
Cla4p Kinase Activity Is Down-Regulated by Fus3p during Yeast Mating


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains, Plasmids, and General Methods
2.2. Microscopy and Imaging Analysis
2.3. Screening for Pheromone and Fus3p-Specific Phosphorylation Sites of Cla4p
2.4. In Vitro Cla4p Kinase Assay
3. Results
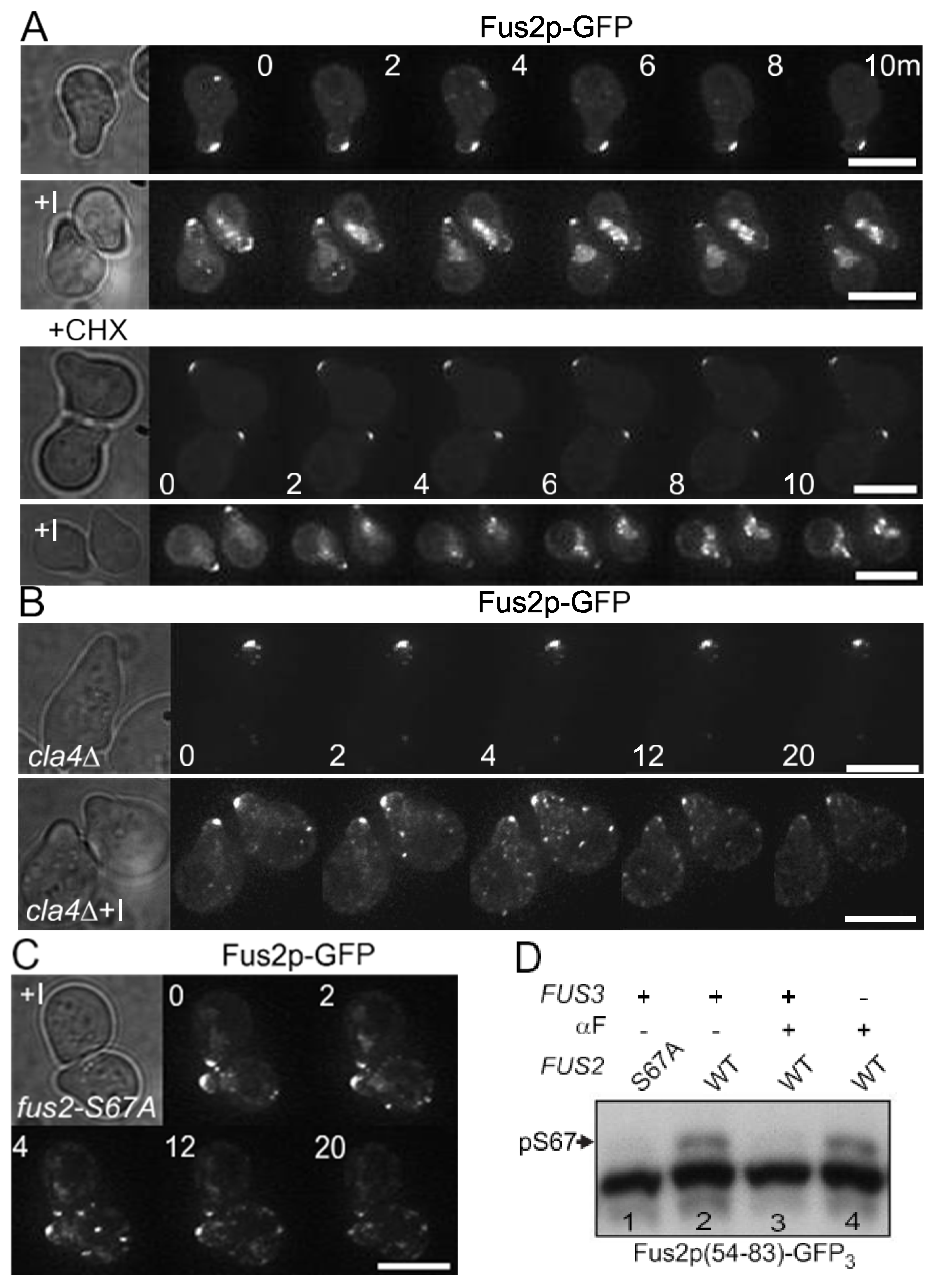
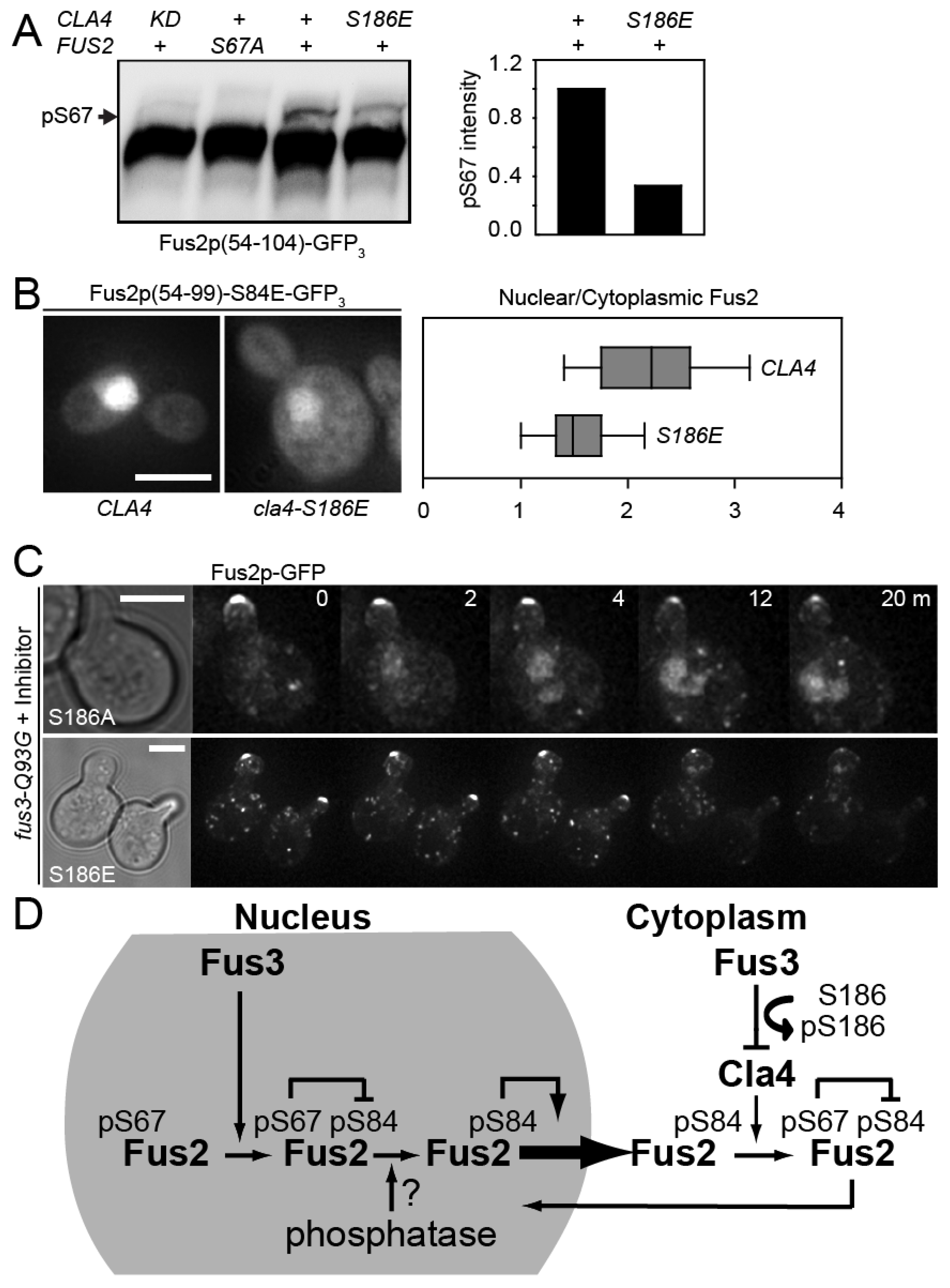
3.1. Fus3p Down-Regulates Fus2p-S67 Phosphorylation by Cla4p during Mating
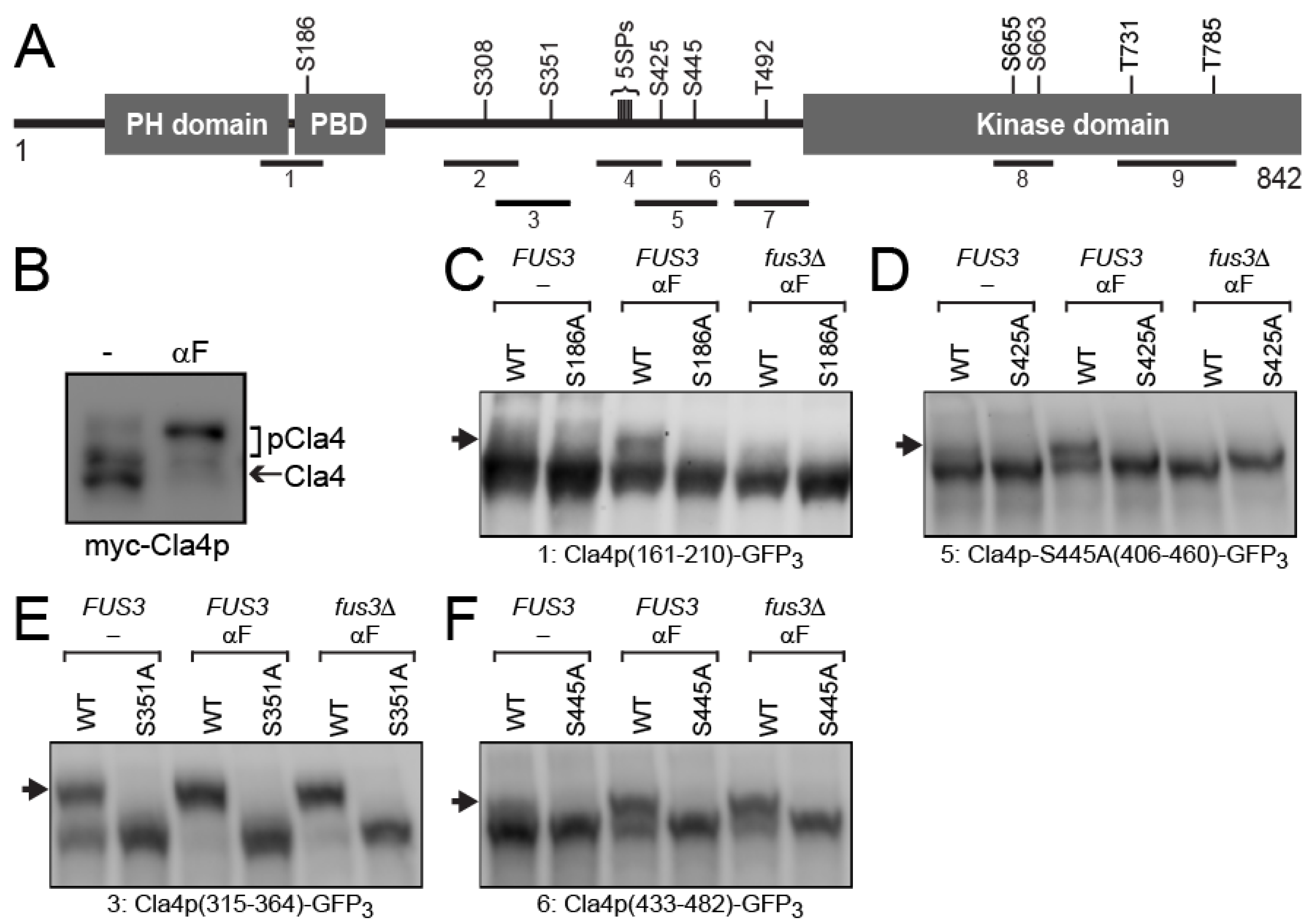
3.2. Cla4p-S186 and -S425 Are Phosphorylated Specifically by Fus3p
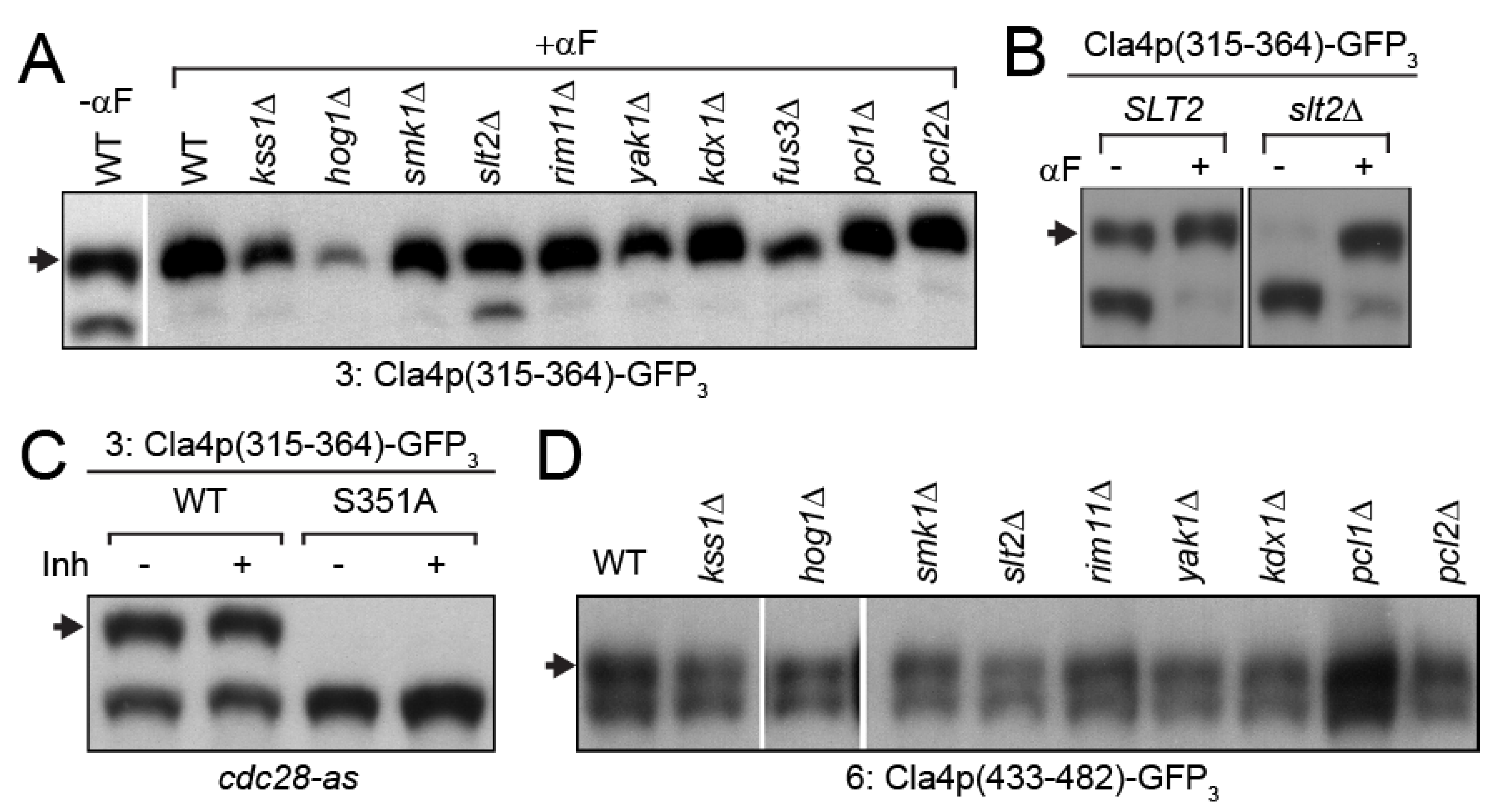
3.3. MAP-Kinase Slt2p Phosphorylates Cla4p-S351
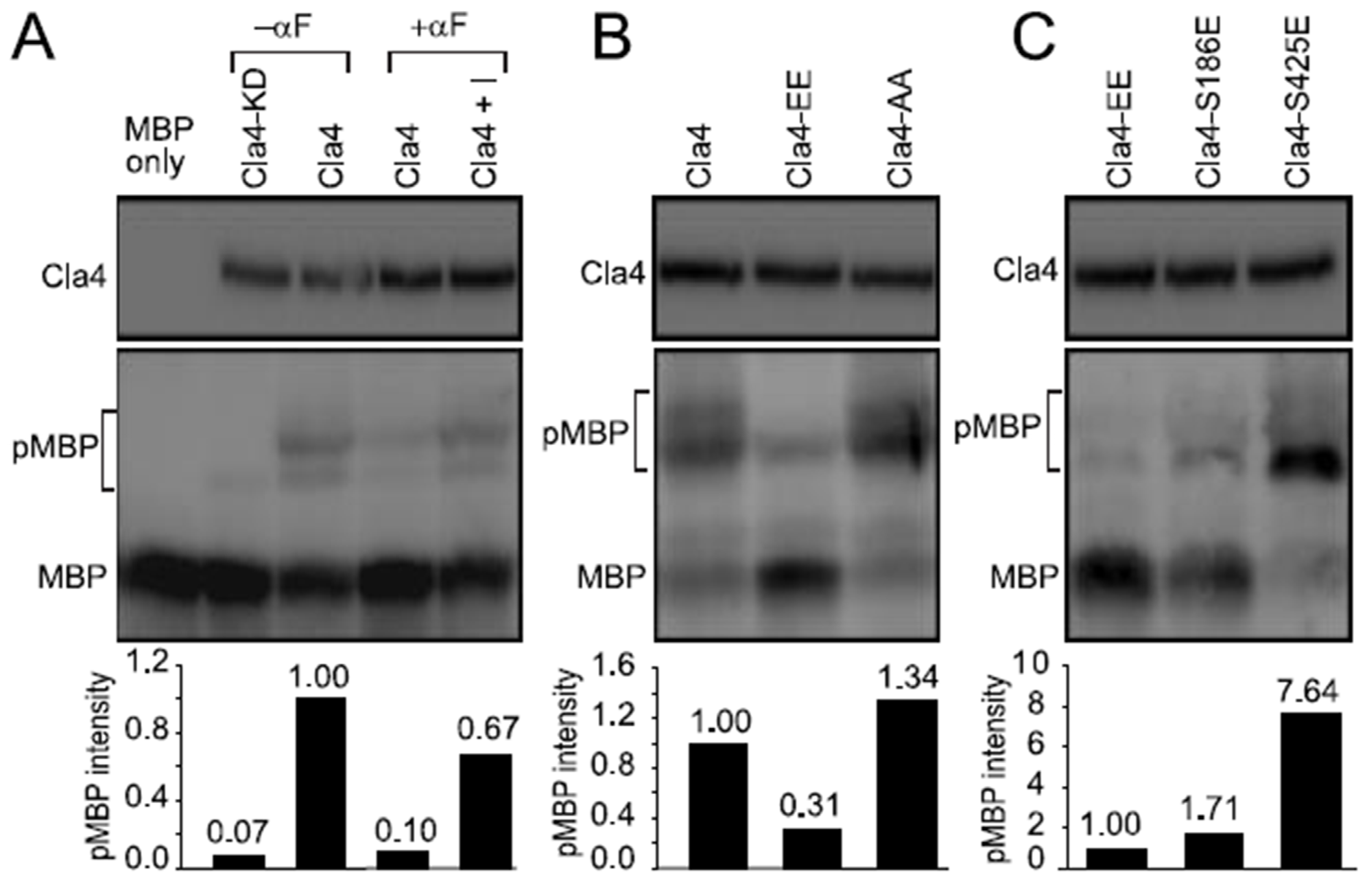
3.4. Phosphorylation of Cla4p-S186 Negatively Regulates Cla4p Kinase Activity In Vitro
3.5. Cla4p-S186 Phosphorylation Regulates Fus2p Phosphorylation and Nuclear Localization In Vivo
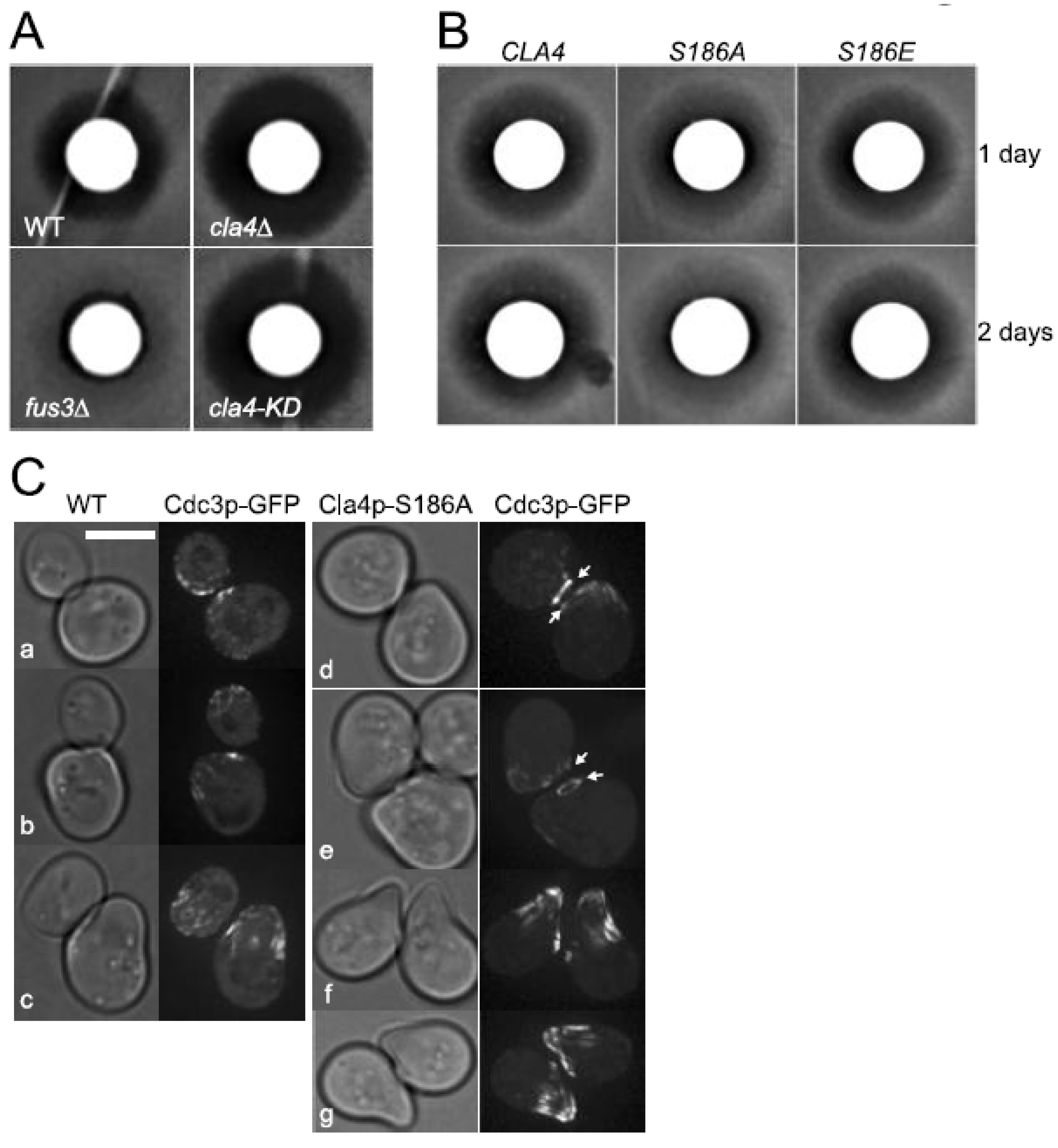
3.6. Biological Function of the Inhibitory Phosphorylation of Cla4p by Fus3p Is Not Limited to Fus2p Localization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hofmann, C.; Shepelev, M.; Chernoff, J. The genetics of Pak. J. Cell Sci. 2004, 117, 4343–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardwell, L. A walk-through of the yeast mating pheromone response pathway. Peptides 2005, 26, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Gancedo, J.M. Control of pseudohyphae formation in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2001, 25, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Raitt, D.C.; Posas, F.; Saito, H. Yeast Cdc42 GTPase and Ste20 PAK-like kinase regulate Sho1-dependent activation of the Hog1 MAPK pathway. EMBO J. 2000, 19, 4623–4631. [Google Scholar] [CrossRef] [PubMed]
- Benton, B.K.; Tinkelenberg, A.; Gonzalez, I.; Cross, F.R. Cla4p, a Saccharomyces cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol. Cell. Biol. 1997, 17, 5067–5076. [Google Scholar] [CrossRef] [Green Version]
- Cvrckova, F.; De Virgilio, C.; Manser, E.; Pringle, J.R.; Nasmyth, K. Ste20-like protein kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes Dev. 1995, 9, 1817–1830. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Lee, S.F.; Furmaniak-Kazmierczak, E.; Cote, G.P.; Thomas, D.Y.; Leberer, E. Activation of myosin-I by members of the Ste20p protein kinase family. J. Biol. Chem. 1996, 271, 31787–31790. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Lytvyn, V.; Thomas, D.Y.; Leberer, E. The phosphorylation site for Ste20p-like protein kinases is essential for the function of myosin-I in yeast. J. Biol. Chem. 1997, 272, 30623–30626. [Google Scholar] [CrossRef] [Green Version]
- Wild, A.C.; Yu, J.W.; Lemmon, M.A.; Blumer, K.J. The p21-activated protein kinase-related kinase Cla4 is a coincidence detector of signaling by Cdc42 and phosphatidylinositol 4-phosphate. J. Biol. Chem. 2004, 279, 17101–17110. [Google Scholar] [CrossRef] [Green Version]
- Kozubowski, L.; Saito, K.; Johnson, J.M.; Howell, A.S.; Zyla, T.R.; Lew, D.J. Symmetry-breaking polarization driven by a Cdc42p GEF-PAK complex. Curr. Biol. CB 2008, 18, 1719–1726. [Google Scholar] [CrossRef] [Green Version]
- Moran, K.D.; Kang, H.; Araujo, A.V.; Zyla, T.R.; Saito, K.; Tsygankov, D.; Lew, D.J. Cell-cycle control of cell polarity in yeast. J. Cell Biol. 2019, 218, 171–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, I.; Irazoqui, J.E.; Moskow, J.J.; Bardes, E.S.; Zyla, T.R.; Lew, D.J. Assembly of scaffold-mediated complexes containing Cdc42p, the exchange factor Cdc24p, and the effector Cla4p required for cell cycle-regulated phosphorylation of Cdc24p. J. Biol. Chem. 2001, 276, 7176–7186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulli, M.P.; Jaquenoud, M.; Shimada, Y.; Niederhauser, G.; Wiget, P.; Peter, M. Phosphorylation of the Cdc42 exchange factor Cdc24 by the PAK-like kinase Cla4 may regulate polarized growth in yeast. Mol. Cell 2000, 6, 1155–1167. [Google Scholar] [CrossRef]
- Dobbelaere, J.; Gentry, M.S.; Hallberg, R.L.; Barral, Y. Phosphorylation-dependent regulation of septin dynamics during the cell cycle. Dev. Cell 2003, 4, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Kadota, J.; Yamamoto, T.; Yoshiuchi, S.; Bi, E.; Tanaka, K. Septin ring assembly requires concerted action of polarisome components, a PAK kinase Cla4p, and the actin cytoskeleton in Saccharomyces cerevisiae. Mol. Biol. Cell 2004, 15, 5329–5345. [Google Scholar] [CrossRef]
- Schmidt, M.; Varma, A.; Drgon, T.; Bowers, B.; Cabib, E. Septins, under Cla4p regulation, and the chitin ring are required for neck integrity in budding yeast. Mol. Biol. Cell 2003, 14, 2128–2141. [Google Scholar] [CrossRef] [Green Version]
- Versele, M.; Thorner, J. Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J. Cell Biol. 2004, 164, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.L.; Bishop, A.C.; Shokat, K.M.; Drubin, D.G. Chemical genetic analysis of the budding-yeast p21-activated kinase Cla4p. Nat. Cell Biol. 2000, 2, 677–685. [Google Scholar] [CrossRef]
- Sakchaisri, K.; Asano, S.; Yu, L.R.; Shulewitz, M.J.; Park, C.J.; Park, J.E.; Cho, Y.W.; Veenstra, T.D.; Thorner, J.; Lee, K.S. Coupling morphogenesis to mitotic entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4124–4129. [Google Scholar] [CrossRef] [Green Version]
- Hofken, T.; Schiebel, E. A role for cell polarity proteins in mitotic exit. EMBO J. 2002, 21, 4851–4862. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.; Geymonat, M.; Johnson, A.L.; Segal, M.; Johnston, L.H. Spatial regulation of the guanine nucleotide exchange factor Lte1 in Saccharomyces cerevisiae. J. Cell Sci. 2002, 115, 4977–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshan, A.; Bardin, A.J.; Amon, A. Control of Lte1 localization by cell polarity determinants and Cdc14. Curr. Biol. CB 2002, 12, 2098–2110. [Google Scholar] [CrossRef] [Green Version]
- Tjandra, H.; Compton, J.; Kellogg, D. Control of mitotic events by the Cdc42 GTPase, the Clb2 cyclin and a member of the PAK kinase family. Curr. Biol. CB 1998, 8, 991–1000. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Rose, M.D. A mechanism for the coordination of proliferation and differentiation by spatial regulation of Fus2p in budding yeast. Genes Dev. 2012, 26, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Paterson, J.M.; Ydenberg, C.A.; Rose, M.D. Dynamic localization of yeast Fus2p to an expanding ring at the cell fusion junction during mating. J. Cell Biol. 2008, 181, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Ydenberg, C.A.; Rose, M.D. Antagonistic regulation of Fus2p nuclear localization by pheromone signaling and the cell cycle. J. Cell Biol. 2009, 184, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Brizzio, V.; Gammie, A.E.; Rose, M.D. Rvs161p interacts with Fus2p to promote cell fusion in Saccharomyces cerevisiae. J. Cell Biol. 1998, 141, 567–584. [Google Scholar] [CrossRef] [Green Version]
- Ydenberg, C.A.; Stein, R.A.; Rose, M.D. Cdc42p and Fus2p act together late in yeast cell fusion. Mol. Biol. Cell 2012, 23, 1208–1218. [Google Scholar] [CrossRef]
- Smith, J.A.; Hall, A.E.; Rose, M.D. Membrane curvature directs the localization of Cdc42p to novel foci required for cell-cell fusion. J. Cell Biol. 2017, 216, 3971–3980. [Google Scholar] [CrossRef] [Green Version]
- Longtine, M.S.; McKenzie, A., 3rd; Demarini, D.J.; Shah, N.G.; Wach, A.; Brachat, A.; Philippsen, P.; Pringle, J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998, 14, 953–961. [Google Scholar] [CrossRef]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.C.; Ubersax, J.A.; Petsch, D.T.; Matheos, D.P.; Gray, N.S.; Blethrow, J.; Shimizu, E.; Tsien, J.Z.; Schultz, P.G.; Rose, M.D.; et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000, 407, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Matheos, D.; Metodiev, M.; Muller, E.; Stone, D.; Rose, M.D. Pheromone-induced polarization is dependent on the Fus3p MAPK acting through the formin Bni1p. J. Cell Biol. 2004, 165, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elion, E.A. Pheromone response, mating and cell biology. Curr. Opin. Microbiol. 2000, 3, 573–581. [Google Scholar] [CrossRef]
- Brinkworth, R.I.; Munn, A.L.; Kobe, B. Protein kinases associated with the yeast phosphoproteome. BMC Bioinform. 2006, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Mok, J.; Kim, P.M.; Lam, H.Y.; Piccirillo, S.; Zhou, X.; Jeschke, G.R.; Sheridan, D.L.; Parker, S.A.; Desai, V.; Jwa, M.; et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 2010, 3, ra12. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.S.; O’Shea, E.K. Pho85 and signaling environmental conditions. Trends Biochem. Sci. 2002, 27, 87–93. [Google Scholar] [CrossRef]
- Huang, D.; Friesen, H.; Andrews, B. Pho85, a multifunctional cyclin-dependent protein kinase in budding yeast. Mol. Microbiol. 2007, 66, 303–314. [Google Scholar] [CrossRef]
- Gladfelter, A.S.; Moskow, J.J.; Zyla, T.R.; Lew, D.J. Isolation and characterization of effector-loop mutants of CDC42 in yeast. Mol. Biol. Cell 2001, 12, 1239–1255. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.; Kohler, T.; Mosch, H.U. Role of Cdc42-Cla4 interaction in the pheromone response of Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Ford, S.K.; Pringle, J.R. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: Localization of the CDC11 gene product and the timing of events at the budding site. Dev. Genet. 1991, 12, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haarer, B.K.; Pringle, J.R. Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol. Cell. Biol. 1987, 7, 3678–3687. [Google Scholar] [CrossRef]
- Kim, H.B.; Haarer, B.K.; Pringle, J.R. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: Localization of the CDC3 gene product and the timing of events at the budding site. J. Cell Biol. 1991, 112, 535–544. [Google Scholar] [CrossRef]
- Longtine, M.S.; Fares, H.; Pringle, J.R. Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J. Cell Biol. 1998, 143, 719–736. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Rose, M.D. Yeast mating: Getting close to membrane merger. Curr. Biol. CB 2001, 11, R16–R20. [Google Scholar] [CrossRef] [Green Version]
- Sreenivasan, A.; Kellogg, D. The elm1 kinase functions in a mitotic signaling network in budding yeast. Mol. Cell. Biol. 1999, 19, 7983–7994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, D.; Braberg, H.; Mehta, M.; Chechik, G.; Cagney, G.; Mukherjee, P.; Silva, A.C.; Shales, M.; Collins, S.R.; van Wageningen, S.; et al. Functional organization of the S. cerevisiae phosphorylation network. Cell 2009, 136, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Goehring, A.S.; Mitchell, D.A.; Tong, A.H.; Keniry, M.E.; Boone, C.; Sprague, G.F., Jr. Synthetic lethal analysis implicates Ste20p, a p21-activated potein kinase, in polarisome activation. Mol. Biol. Cell 2003, 14, 1501–1516. [Google Scholar] [CrossRef]
- Tong, A.H.; Lesage, G.; Bader, G.D.; Ding, H.; Xu, H.; Xin, X.; Young, J.; Berriz, G.F.; Brost, R.L.; Chang, M.; et al. Global mapping of the yeast genetic interaction network. Science 2004, 303, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, C.; Zarov, P.; Rambourg, A.; Mann, C. The SLT2 (MPK1) MAP kinase homolog is involved in polarized cell growth in Saccharomyces cerevisiae. J. Cell Biol. 1993, 123, 1821–1833. [Google Scholar] [CrossRef]
- Zarzov, P.; Mazzoni, C.; Mann, C. The SLT2(MPK1) MAP kinase is activated during periods of polarized cell growth in yeast. EMBO J. 1996, 15, 83–91. [Google Scholar] [CrossRef]
- Holt, L.J.; Tuch, B.B.; Villen, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, A.; Bodenmiller, B.; Uotila, A.; Stahl, M.; Wanka, S.; Gerrits, B.; Aebersold, R.; Loewith, R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009, 23, 1929–1943. [Google Scholar] [CrossRef] [Green Version]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villen, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Bi, E.; Chiavetta, J.B.; Chen, H.; Chen, G.C.; Chan, C.S.; Pringle, J.R. Identification of novel, evolutionarily conserved Cdc42p-interacting proteins and of redundant pathways linking Cdc24p and Cdc42p to actin polarization in yeast. Mol. Biol. Cell 2000, 11, 773–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Rose, M.D. Cla4p Kinase Activity Is Down-Regulated by Fus3p during Yeast Mating. Biomolecules 2022, 12, 598. https://doi.org/10.3390/biom12040598
Kim J, Rose MD. Cla4p Kinase Activity Is Down-Regulated by Fus3p during Yeast Mating. Biomolecules. 2022; 12(4):598. https://doi.org/10.3390/biom12040598
Chicago/Turabian StyleKim, Junwon, and Mark D. Rose. 2022. "Cla4p Kinase Activity Is Down-Regulated by Fus3p during Yeast Mating" Biomolecules 12, no. 4: 598. https://doi.org/10.3390/biom12040598