
Mutational Effect of Some Major COVID-19 Variants on Binding of the S Protein to ACE2


Abstract
:1. Introduction
2. Theoretical Method
2.1. Molecular Dynamics Simulations
2.2. Hotspot Prediction by ASGB Method
3. Results and Discussion
3.1. ASGB Analysis for Single Point Mutations in Alpha, Beta, Gamma and Delta
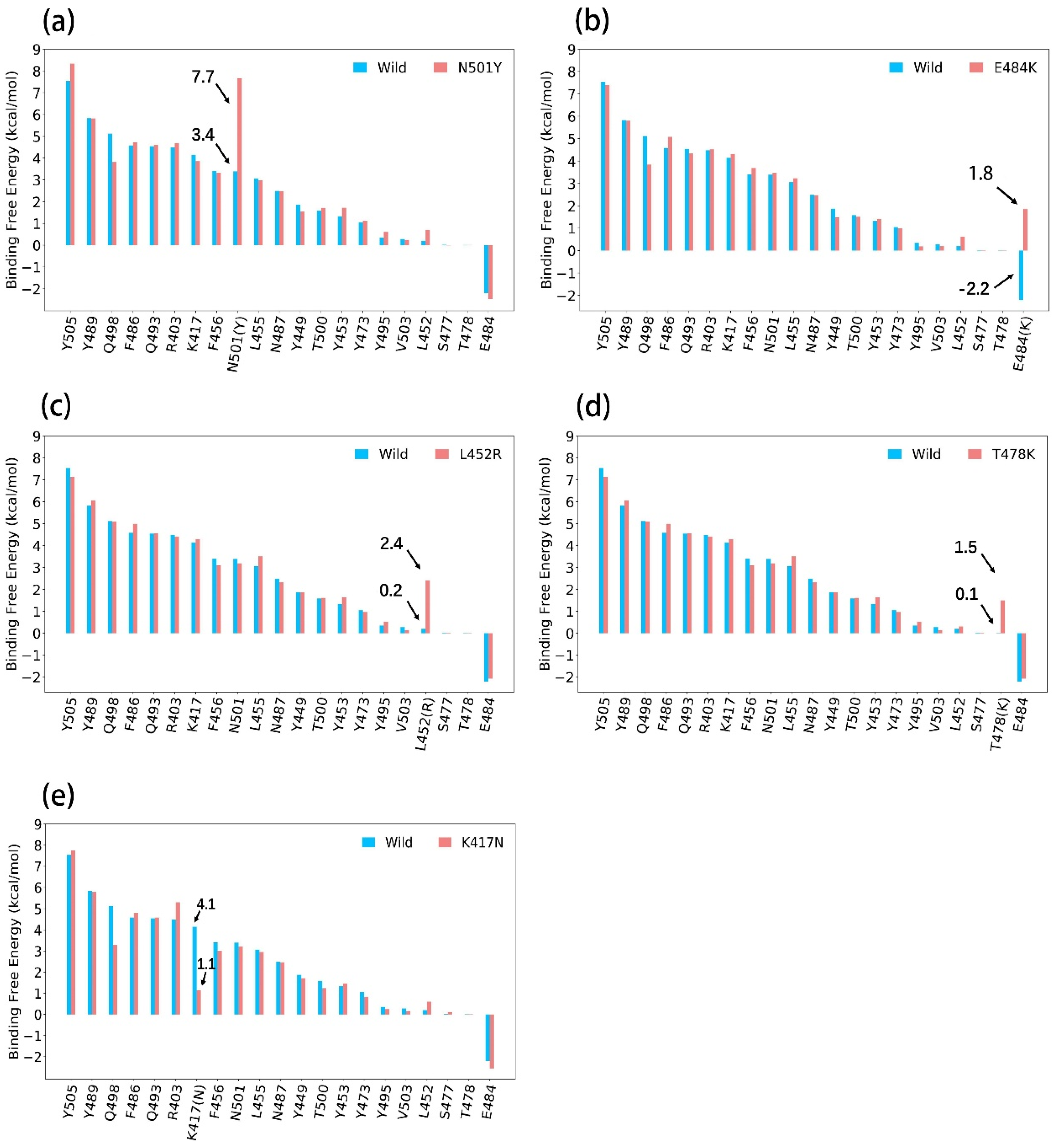
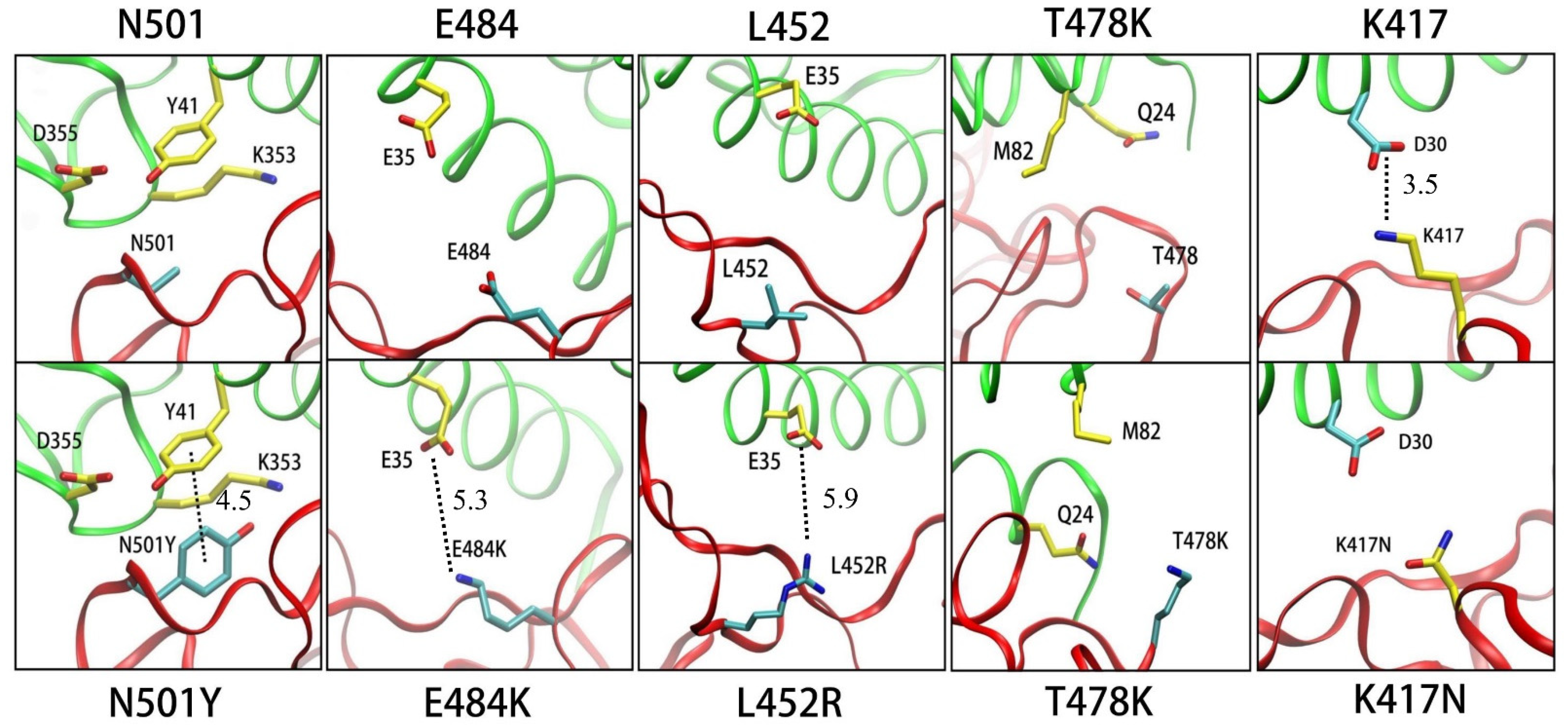
3.1.1. N501Y
3.1.2. E484K
3.1.3. L452R
3.1.4. T478K
3.1.5. K417N
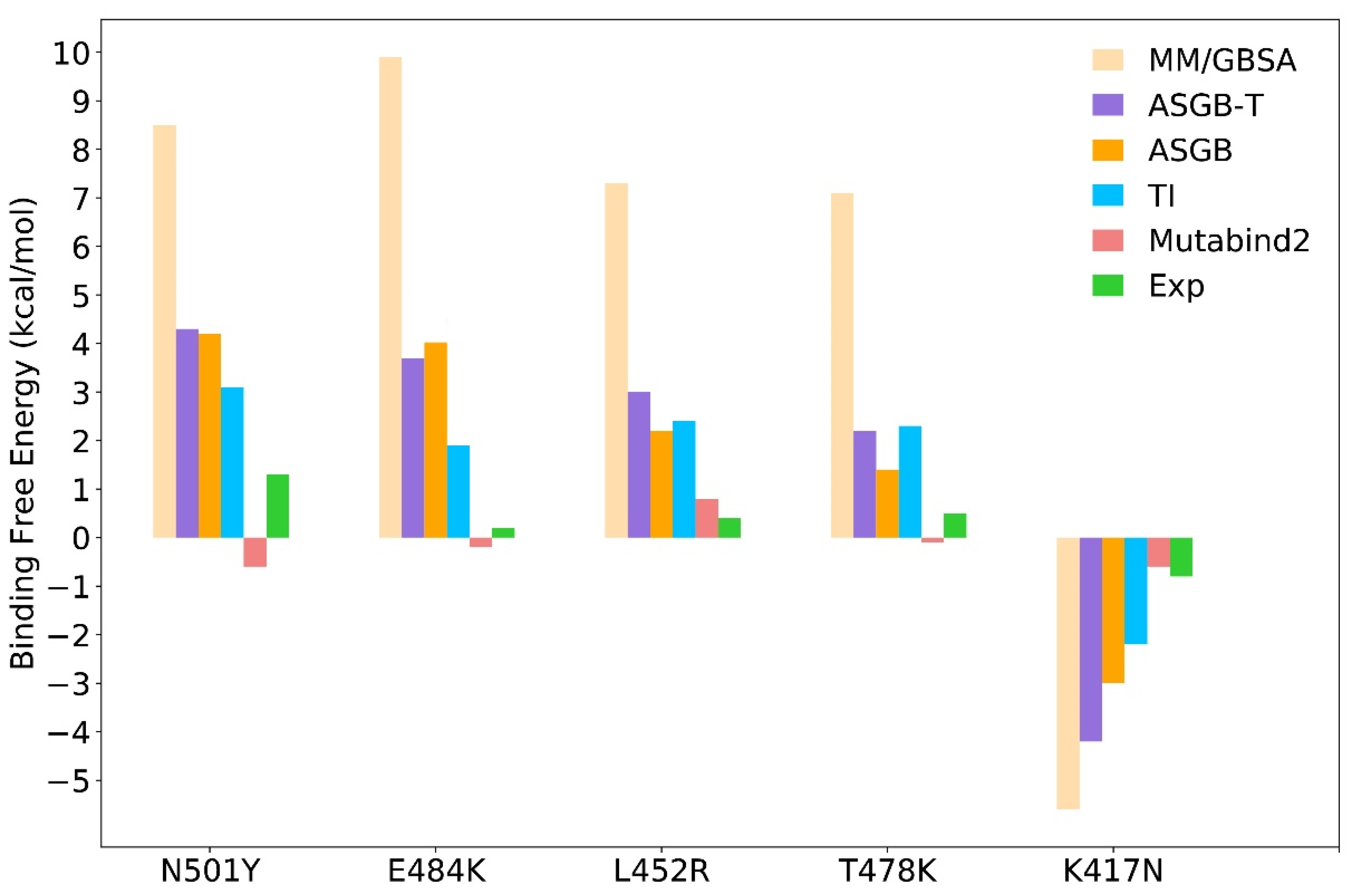
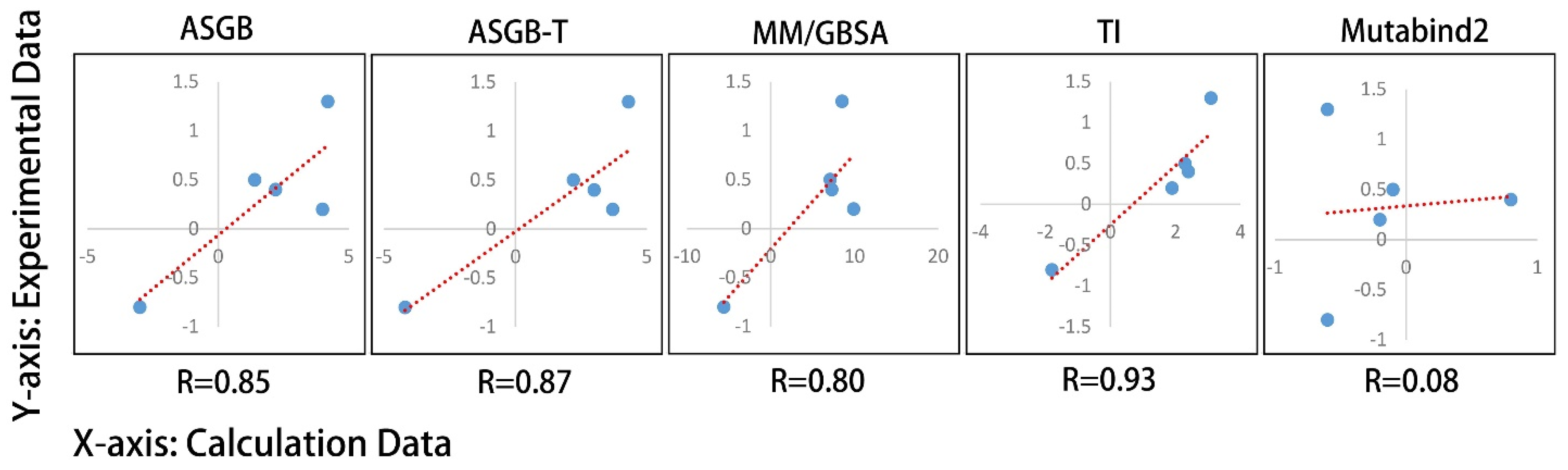
3.2. Comparison of Results Using Different Methods
3.2.1. MM/GBSA Method
3.2.2. ASGB-T Method
3.2.3. TI Method
3.2.4. Mutabind Method
3.3. ASGB Analysis for Single Point Mutations in Omicron Variants
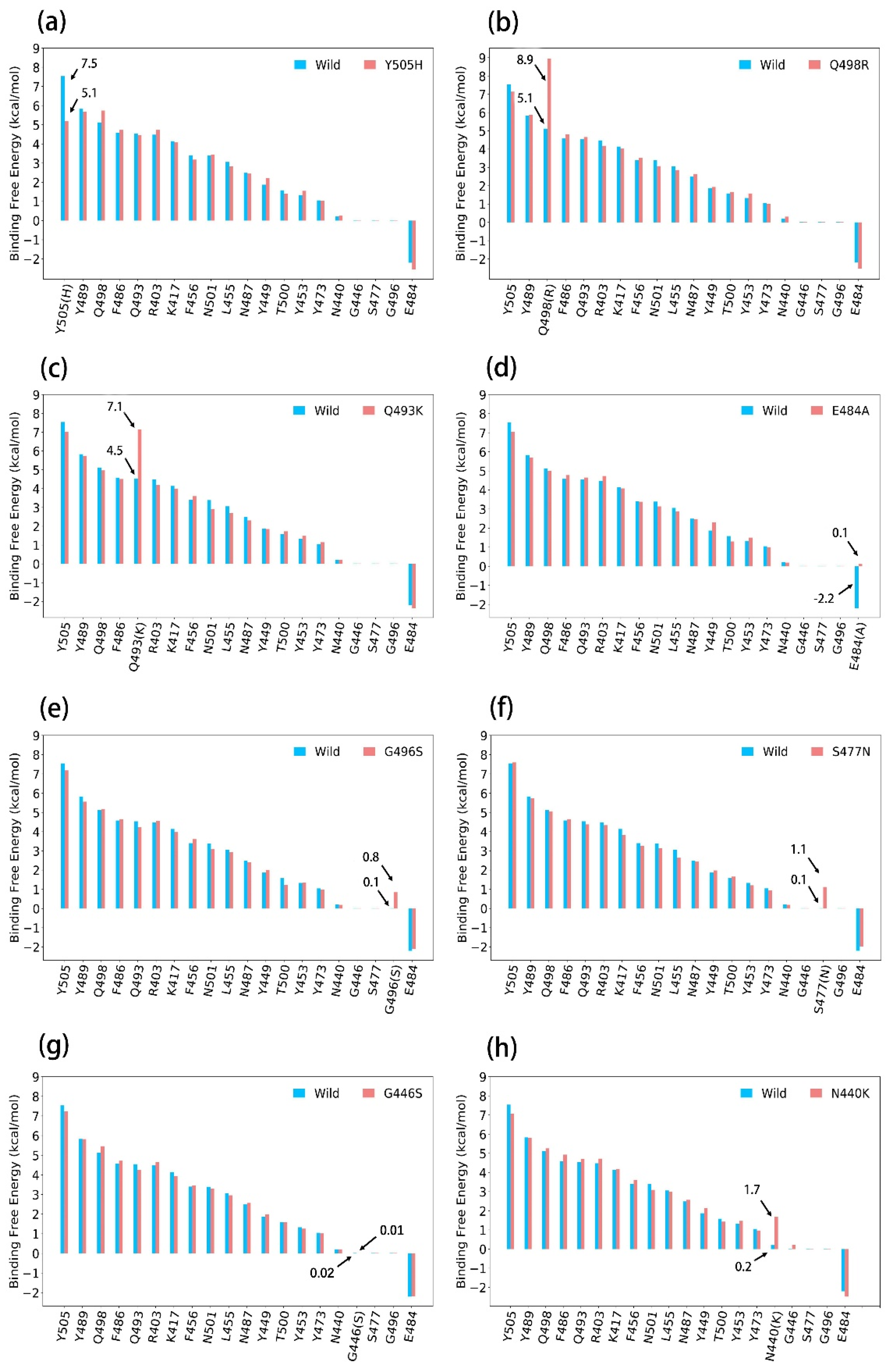
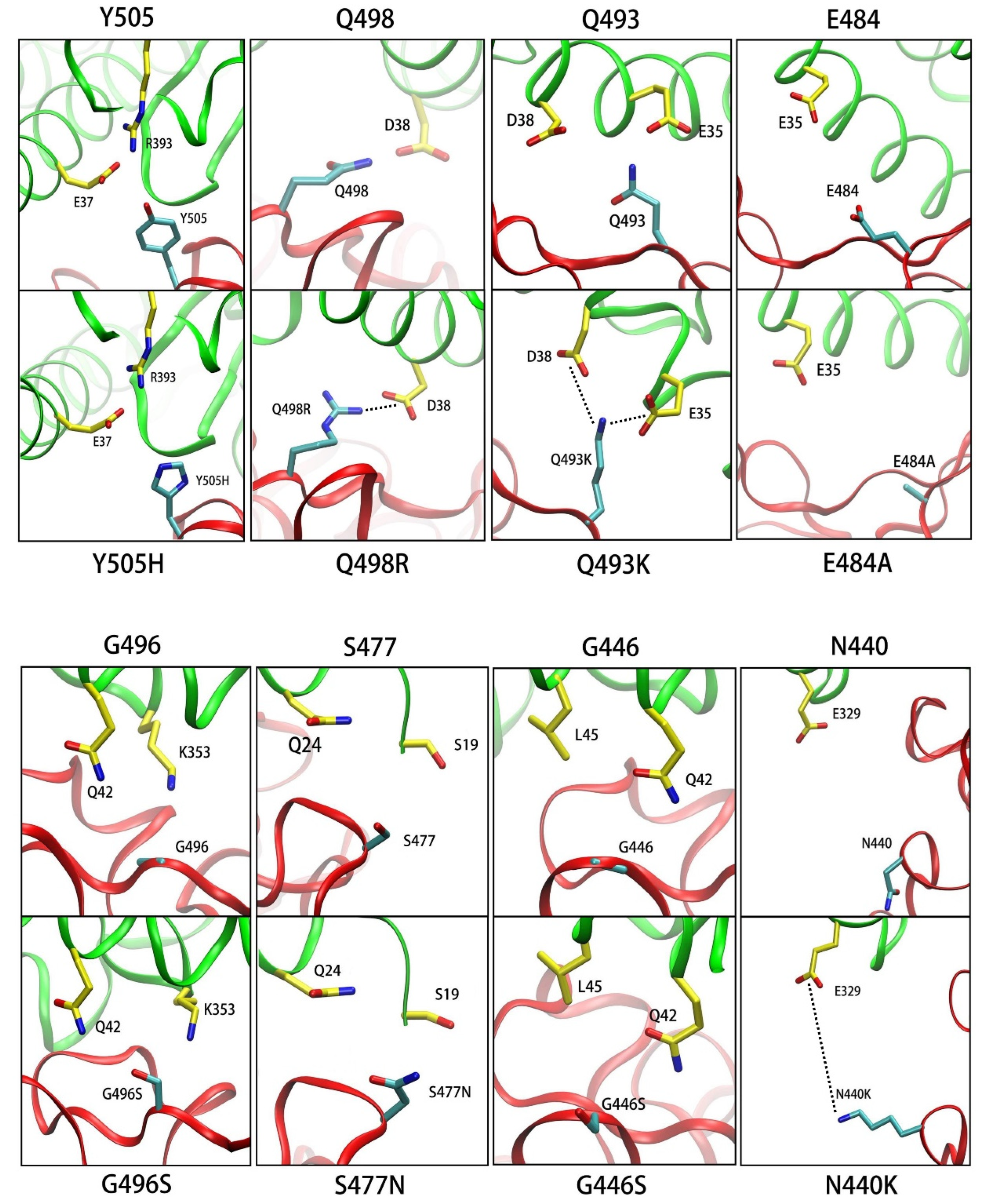
3.3.1. Y505H
3.3.2. Q498R
3.3.3. Q493K
3.3.4. E484A
3.3.5. G496S
3.3.6. S477N
3.3.7. G446S
3.3.8. N440K
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, W.; Veeresha, P.; Baskonus, H.M.; Prakasha, D.G.; Kumar, P. A new study of unreported cases of 2019-nCOV epidemic outbreaks. Chaos Soliton Fractals 2020, 138, 109929. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Menachery, V.D. Return of the Coronavirus: 2019-nCoV. Viruses 2020, 12, 135. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.W.; Yu, J.F.; Shan, S.S.; Zhou, H.; Fan, S.L.; Zhang, Q.; Shi, X.L.; Wang, Q.S.; Zhang, L.Q.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215. [Google Scholar] [CrossRef] [Green Version]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Haider, N.; Abbasi, A.F.; Jaferi, U.; Prakash, S.; Balendra, V. The emerging SARS-CoV-2 variants of concern. Ther. Adv. Infect. Dis. 2021, 8, 20499361211024372. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.S.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.d.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D.J.T.L. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–954. [Google Scholar] [CrossRef]
- Nelson, G.; Buzko, O.; Spilman, P.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. BioRxiv 2021. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Torjesen, I. COVID-19: Omicron may be more transmissible than other variants and partly resistant to existing vaccines, scientists fear. BMJ 2021, 375, n2943. [Google Scholar] [CrossRef]
- Gao, S.J.; Guo, H.; Luo, G. Omicron variant (B.1.1.529) of SARS-CoV-2, a global urgent public health alert! J. Med. Virol. 2021, 94, 1255–1256. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Khateeb, J.; Li, Y.; Zhang, H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Crit. Care 2021, 25, 244. [Google Scholar] [CrossRef]
- Tanaka, S.; Nelson, G.; Olson, C.A.; Buzko, O.; Higashide, W.; Shin, A.; Gonzalez, M.; Taft, J.; Patel, R.; Buta, S.; et al. An ACE2 Triple Decoy that neutralizes SARS-CoV-2 shows enhanced affinity for virus variants. Sci. Rep. 2021, 11, 12740. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Luan, B.; Wang, H.; Huynh, T. Molecular Mechanism of the N501Y Mutation for Enhanced Binding between SARS-CoV-R s Spike Protein and Human ACE2 Receptor. BioRxiv 2021. [Google Scholar] [CrossRef]
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Miotto, M.; Di Rienzo, L.; Gosti, G.; Bo’, L.; Parisi, G.; Piacentini, R.; Boffi, A.; Ruocco, G.; Milanetti, E. Inferring the stabilization effects of SARS-CoV-2 variants on the binding with ACE2 receptor. Commun. Biol. 2022, 5, 20221. [Google Scholar] [CrossRef]
- Koehler, M.; Ray, A.; Moreira, R.A.; Juniku, B.; Poma, A.B.; Alsteens, D. Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6977. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Modeling 2021, 61, 4425–4441. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]
- Rath, S.L.; Padhi, A.K.; Mandal, N. Scanning the RBD-ACE2 molecular interactions in Omicron variant. Biochem. Biophys. Res. Commun. 2022, 592, 18–23. [Google Scholar] [CrossRef]
- Santos, J.C.; Passos, G.A. The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation. BioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, X.; Peng, L.; Zhou, Y.F.; Zhang, Y.Z.; Zhang, J.Z. Computational Alanine Scanning with Interaction Entropy for Protein-Ligand Binding Free Energies. J. Chem. Theory Comput. 2018, 14, 1772–1780. [Google Scholar] [CrossRef]
- He, L.P.; Bao, J.X.; Yang, Y.P.; Dong, S.Z.; Zhang, L.J.; Qi, Y.F.; Zhang, J.Z. Study of SHMT2 Inhibitors and Their Binding Mechanism by Computational Alanine Scanning. J. Chem. Inf. Modeling 2019, 59, 3871–3878. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yang, M.Y.; Ji, C.G.; Zhang, J.Z.H. Interaction Entropy for Computational Alanine Scanning. J. Chem. Inf. Model. 2017, 57, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Qi, Y.; Song, J.; Zhang, J.Z. Calculation of hot spots for protein–protein interaction in p53/PMI-MDM2/MDMX complexes. J. Comput. Chem. 2019, 40, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.D.; Wen, W.; Liu, X.; Li, Y.; Zhang, J.Z. Computational analysis of hot spots and binding mechanism in the PD-1/PD-L1 interaction. RSC Adv. 2019, 9, 14944–14956. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Yan, Y.; Sun, Z.; Song, J.; Zhang, J.Z. Interaction entropy for computational alanine scanning in protein–protein binding. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1342. [Google Scholar] [CrossRef]
- Li, Z.; Bao, J.; Qi, Y.; Zhang, J.Z. Computational approaches to studying methylated H4K20 recognition by DNA repair factor 53BP1. Phys. Chem. Chem. Phys. 2020, 22, 6136–6144. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.Z. Quantitative analysis of ACE2 bindings to coronavirus spike proteins: SARS-CoV-2 vs SARS-CoV and RaTG13. Phys. Chem. Chem. Phys. 2021, 23, 13926–13933. [Google Scholar] [CrossRef]
- Cong, Y.; Feng, Y.; Ni, H.; Zhi, F.; Miao, Y.; Fang, B.; Zhang, L.; Zhang, J.Z. Anchor-Locker Binding Mechanism of the Coronavirus Spike Protein to Human ACE2: Insights from Computational Analysis. J. Chem. Inf. Modeling 2021, 61, 3529–3542. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Method Enzymol. 2011, 487, 545–574. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C.J. computation, ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L.J. Particle mesh Ewald: An pÅlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Peng, L.; Zhang, J.Z. Accurate and Efficient Calculation of Protein–Protein Binding Free Energy-Interaction Entropy with Residue Type-Specific Dielectric Constants. J. Chem. Inf. Modeling 2018, 59, 272–281. [Google Scholar] [CrossRef]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikainen, O.T. MMGBSA As a Tool To Understand the Binding Affinities of Filamin-Peptide Interactions. J. Chem. Inf. Modeling 2013, 53, 2626–2633. [Google Scholar] [CrossRef]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Wang, E.; Weng, G.; Sun, H.; Du, H.; Zhu, F.; Chen, F.; Wang, Z.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein-protein Interactions. Phys. Chem. Chem. Phys. 2019, 21, 18958–18969. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Modeling 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Modeling 2018, 58, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Hu, Y.; Sherborne, B.; Guo, Z.; York, D.M. Toward Fast and Accurate Binding Affinity Prediction with pmemdGTl: An Efficient Implementation of GPU-Accelerated Thermodynamic Integration. J. Chem. Theory Comput. 2017, 13, 3077–3084. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Sites | Alpha | Beta | Gamma | Delta | Omicron |
|---|---|---|---|---|---|
| K417N | √ | √ | |||
| N440K | √ | ||||
| G446S | √ | ||||
| L452R | √ | ||||
| S477N | √ | ||||
| T478K | √ | √ | |||
| E484A | √ | ||||
| E484K | √ | √ | |||
| Q493K | √ | ||||
| G496S | √ | ||||
| Q498R | √ | ||||
| N501Y | √ | √ | √ | √ | |
| Y505H | √ |
| Mutation | ΔΔEvdw | ΔΔEele | ΔΔEgb | ΔΔEnp | ΔΔGcal | ΔΔGexp a |
|---|---|---|---|---|---|---|
| N501Y | 3.8 | 0.4 | −0.1 | 0.3 | 4.2 | 1.3 |
| E484K | 0.1 | 84.8 | −80.7 | −0.1 | 4.0 | 0.2 |
| L452R | 0.3 | 32.4 | −30.6 | 0.1 | 2.2 | 0.4 |
| T478K | 0.9 | 10.1 | −9.7 | 0.1 | 1.4 | 0.5 |
| K417N | −0.5 | −56.8 | 54.3 | −0.2 | −3.0 | −0.8 |
| Systems | ASGB | ASGB-T | MM/GBSA | TI | Mutabind2 | ΔΔGexp a |
|---|---|---|---|---|---|---|
| N501Y | 4.2 | 4.3 | 8.5 | 3.1 | −0.6 | 1.3 |
| E484K | 4.0 | 3.7 | 9.9 | 1.9 | −0.2 | 0.2 |
| L452R | 2.2 | 3.0 | 7.3 | 2.4 | 0.8 | 0.4 |
| T478K | 1.4 | 2.2 | 7.1 | 2.3 | −0.1 | 0.5 |
| K417N | −3.0 | −4.2 | −5.6 | −1.8 | −0.6 | −0.8 |
| Mutation | ΔΔEvdw | ΔΔEele | ΔΔEgb | ΔΔEnp | ΔΔGcal |
|---|---|---|---|---|---|
| Y505H ↓ | −1.5 | −1.0 | 0.3 | −0.2 | −2.3 |
| Q498R ↑ | 2.2 | 61.2 | −60.1 | 0.5 | 3.8 |
| Q493K ↑ | −2.1 | 62.8 | −58.1 | 0.1 | 2.7 |
| E484A ↑ | −0.1 | −37.1 | 39.2 | −0.1 | 2.3 |
| G496S ↑ | 0.7 | 0.4 | −0.5 | 0.2 | 0.8 |
| S477N ↑ | 0.9 | 0.1 | 0.2 | −0.1 | 1.1 |
| G446S = | 0.1 | 0.1 | −0.2 | 0 | 0 |
| N440K ↑ | 0.5 | 45.5 | −44.7 | 0.2 | 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Zhang, J.Z.H. Mutational Effect of Some Major COVID-19 Variants on Binding of the S Protein to ACE2. Biomolecules 2022, 12, 572. https://doi.org/10.3390/biom12040572
Li Z, Zhang JZH. Mutational Effect of Some Major COVID-19 Variants on Binding of the S Protein to ACE2. Biomolecules. 2022; 12(4):572. https://doi.org/10.3390/biom12040572
Chicago/Turabian StyleLi, Zhendong, and John Z. H. Zhang. 2022. "Mutational Effect of Some Major COVID-19 Variants on Binding of the S Protein to ACE2" Biomolecules 12, no. 4: 572. https://doi.org/10.3390/biom12040572