
The Molecular Mechanism of Human Voltage-Dependent Anion Channel 1 Blockade by the Metallofullerenol Gd@C82(OH)22: An In Silico Study

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Molecular Dynamics Simulation
2.2. Potential of Mean Force (PMF)
3. Results and Discussion
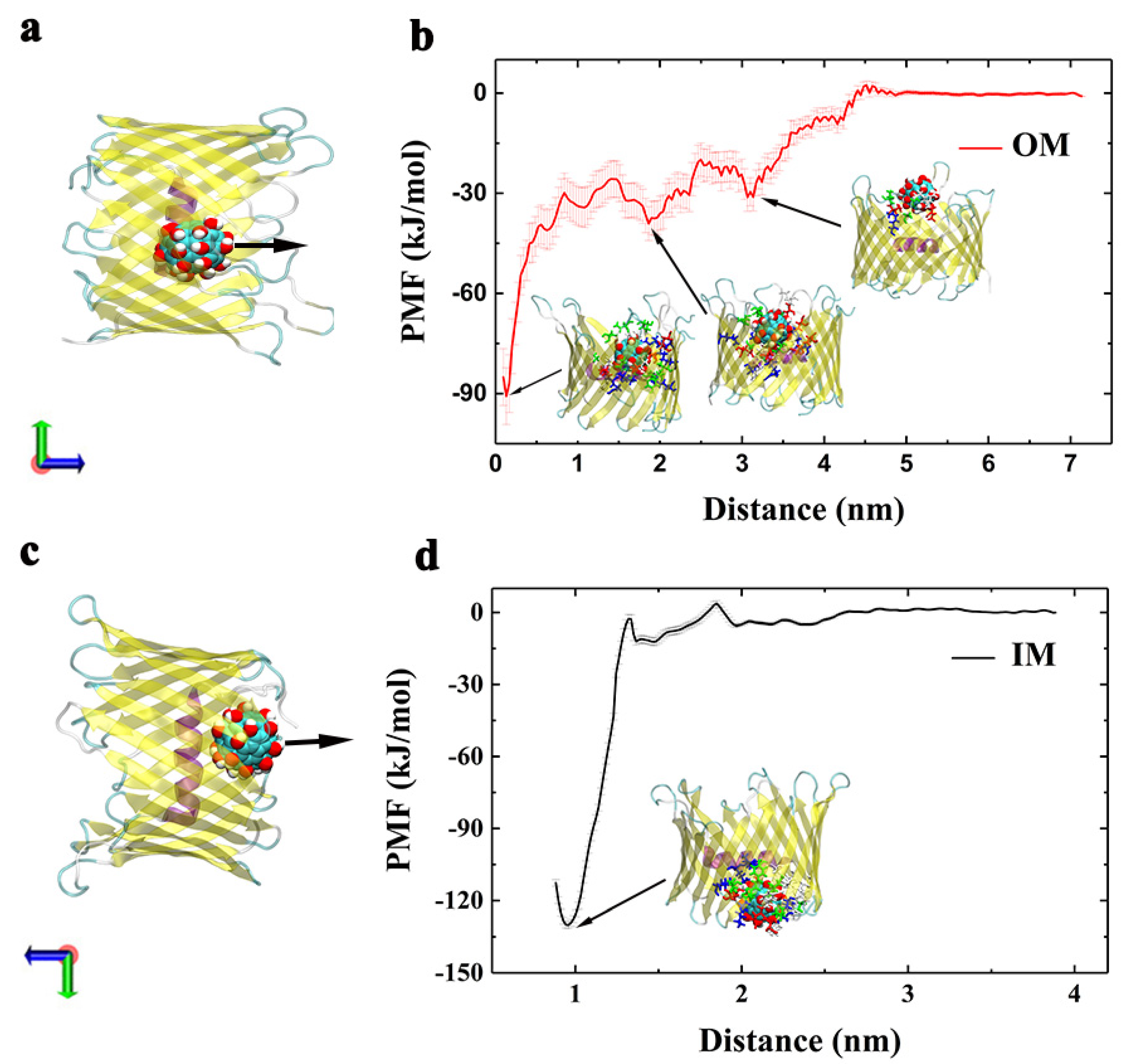
3.1. Binding Interactions and Kinetics of Gd@C82(OH)22 Entering the Lumen of hVDAC1 from the OM
- (1)
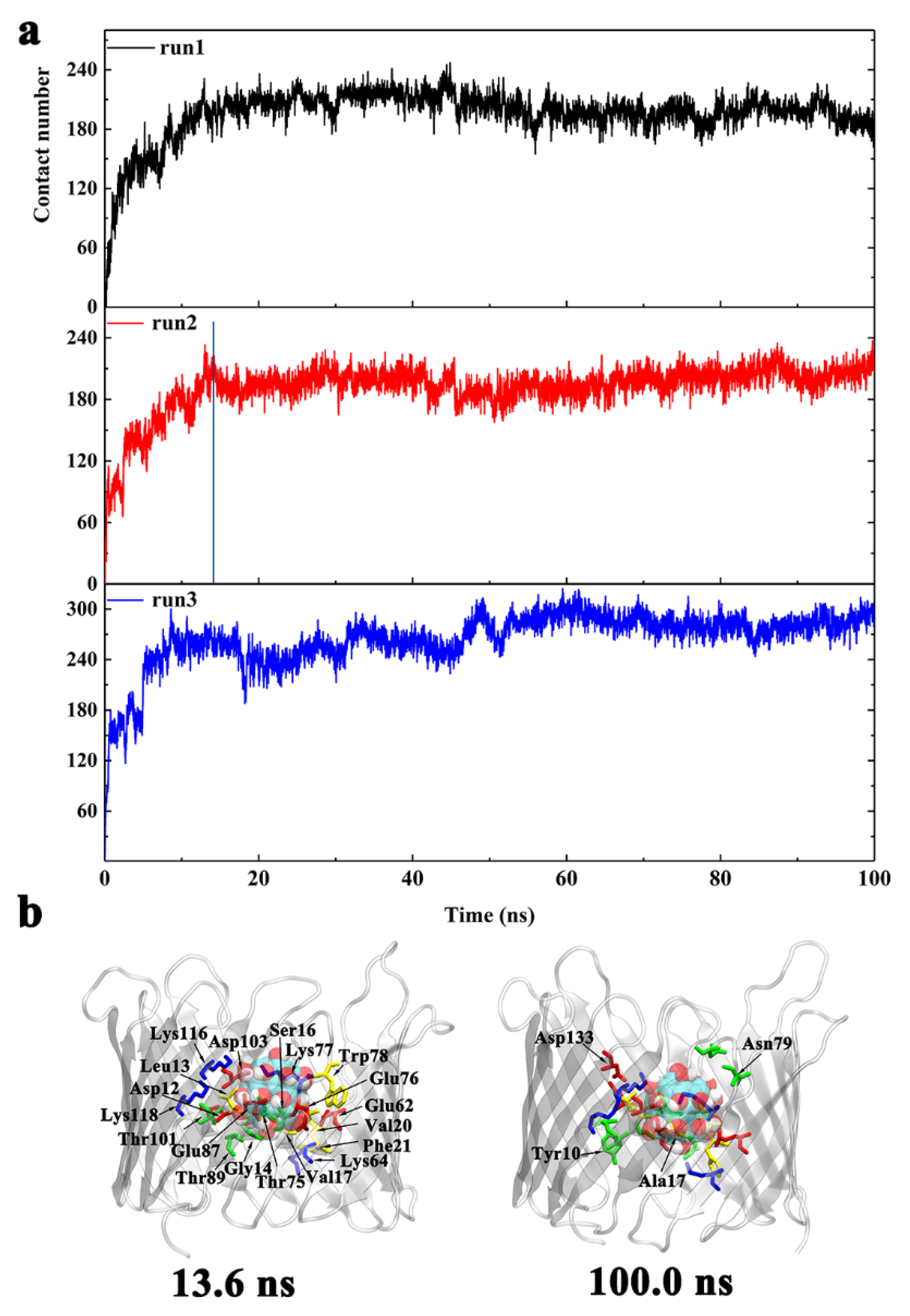
- From t = 0 to 13.6 ns, Gd@C82(OH)22 promptly entered into the porin with the total number of atomic contacts sharply increasing to ~190. At this stage, Gd@C82(OH)22 interacted with residues D12, L13, G14, S16, V17, V20, F21, E62, K64, E87, T89, T101, D103, K116, and K118 (Figure 4b). Of these residues, D12 to F21 are located at the inner helix, comprising 50% of the helical residues. Statistics of the contact residue types showed there are nine charged (five acidic, four basic), five hydrophilic, and five hydrophobic/aromatic residues, indicating the diversity of residues that Gd@C82(OH)22 can interact with in the protein tertiary structure. Gd@C82(OH)22 molecules contain both abundant hydroxyl groups and exposed aromatic rings on the surface; therefore, it has the capacity to form hydrogen bonds and hydrophobic interactions with local surrounded protein residues, making it a ‘versatile’ molecule.
- (2)
- From t = 13.6 to 100 ns, the total contact number reached a long plateau and fluctuated around 200. At this stage, the Gd@C82(OH)22 molecule was observed to interact with four additional residues: Y10, A17, N79, and D133 (Figure 4b). Of the four residues, Y10 and A17 are from the inner helix, indicating a deeper insertion of Gd@C82(OH)22 into the lumen of the hVDAC1. Now, Gd@C82(OH)22 is positioned at the interspace of the helix and β-barrel and fully blocks the hVDAC1 porin. The RMSD of hVDAC1 backbone stabilized at around 0.35 nm during this stage (Figure S2), implying that an equilibrated binding mode had formed between Gd@C82(OH)22 and the protein interface.
3.2. Binding Interactions and Kinetics of Gd@C82(OH)22 Entering the Lumen of hVDAC1 from the IM
- (1)
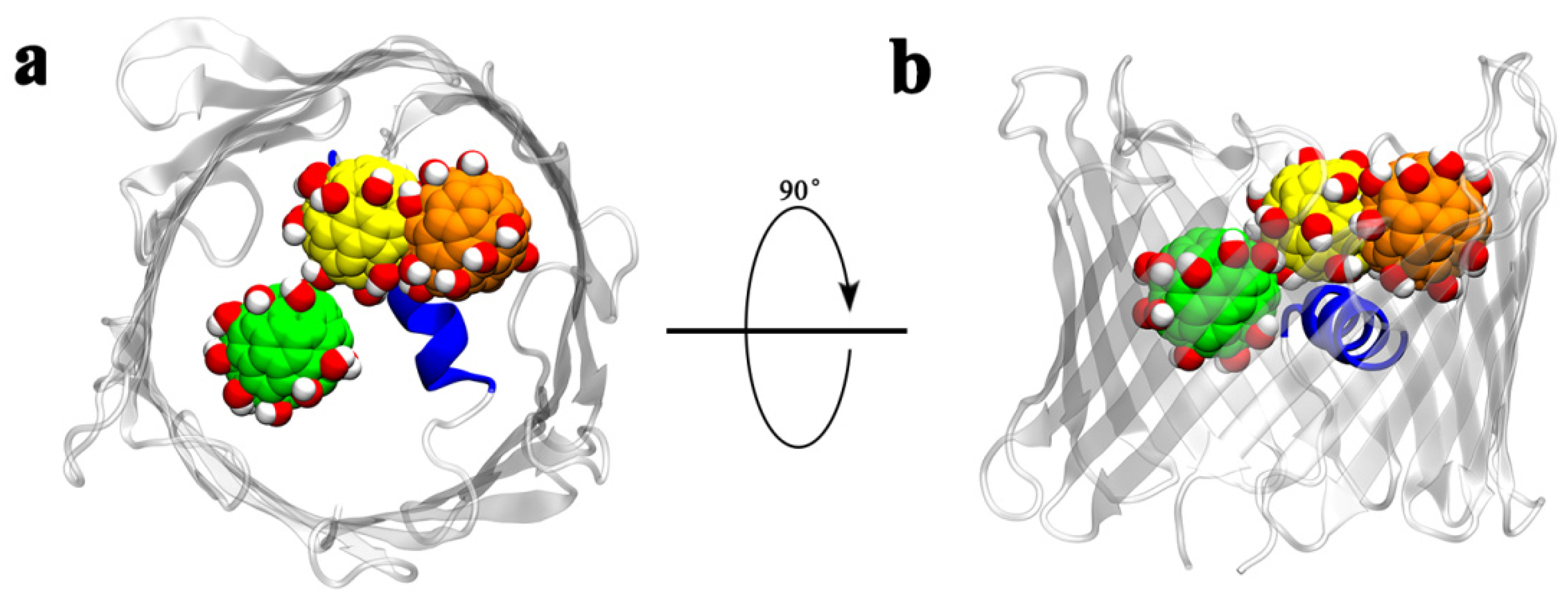
- From t = 0 to 6.4 ns, a transient plateau was formed with total contact number staying at around 200, indicating a relatively stable conformation with one Gd@C82(OH)22 molecule contacting with the protein. The intimate contacts were formed between the molecule and M1, R2, G3, S4, P8, K15, R18, K177, T178, D179, E180, F181, Y198, K200, and K227. Of these residues, M1 to P8 are located on the N-terminus, K15 and R18 are located on the inner helix, and K177 to F181 are located at the loop connecting β-strand 11 and β-strand 12. At this time point, Gd@C82(OH)22 mainly interacted with the intracellular residues and had not fully entered the central pore.
- (2)
- From t = 6.4 to 12.9 ns, the first Gd@C82(OH)22 inserted further; meanwhile, the second Gd@C82(OH)22 engaged in contacting with the protein. Accordingly, the total contact numbers sharply increased from 200 to 400. This increase corresponded to 21 additional residues forming contacts with Gd@C82(OH)22: A5, V6, P7, P8, Y10, A11, G14, D19, F21, E39, E43, K64, R66, E69, Y70, E91, Q93, Q182, Q199, E206, and A208. In this list, A5 to F21 comprise 42.8% of residues and are on the N-terminus and the inner helix, indicating a deeper insertion of Gd@C82(OH)22 into the pore of hVDAC1.
- (3)
- From t = 12.9 ns to the end of the simulation time, the total contact number stabilized at around 430. At this stage, two molecules of Gd@C82(OH)22 fully blocked the pore of the channel. Ten additional residues formed contacts with Gd@C82(OH)22: K15, R18, K37, T73, D92, L94, K99, K122, T207, and Q229. The statistics of residue types showed that there are 14 hydrophobic, 14 hydrophilic, 10 basic, and 10 acidic residues that interact with the Gd@C82(OH)22 cluster in the final conformation, again indicating the amphiphilicity of the Gd@C82(OH)22 molecules that have the capability to contact a variety of amino acid residues.
3.3. Interaction Energy Calculations between hVDAC1 Protein and Gd@C82(OH)22
3.4. PMF Calculation of Gd@C82(OH)22 Interactions with hVDAC1 Protein
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weinmann, H.J.; Brasch, R.C.; Press, W.R.; Wesbey, G.E. Characteristics of gadolinium-DTPA complex: A potential NMR contrast agent. AJR 1984, 142, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Brasch, R.C.; Weinmann, H.J.; Wesbey, G.E. Contrast-enhanced NMR imaging—Animal studies using gadolinium-dtpa complex. Am. J. Roentgenol. 1984, 142, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xing, G.M.; Sun, B.Y.; Zhao, F.; Lei, H.; Li, W.; Song, Y.; Chen, Z.; Yuan, H.; Wang, X.X.; et al. Potent Angiogenesis Inhibition by the Particulate Form of Fullerene Derivatives. Acs Nano 2010, 4, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Kanazawa, Y.; Okumura, M.; Taninaka, A.; Yokawa, T.; Shinohara, H. Lanthanoid endohedral metallofullerenols for MRI contrast agents. J. Am. Chem. Soc. 2003, 125, 4391–4397. [Google Scholar] [CrossRef]
- Meng, J.; Liang, X.; Chen, X.; Zhao, Y. Biological characterizations of (Gd@C82(OH)22)n nanoparticles as fullerene derivatives for cancer therapy. Integr. Biol. 2012, 5, 43–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tian, Y.; Nie, G. Antineoplastic activities of Gd@C82(OH)22 nanoparticles: Tumor microenvironment regulation. Sci. China Life Sci. 2012, 55, 884–890. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xing, G.; Wang, J.; Zhao, Y.; Li, B.; Tang, J.; Jia, G.; Wang, T.; Sun, J.; Xing, L.; et al. Multihydroxylated (Gd@C82(OH)22)n nanoparticles: Antineoplastic activity of high efficiency and low toxicity. Nano Lett. 2005, 5, 2050–2057. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, B.; Zhang, L.; Zhao, B.; Nie, G.; Zhao, Y. Biosafety assessment of Gd@C82(OH)22 nanoparticles on Caenorhabditis elegans. Nanoscale 2011, 3, 2636–2641. [Google Scholar] [CrossRef]
- Wang, J.; Chen, C.; Li, B.; Yu, H.; Zhao, Y.; Sun, J.; Li, Y.; Xing, G.; Yuan, H.; Tang, J.; et al. Antioxidative function and biodistribution of (Gd@C82(OH)22)n nanoparticles in tumor-bearing mice. Biochem. Pharmacol. 2006, 71, 872–881. [Google Scholar] [CrossRef]
- Yin, J.J.; Lao, F.; Meng, J.; Fu, P.P.; Zhao, Y.L.; Xing, G.M.; Gao, X.Y.; Sun, B.Y.; Wang, P.C.; Chen, C.Y.; et al. Inhibition of tumor growth by endohedral metallofullerenol nanoparticles optimized as reactive oxygen species scavenger. Mol. Pharmacol. 2008, 74, 1132–1140. [Google Scholar] [CrossRef]
- Liu, Y.; Jiao, F.; Qiu, Y.; Li, W.; Lao, F.; Zhou, G.; Sun, B.; Xing, G.; Dong, J.; Zhao, Y.; et al. The effect of Gd@C82(OH)22 nanoparticles on the release of Th1/Th2 cytokines and induction of TNF-alpha mediated cellular immunity. Biomaterials 2009, 30, 3934–3945. [Google Scholar] [CrossRef]
- Kang, S.G.; Zhou, G.Q.; Yang, P.; Liu, Y.; Sun, B.Y.; Huynh, T.; Meng, H.; Zhao, L.N.; Xing, G.M.; Chen, C.Y.; et al. Molecular mechanism of pancreatic tumor metastasis inhibition by Gd@C82(OH)22 and its implication for de novo design of nanomedicine. Proc. Natl. Acad. Sci. USA 2012, 109, 15431–15436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, F.; Qu, Y.; Zhou, G.; Liu, Y.; Li, W.; Ge, C.; Li, Y.; Hu, W.; Li, B.; Gao, Y.; et al. Modulation of oxidative stress by functionalized fullerene materials in the lung tissues of female C57/BL mice with a metastatic Lewis lung carcinoma. J. Nanosci. Nanotechnol. 2010, 10, 8632–8637. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Lao, F.; Fu, P.P.; Wamer, W.G.; Zhao, Y.; Wang, P.C.; Qiu, Y.; Sun, B.; Xing, G.; Dong, J.; et al. The scavenging of reactive oxygen species and the potential for cell protection by functionalized fullerene materials. Biomaterials 2009, 30, 611–621. [Google Scholar] [CrossRef]
- Kang, S.G.; Araya-Secchi, R.; Wang, D.; Wang, B.; Huynh, T.; Zhou, R. Dual inhibitory pathways of metallofullerenol Gd@C82(OH)22 on matrix metalloproteinase-2: Molecular insight into drug-like nanomedicine. Sci. Rep. 2014, 4, 4775. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Xing, G.M.; Blanco, E.; Song, Y.; Zhao, L.N.; Sun, B.Y.; Li, X.D.; Wang, P.C.; Korotcov, A.; Li, W.; et al. Gadolinium metallofullerenol nanoparticles inhibit cancer metastasis through matrix metalloproteinase inhibition: Imprisoning instead of poisoning cancer cells. Nanomed. Nanotechnol. 2012, 8, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Zhao, Y.; Guo, H.; Li, Y.; Tewary, P.; Xing, G.; Hou, W.; Oppenheim, J.J.; Zhang, N. (Gd@C82(OH)22)n nanoparticles induce dendritic cell maturation and activate Th1 immune responses. Acs Nano 2010, 4, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kang, S.G.; Wang, P.; Wang, Y.; Lv, X.; Liu, Y.; Wang, F.; Gu, Z.; Yang, Z.; Weber, J.K.; et al. Molecular mechanism of Gd@C82(OH)22 increasing collagen expression: Implication for encaging tumor. Biomaterials 2018, 152, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, Y.; Sun, B.; Li, H.; Dong, J.; Zhang, L.; Wang, L.; Wang, P.; Zhao, Y.; Chen, C. Polyhydroxylated metallofullerenols stimulate il-1β secretion of macrophage through tlrs/myd88/nf-κb pathway and nlrp3 inflammasome activation. Small 2014, 10, 2362–2372. [Google Scholar] [CrossRef]
- Hadad, A.; Azevedo, D.L.; Caetano, E.W.S.; Freire, V.N.; Mendonca, G.L.F.; Neto, P.L.; Albuquerque, E.L.; Margis, R.; Gottfried, C. Two-Level Adsorption of Ibuprofen on C-60 Fullerene for Transdermal Delivery: Classical Molecular Dynamics and Density Functional Theory Computations. J. Phys. Chem. C 2011, 115, 24501–24511. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, Y.; Liang, X.J. Theranostic nanoparticles engineered for clinic and pharmaceutics. Acc. Chem. Res. 2011, 44, 1114–1122. [Google Scholar] [CrossRef]
- Meng, H.; Xue, M.; Xia, T.; Ji, Z.X.; Tarn, D.Y.; Zink, J.I.; Nel, A.E. Use of Size and a Copolymer Design Feature To Improve the Biodistribution and the Enhanced Permeability and Retention Effect of Doxorubicin-Loaded Mesoporous Silica Nanoparticles in a Murine Xenograft Tumor Model. Acs Nano 2011, 5, 4131–4144. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.X.; Zhang, J.; Dong, J.Q.; Yuan, B.K.; Qiu, X.H.; Yang, S.Y.; Hao, J.A.; Zhang, H.; Yuan, H.; Xing, G.M.; et al. Scanning Tunneling Microscopy Investigation of Substrate-Dependent Adsorption and Assembly of Metallofullerene Gd@C-82 on Cu(111) and Cu(100). J. Phys. Chem. C 2011, 115, 6265–6268. [Google Scholar] [CrossRef]
- Abdullah, A.A.A.; Amr Ahmed, W.; Sherif Abdelaziz, I. Innovative Approaches for Nanobiotechnology in Healthcare Systems; Touseef, A., Shamshi Hassan, M., Eds.; IGI Global: Hershey, PA, USA, 2022; pp. 52–113. [Google Scholar]
- Kang, S.-g.; Tien, H.; Zhou, R. Metallofullerenol Gd@C82(OH)22 distracts the proline-rich-motif from putative binding on the SH3 domain. Nanoscale 2013, 5, 2703–2712. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Huynh, T.; Zhou, R. Non-destructive inhibition of metallofullerenol Gd@C82(OH)22)on WW domain: Implication on signal transduction pathway. Sci. Rep. 2012, 2, 957. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Meng, X.-Y.; Bell, D.R.; Liu, S.; Zhou, R. Inhibition of CYP2C8 by metallofullerenol Gd@C82(OH)22 through blocking substrate channels and substrate recognition sites. Carbon 2018, 127, 667–675. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [Green Version]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as pharmacological target in cancer and neurodegeneration: Focus on its role in apoptosis. Front. Chem. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 2008, 321, 1206–1210. [Google Scholar] [CrossRef] [Green Version]
- Ujwal, R.; Cascio, D.; Colletier, J.P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schredelseker, J.; Paz, A.; Lopez, C.J.; Altenbach, C.; Leung, C.S.; Drexler, M.K.; Chen, J.N.; Hubbell, W.L.; Abramson, J. High resolution structure and double electron-electron resonance of the zebrafish voltage-dependent anion channel 2 reveal an oligomeric population. J. Biol. Chem. 2014, 289, 12566–12577. [Google Scholar] [CrossRef] [Green Version]
- Camara, A.K.S.; Zhou, Y.F.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial vdac1: A key gatekeeper as potential therapeutic target. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.W.; Smith, P.R.; Cognon, B.; D’Arcangelis, D.; Dolginova, E.; Mannella, C.A. Molecular design of the voltage-dependent, anion-selective channel in the mitochondrial outer membrane. J. Struct. Biol. 1995, 114, 41–59. [Google Scholar] [CrossRef]
- Mannella, C.A. Conformational changes in the mitochondrial channel protein, VDAC, and their functional implications. J. Struct. Biol. 1998, 121, 207–218. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kroemer, G. Mitochondrial apoptosis without VDAC. Nat. Cell. Biol. 2007, 9, 487–489. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Zakar, M.; Rosenthal, K.; Abu-Hamad, S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Bba-Bioenergetics 2009, 1787, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell. Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Anand, U.; Nahon-Crystal, E.; Di Carlo, M.; Shteinfer-Kuzmine, A. Adverse Effects of Metformin From Diabetes to COVID-19, Cancer, Neurodegenerative Diseases, and Aging: Is VDAC1 a Common Target? Front. Physiol. 2021, 12. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Villinger, S.; Briones, R.; Giller, K.; Zachariae, U.; Lange, A.; de Groot, B.L.; Griesinger, C.; Becker, S.; Zweckstetter, M. Functional dynamics in the voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2010, 107, 22546–22551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 96, 41a. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.N.; Wang, M.; Zhao, L.C.; Sun, B.Y.; Wang, B.; Chen, H.Q.; Zhao, Y.L.; Chai, Z.F.; Feng, W.Y. Quantitative analysis of Gd@C82(OH)22 and cisplatin uptake in single cells by inductively coupled plasma mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2383–2391. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison Of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 27–38. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single-crystals—a new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle—an Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Roux, B. The Calculation of the Potential of Mean Force Using Computer-Simulations. Comput. Phys. Commun. 1995, 91, 275–282. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. Multidimensional Free-Energy Calculations Using the Weighted Histogram Analysis Method. J. Comput. Chem. 1995, 16, 1339–1350. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Non-physical sampling distributions in monte-carlo free-energy estimation—umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Hub, J.S.; de Groot, B.L.; van der Spoel, D. g_wham-A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.; Sima, C.; Braga-Neto, U.M.; Dougherty, E.R. Unbiased bootstrap error estimation for linear discriminant analysis. EURASIP J. Bioinform. Syst. Biol. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Holst, M. Adaptive numerical treatment of elliptic systems on manifolds. Adv. Comput. Math. 2001, 15, 139–191. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- Calvaresi, M.; Furini, S.; Domene, C.; Bottoni, A.; Zerbetto, F. Blocking the passage: C60 geometrically clogs K(+) channels. ACS Nano 2015, 9, 4827–4834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, N.; Zheng, Z.; Cerutti, D.S.; Merz, K.M. On the fly estimation of host-guest binding free energies using the movable type method: Participation in the SAMPL5 blind challenge. J. Comput. Aid. Mol. Des. 2017, 31, 47–60. [Google Scholar] [CrossRef]
- Deng, N.; Cui, D.; Zhang, B.W.; Xia, J.; Cruz, J.; Levy, R. Comparing alchemical and physical pathway methods for computing the absolute binding free energy of charged ligands. Phys. Chem. Chem. Phys. 2018, 20, 17081–17092. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Tian, S.; Zhou, S.; Li, Y.; Li, D.; Xu, L.; Shen, M.; Pan, P.; Hou, T. Revealing the favorable dissociation pathway of type II kinase inhibitors via enhanced sampling simulations and two-end-state calculations. Sci. Rep. 2015, 5, 8457. [Google Scholar] [CrossRef]
- Zhou, J.K.; Yang, D.Y.; Sheu, S.Y. The molecular mechanism of ligand unbinding from the human telomeric G-quadruplex by steered molecular dynamics and umbrella sampling simulations. Phys. Chem. Chem. Phys. 2015, 17, 12857–12869. [Google Scholar] [CrossRef]
- Pathak, A.K.; Bandyopadhyay, T. Unbinding free energy of acetylcholinesterase bound oxime drugs along the gorge pathway from metadynamics-umbrella sampling investigation. Proteins 2014, 82, 1799–1818. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.J.; Roux, B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proc. Natl. Acad. Sci. USA 2005, 102, 6825–6830. [Google Scholar] [CrossRef] [Green Version]
- Lan, N.T.; Vu, K.B.; Ngoc, M.K.D.; Tran, P.T.; Hiep, D.M.; Tung, N.T.; Ngo, S.T. Prediction of AChE-ligand affinity using the umbrella sampling simulation. J. Mol. Graph. Model. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, O.P.; Paz, A.; Adelman, J.L.; Colletier, J.P.; Abramson, J.; Grabe, M. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat. Struct. Mol. Biol. 2014, 21, 626–632. [Google Scholar] [CrossRef] [Green Version]
- Yehezkel, G.; Hadad, N.; Zaid, H.; Sivan, S.; Shoshan-Barmatz, V. Nucleotide-binding sites in the voltage-dependent anion channel: Characterization and localization. J. Biol. Chem. 2006, 281, 5938–5946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehezkel, G.; Abu-Hamad, S.; Shoshan-Barmatz, V. An N-terminal nucleotide-binding site in VDAC1: Involvement in regulating mitochondrial function. J. Cell. Physiol. 2007, 212, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett 2011, 585, 2363–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, E.M.; Vu, G.T.; Homble, F.; Prevost, M. Dual Mechanism of Ion Permeation through VDAC Revealed with Inorganic Phosphate Ions and Phosphate Metabolites. PLoS ONE 2015, 10, e0121746. [Google Scholar] [CrossRef] [Green Version]
- Noskov, S.Y.; Rostovtseva, T.K.; Bezrukov, S.M. ATP Transport through VDAC and the VDAC-Tubulin Complex Probed by Equilibrium and Nonequilibrium MD Simulations. Biochemistry 2013, 52, 9246–9256. [Google Scholar] [CrossRef]
- Villinger, S.; Giller, K.; Bayrhuber, M.; Lange, A.; Griesinger, C.; Becker, S.; Zweckstetter, M. Nucleotide interactions of the human voltage-dependent anion channel. J. Biol. Chem. 2014, 289, 13397–13406. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Yang, N.; Su, J.; Wu, C.; Liu, S.; Chang, L.; Plant, L.D.; Meng, X. The Molecular Mechanism of Human Voltage-Dependent Anion Channel 1 Blockade by the Metallofullerenol Gd@C82(OH)22: An In Silico Study. Biomolecules 2022, 12, 123. https://doi.org/10.3390/biom12010123
Wang X, Yang N, Su J, Wu C, Liu S, Chang L, Plant LD, Meng X. The Molecular Mechanism of Human Voltage-Dependent Anion Channel 1 Blockade by the Metallofullerenol Gd@C82(OH)22: An In Silico Study. Biomolecules. 2022; 12(1):123. https://doi.org/10.3390/biom12010123
Chicago/Turabian StyleWang, Xiuxiu, Nan Yang, Juan Su, Chenchen Wu, Shengtang Liu, Lei Chang, Leigh D. Plant, and Xuanyu Meng. 2022. "The Molecular Mechanism of Human Voltage-Dependent Anion Channel 1 Blockade by the Metallofullerenol Gd@C82(OH)22: An In Silico Study" Biomolecules 12, no. 1: 123. https://doi.org/10.3390/biom12010123