
Insulin Receptors and Insulin Action in the Heart: The Effects of Left Ventricular Assist Devices

 , , , ,
, , , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Diabetic Cardiomyopathy
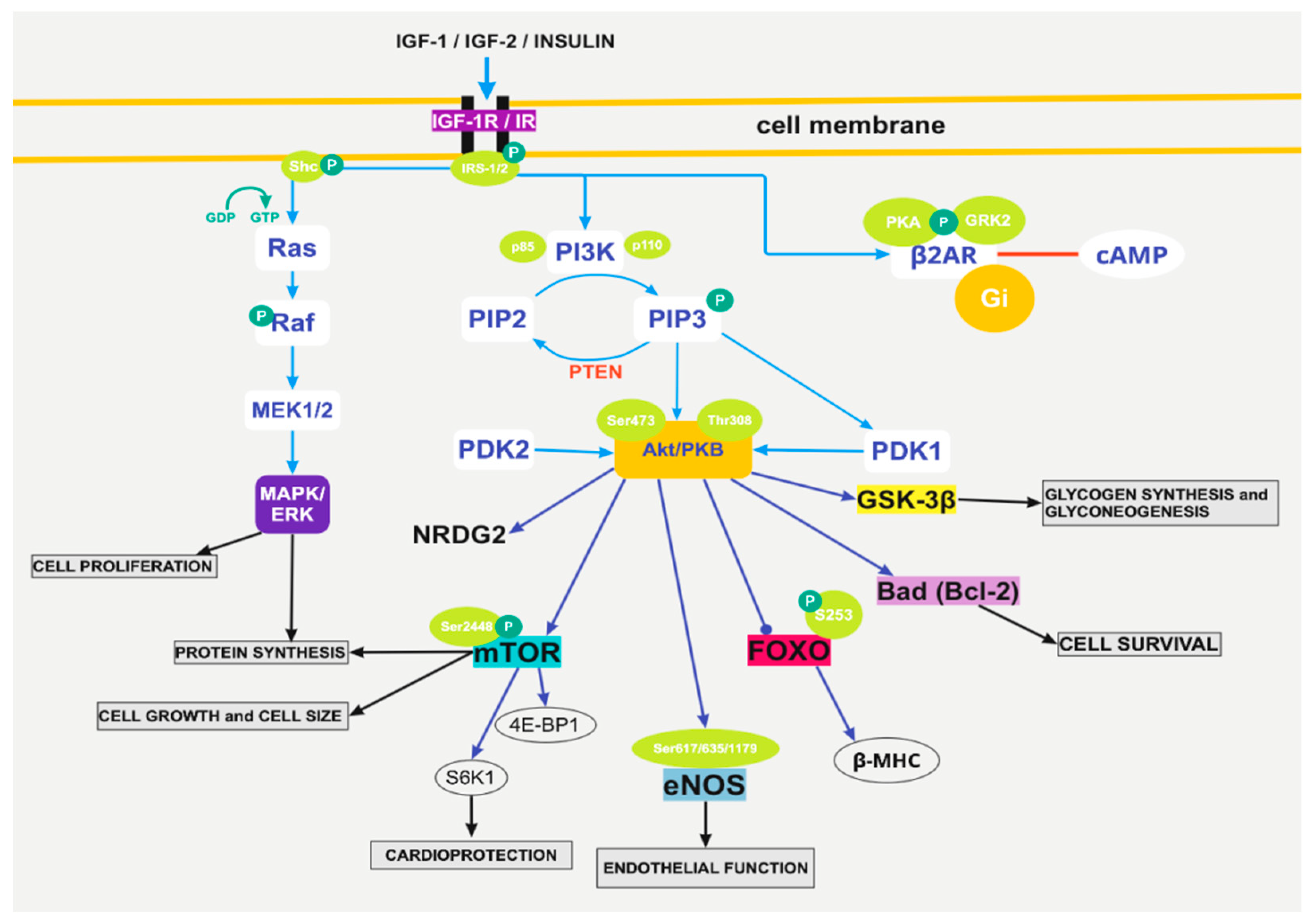
3. The Pathophysiological Pathways of Insulin Action Implicate Akt-mTOR, eNOS, and grk2
3.1. The Activation of PI3K/Akt
3.2. The Pathway of Foxo1
3.3. Insulin Growth Factor 1
3.4. The Pathway of eNOS
3.5. The Action of G-Protein-Coupled Kinase 2
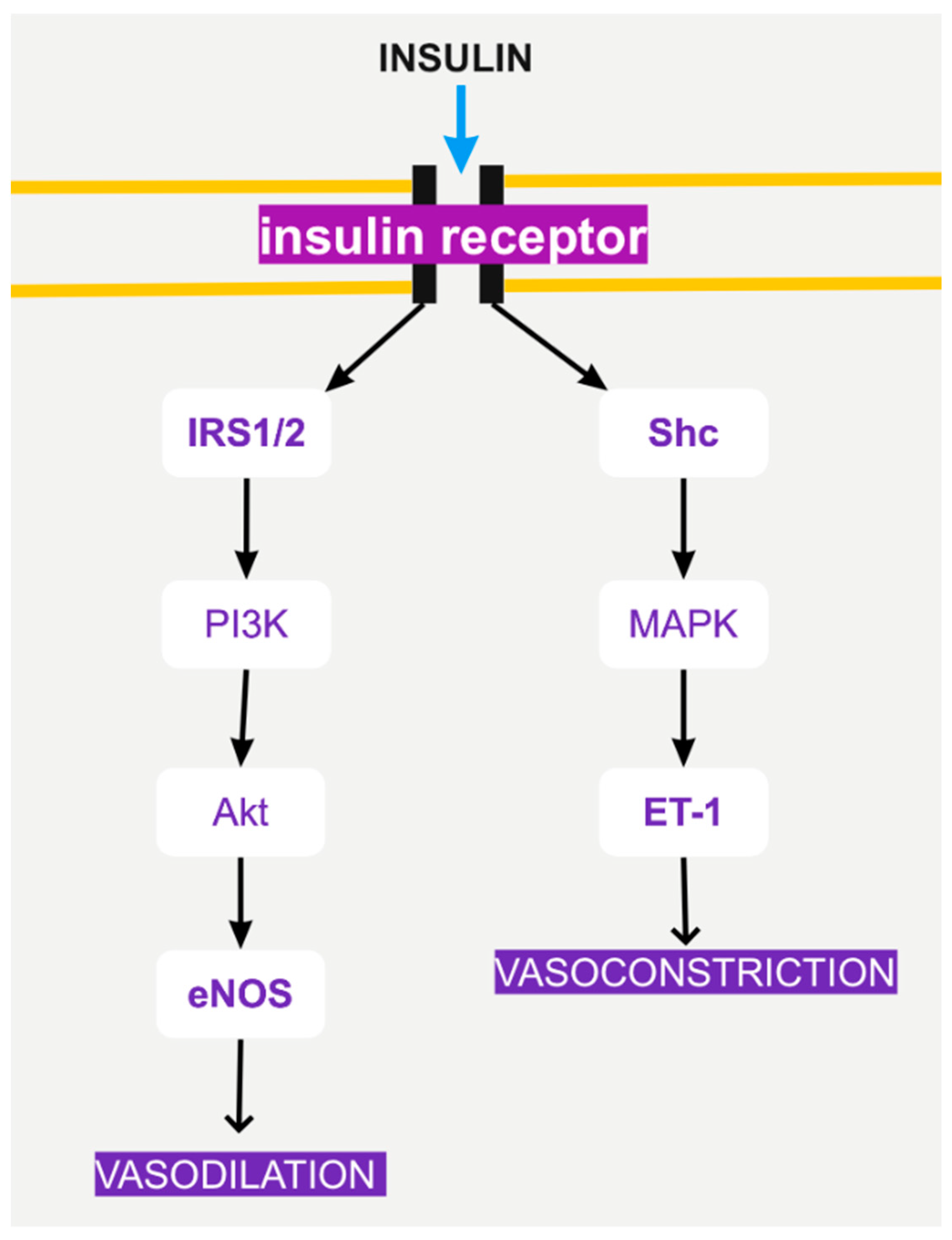
4. Insulin Signaling and Endothelial Function
5. Genetic Loss of Insulin Receptor
6. The Role of Sacro/Endoplasmic Reticulum Ca2+-ATPase (SERCA)
7. Chronic Hyperinsulinemia and Angiotensin II (Ang II)
8. Insulin-Mediated Glucose Transport via Glut
8.1. GLUT-1
8.2. GLUT-4
9. Adaptations in Cardiomyocytes under Conditions of Insulin Resistance
10. Ventricular Assist Device Implantation Reverses Insulin Resistance
10.1. HF and DM Association
10.2. Pharmacological Treatment of DM Type 2 in Patients with Cardiovascular Disease
10.3. LVADs
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iliadis, F.; Kadoglou, N.; Didangelos, T. Insulin and the heart. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S86–S91. [Google Scholar] [CrossRef]
- Velloso, L.A.; Carvalho, C.R.; Rojas, F.A.; Folli, F.; Saad, M.J. Insulin signalling in heart involves insulin receptor substrates-1 and -2, activation of phosphatidylinositol 3-kinase and the JAK 2-growth related pathway. Cardiovasc. Res. 1998, 40, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.A.; Guo, S. Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure. J. Endocrinol. 2017, 233, R131–R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Liu, B.; Sun, Y.; Du, Y.; Snetselaar, L.G.; Hu, F.B.; Bao, W. Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: Population based study. BMJ 2018, 362, k1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didangelos, T.; Kantartzis, K. Diabetes and Heart Failure: Is it Hyperglycemia or Hyperinsulinemia? Curr. Vasc. Pharmacol. 2020, 18, 148–157. [Google Scholar] [CrossRef]
- Patel, N.; Gluck, J.A.; Radojevic, J.; Coleman, C.; Baker, W.L. Left ventricular assist device implantation improves glycaemic control: A systematic review and meta-analysis. ESC Heart. Fail. 2018, 5, 1141–1149. [Google Scholar] [CrossRef]
- Goetz, M.E.; Charnigo, R.; Guglin, M. Implantation of Left Ventricular Assist Device Results in Immediate Improvement of Glucose Metabolism in Patients with and Without Diabetes Mellitus. Heart Lung Circ. 2020, 29, 931–935. [Google Scholar] [CrossRef]
- Didangelos, T.P.; Arsos, G.A.; Karamitsos, D.T.; Athyros, V.G.; Karatzas, N.D. Left Ventricular Systolic and Diastolic Function in Normotensive Type 1 Diabetic Patients with or Without Autonomic Neuropathy. Diabetes Care 2003, 26, 1955–1960. [Google Scholar] [CrossRef] [Green Version]
- Didangelos, T.P.; Arsos, G.; Karamitsos, T.; Iliadis, F.; Papageorgiou, A.; Moralidis, E.; Athyros, V. Left Ventricular Systolic and Diastolic Function in Normotensive Type 2 Diabetic Patients with or Without Autonomic Neuropathy. Angiology 2013, 65, 877–882. [Google Scholar] [CrossRef]
- Yao, H.; Han, X.; Han, X. The Cardioprotection of the Insulin-Mediated PI3K/Akt/mTOR Signaling Pathway. Am. J. Cardiovasc. Drugs 2014, 14, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ragolia, L. AKT phosphorylation is essential for insulin-induced relaxation of rat vascular smooth muscle cells. Am. J. Physiol. Physiol. 2006, 291, C1355–C1365. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Guthrie, P.H.; Chan, S.S.; Haq, S.; Taegtmeyer, H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc. Res. 2007, 76, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulakat, L.; Demarco, V.G.; Whaley-Connell, A.; Sowers, J.R. The Impact of Overnutrition on Insulin Metabolic Signaling in the Heart and the Kidney. Cardiorenal Med. 2011, 1, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Zhu, Q.; Zhang, K.; Thomas, C.; Wu, Y.; Kumar, R.; Baker, K.M.; Xu, Z.; Chen, S.; Guo, S. Activation of Foxo1 by Insulin Resistance Promotes Cardiac Dysfunction and β–Myosin Heavy Chain Gene Expression. Circ. Heart Fail. 2015, 8, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Xu, Z.; Zhu, Q.; Thomas, C.; Kumar, R.; Feng, H.; Dostal, D.E.; White, M.F.; Baker, K.M.; Guo, S. Myocardial Loss of IRS1 and IRS2 Causes Heart Failure and Is Controlled by p38α MAPK During Insulin Resistance. Diabetes 2013, 62, 3887–3900. [Google Scholar] [CrossRef] [Green Version]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef]
- Ren, J.; Samson, W.K.; Sowers, J.R. Insulin-like Growth Factor I as aÈCardiac Hormone: Physiological and Pathophysiological Implications in Heart Disease. J. Mol. Cell. Cardiol. 1999, 31, 2049–2061. [Google Scholar] [CrossRef]
- Leroith, D. Insulin-like growth factor receptors and binding proteins. Bailliere’s clinical endocrinology and metabolism. 1996, 10, 49–73. [Google Scholar] [CrossRef]
- Jones, J.I.; Clemmons, D.R. Insulin-Like Growth Factors and Their Binding Proteins: Biological Actions. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [CrossRef]
- Fuller, S.J.; Mynett, J.R.; Sugden, P.H. Stimulation of cardiac protein synthesis by insulin-like growth factors. Biochem. J. 1992, 282 Pt 1, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Cheng, W.; Ferber, A.; Kajstura, J.; Li, P.; Li, B.; Olivetti, G.; Homcy, C.J.; Baserga, R.; Anversa, P. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8630–8635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florini, J.R.; Ewton, D.Z.; Coolican, S.A. Growth Hormone and the Insulin-Like Growth Factor System in Myogenesis. Endocr. Rev. 1996, 17, 481–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piedras-Rentería, E.S.; Chen, C.-C.; Best, P.M. Antisense oligonucleotides against rat brain α1E DNA and its atrial homologue decrease T-type calcium current in atrial myocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 14936–14941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Kada, K.; Kamiya, K.; Toyama, J. IGF-I regulates K(+)-channel expression of cultured neonatal rat ventricular myocytes. Am. J. Physiol. Content 1997, 272 Pt 2, H2599–H2606. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, W.; Markovich, R.; Chen, J.-W.; Wang, P.H. Regulation of Cardiomyocyte Apoptotic Signaling by Insulin-like Growth Factor I. Circ. Res. 1998, 83, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Andronico, G.; Mangano, M.T.; Nardi, E.; Mulè, G.; Piazza, G.; Cerasola, G. Insulin-like growth factor 1 and sodium—Lithium countertransport in essential hypertension and in hypertensive left ventricular hypertrophy. J. Hypertens. 1993, 11, 1097–1101. [Google Scholar] [CrossRef]
- Wickman, A.; Friberg, P.; Adams, M.A.; Matejka, G.L.; Brantsing, C.; Guron, G.; Isgaard, J. Induction of Growth Hormone Receptor and Insulin-Like Growth Factor-I mRNA in Aorta and Caval Vein During Hemodynamic Challenge. Hypertension 1997, 29, 123–130. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. eNOS phosphorylation in health and disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef]
- Marletta, M.A. Nitric oxide synthase: Aspects concerning structure and catalysis. Cell 1994, 78, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.; Bauersachs, J.; Fisslthaler, B.; Busse, R. Ca2+ -Independent Activation of the Endothelial Nitric Oxide Synthase in Response to Tyrosine Phosphatase Inhibitors and Fluid Shear Stress. Circ. Res. 1998, 82, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mombouli, J.V.; Vanhoutte, P.M. Kinins and Endothelial Control of Vascular Smooth Muscle. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 679–705. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Smith, R.S.; Hsieh, C.-M.; Sun, J.; Chao, J.; Liao, J.K. Activation of the Phosphatidylinositol 3-Kinase/Protein Kinase Akt Pathway Mediates Nitric Oxide-Induced Endothelial Cell Migration and Angiogenesis. Mol. Cell. Biol. 2003, 23, 5726–5737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.-N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles for Insulin Receptor, PI3-Kinase, and Akt in Insulin-Signaling Pathways Related to Production of Nitric Oxide in Human Vascular Endothelial Cells. Circulation 2000, 101, 1539–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, K.S.B.; Lipke, E.A.; Rice, E.E.H.; Liel, M.S.; Myler, H.A.; Zygourakis, C.; Tulis, D.A.; West, J.L. Nitric oxide-generating hydrogels inhibit neointima formation. J. Biomater. Sci. Polym. Ed. 2005, 16, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Xu, B.; Liu, Y.; Parikh, D.; Li, J.; Li, Y.; Zhang, Y.; Riehle, C.; Zhu, Y.; Rawlings, T.; et al. Insulin Inhibits Cardiac Contractility by Inducing a Gi-Biased β2-Adrenergic Signaling in Hearts. Diabetes 2014, 63, 2676–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, E.; Jurado-Pueyo, M.; Fortuño, M.A.; Fernández-Veledo, S.; Vila-Bedmar, R.; Jiménez-Borreguero, L.J.; Lazcano, J.J.; Gao, E.; Gómez-Ambrosi, J.; Frühbeck, G.; et al. Downregulation of G protein-coupled receptor kinase 2 levels enhances cardiac insulin sensitivity and switches on cardioprotective gene expression patterns. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2448–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila-Bedmar, R.; Cruces-Sande, M.; Lucas, E.; Willemen, H.L.D.M.; Heijnen, C.J.; Kavelaars, A.; Mayor, F.; Murga, C. Reversal of diet-induced obesity and insulin resistance by inducible genetic ablation of GRK2. Sci. Signal. 2015, 8, ra73. [Google Scholar] [CrossRef] [Green Version]
- Rask-Madsen, C.; Ihlemann, N.; Krarup, T.; Christiansen, E.; Kober, L.; Kistorp, C.N.; Torp-Pedersen, C. Insulin Therapy Improves Insulin-Stimulated Endothelial Function in Patients with Type 2 Diabetes and Ischemic Heart Disease. Diabetes 2001, 50, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Triggle, C.R.; Ding, H.; Marei, I.; Anderson, T.J.; Hollenberg, M.D. Why the endothelium? The endothelium as a target to reduce diabetes-associated vascular disease. Can. J. Physiol. Pharmacol. 2020, 98, 415–430. [Google Scholar] [CrossRef]
- Fonseca, V.A. The effects of insulin on the endothelium. Endocrinol. Metab. Clin. North Am. 2007, 36, 20–26. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Riehle, C.; Jaishy, B.; Wende, A.R.; Tuinei, J.; Chen, D.; Soto, J.; Pires, K.M.; Boudina, S.; Theobald, H.A.; et al. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J. Mol. Cell. Cardiol. 2012, 52, 1019–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ock, S.; Lee, W.S.; Ahn, J.; Kim, H.M.; Kang, H.; Kim, H.-S.; Jo, D.; Abel, E.D.; Lee, T.J.; Kim, J. Deletion of IGF-1 Receptors in Cardiomyocytes Attenuates Cardiac Aging in Male Mice. Endocrinology 2016, 157, 336–345. [Google Scholar] [CrossRef]
- Wold, L.E.; Dutta, K.; Mason, M.M.; Ren, J.; Cala, S.E.; Schwanke, M.L.; Davidoff, A.J. Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J. Mol. Cell. Cardiol. 2005, 39, 297–307. [Google Scholar] [CrossRef]
- Pierce, G.N.; Russell, J.C. Regulation of intracellular Ca2+ in the heart during diabetes. Cardiovasc. Res. 1997, 34, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Russ, M.; Reinauer, H.; Eckel, J. Diabetes-induced decrease in the mRNA coding for sarcoplasmic reticulum Ca2+-ATPase in adult rat cardiomyocytes. Biochem. Biophys. Res. Commun. 1991, 178, 906–912. [Google Scholar] [CrossRef]
- Abe, T.; Ohga, Y.; Tabayashi, N.; Kobayashi, S.; Sakata, S.; Misawa, H.; Tsuji, T.; Kohzuki, H.; Suga, H.; Taniguchi, S.; et al. Left ventricular diastolic dysfunction in type 2 diabetes mellitus model rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H138–H148. [Google Scholar] [CrossRef]
- Misra, T.; Gilchrist, J.S.C.; Russell, J.C.; Pierce, G.N. Cardiac myofibrillar and sarcoplasmic reticulum function are not depressed in insulin-resistant JCR:LA-cp rats. Am. J. Physiol. Content 1999, 276, H1811–H1817. [Google Scholar] [CrossRef]
- Samuelsson, A.-M.; Bollano, E.; Mobini, R.; Larsson, B.-M.; Omerovic, E.; Fu, M.; Waagstein, F.; Holmäng, A. Hyperinsulinemia: Effect on cardiac mass/function, angiotensin II receptor expression, and insulin signaling pathways. Am. J. Physiol. Circ. Physiol. 2006, 291, H787–H796. [Google Scholar] [CrossRef] [Green Version]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Abel. E.D. Glucose transport in the heart. Front. Biosci. 2004, 9, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Leguisamo, N.M.; Lehnen, A.M.; Machado, U.F.; Okamoto, M.M.; Markoski, M.M.; Pinto, G.H.; Schaan, B.D. GLUT4 content decreases along with insulin resistance and high levels of inflammatory markers in rats with metabolic syndrome. Cardiovasc. Diabetol. 2012, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Obes. Lipotoxicity 2017, 960, 277–304. [Google Scholar] [CrossRef]
- Oellgaard, J.; Gæde, P.; Rossing, P.; Rørth, R.; Køber, L.; Parving, H.-H.; Pedersen, O. Reduced risk of heart failure with intensified multifactorial intervention in individuals with type 2 diabetes and microalbuminuria: 21 years of follow-up in the randomised Steno-2 study. Diabetologia 2018, 61, 1724–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gæde, P.; Oellgaard, J.; Carstensen, B.; Rossing, P.; Lund-Andersen, H.; Parving, H.-H.; Pedersen, O. Years of life gained by multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: 21 years follow-up on the Steno-2 randomised trial. Diabetologia 2016, 59, 2298–2307. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, A.; Puymirat, E.; Danchin, N.; Aissaoui, N. Left ventricular assist device may improve glycemic control in diabetes mellitus patients but the reverse is not true. J. Thorac. Dis. 2018, 10, S4093–S4095. [Google Scholar] [CrossRef]
- Riehle, C.; Weatherford, E.T.; Wende, A.R.; Jaishy, B.P.; Seei, A.W.; McCarty, N.S.; Rech, M.; Shi, Q.; Reddy, G.R.; Kutschke, W.J.; et al. Insulin receptor substrates differentially exacerbate insulin-mediated left ventricular remodeling. JCI Insight 2020, 5, e134920. [Google Scholar] [CrossRef] [Green Version]
- Lehrke, M.; Marx, N. Diabetes Mellitus and Heart Failure. Am. J. Cardiol. 2017, 120, S37–S47. [Google Scholar] [CrossRef] [Green Version]
- Patoulias, D.; Stavropoulos, K.; Imprialos, K.; Athyros, V.; Doumas, M.; Karagiannis, A. Pharmacological Management of Cardiac Disease in Patients with Type 2 Diabetes: Insights into Clinical Practice. Curr. Vasc. Pharmacol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.L.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef] [PubMed]
- Mc Rosano, G.; Vitale, C.; Seferovic, P. Heart Failure in Patients with Diabetes Mellitus. Card. Fail. Rev. 2017, 3, 52–55. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, H.L.; Kent, W.D.T.; MacQueen, K.T.; Miller, R.J.H.; Holloway, D.D.; Hassanabad, A.F. Left ventricular assist devices: A comprehensive review of major clinical trials, devices, and future directions. J. Card. Surg. 2021, 36, 1480–1491. [Google Scholar] [CrossRef]
- Khawaja, T.; Chokshi, A.; Ji, R.; Kato, T.S.; Xu, K.; Zizola, C.; Wu, C.; Forman, D.E.; Ota, T.; Kennel, P.J.; et al. Ventricular assist device implantation improves skeletal muscle function, oxidative capacity, and growth hormone/insulin-like growth factor-1 axis signaling in patients with advanced heart failure. J. Cachex-Sarcopenia Muscle 2014, 5, 297–305. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantazi, K.; Karlafti, E.; Bekiaridou, A.; Didagelos, M.; Ziakas, A.; Didangelos, T. Insulin Receptors and Insulin Action in the Heart: The Effects of Left Ventricular Assist Devices. Biomolecules 2022, 12, 578. https://doi.org/10.3390/biom12040578
Pantazi K, Karlafti E, Bekiaridou A, Didagelos M, Ziakas A, Didangelos T. Insulin Receptors and Insulin Action in the Heart: The Effects of Left Ventricular Assist Devices. Biomolecules. 2022; 12(4):578. https://doi.org/10.3390/biom12040578
Chicago/Turabian StylePantazi, Konstantina, Eleni Karlafti, Alexandra Bekiaridou, Matthaios Didagelos, Antonios Ziakas, and Triantafyllos Didangelos. 2022. "Insulin Receptors and Insulin Action in the Heart: The Effects of Left Ventricular Assist Devices" Biomolecules 12, no. 4: 578. https://doi.org/10.3390/biom12040578