoclonal | ThermoFisher, HEK/1/85a |
| Collagen I-V | rabbit polyclonal | Sanbio BV, PS046 |
| GAPDH | mouse monoclonal | Bio-Rad, 6C5 |
| GFAP | rabbit polyclonal | Abcam, ab7260 |
| PLIN1 | rabbit monoclonal | Abcam, EPR3753(2) |
| -SMA | rabbit monoclonal | Cell Signal. Tech., D4K9N |
| Synthenin | rabbit monoclonal | Abcam, EPR8102 |
| TSG101 | rabbit polyclonal | Sigma, T5701 |
| Vimentin | rabbit monoclonal | Abcam, EPR3776 |
| Anti-mouse-peroxidase | goat polyclonal | Agilent, P044701-2 |
| Anti-rabbit-peroxidase | goat polyclonal | Agilent, P044801-2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koenen, M.T.; Brandt, E.F.; Kaczor, D.M.; Caspers, T.; Heinzmann, A.C.A.; Fischer, P.; Heinrichs, D.; Wirtz, T.H.; Trautwein, C.; Koenen, R.R.; et al. Extracellular Vesicles from Steatotic Hepatocytes Provoke Pro-Fibrotic Responses in Cultured Stellate Cells. Biomolecules 2022, 12, 698. https://doi.org/10.3390/biom12050698
Koenen MT, Brandt EF, Kaczor DM, Caspers T, Heinzmann ACA, Fischer P, Heinrichs D, Wirtz TH, Trautwein C, Koenen RR, et al. Extracellular Vesicles from Steatotic Hepatocytes Provoke Pro-Fibrotic Responses in Cultured Stellate Cells. Biomolecules. 2022; 12(5):698. https://doi.org/10.3390/biom12050698
Chicago/Turabian StyleKoenen, Maria Teresa, Elisa Fabiana Brandt, Dawid Marcin Kaczor, Tim Caspers, Alexandra Catharina Anna Heinzmann, Petra Fischer, Daniel Heinrichs, Theresa Hildegard Wirtz, Christian Trautwein, Rory R Koenen, and et al. 2022. "Extracellular Vesicles from Steatotic Hepatocytes Provoke Pro-Fibrotic Responses in Cultured Stellate Cells" Biomolecules 12, no. 5: 698. https://doi.org/10.3390/biom12050698