
Factors Influencing Gallstone Formation: A Review of the Literature

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Classification and Formation of Gallstones
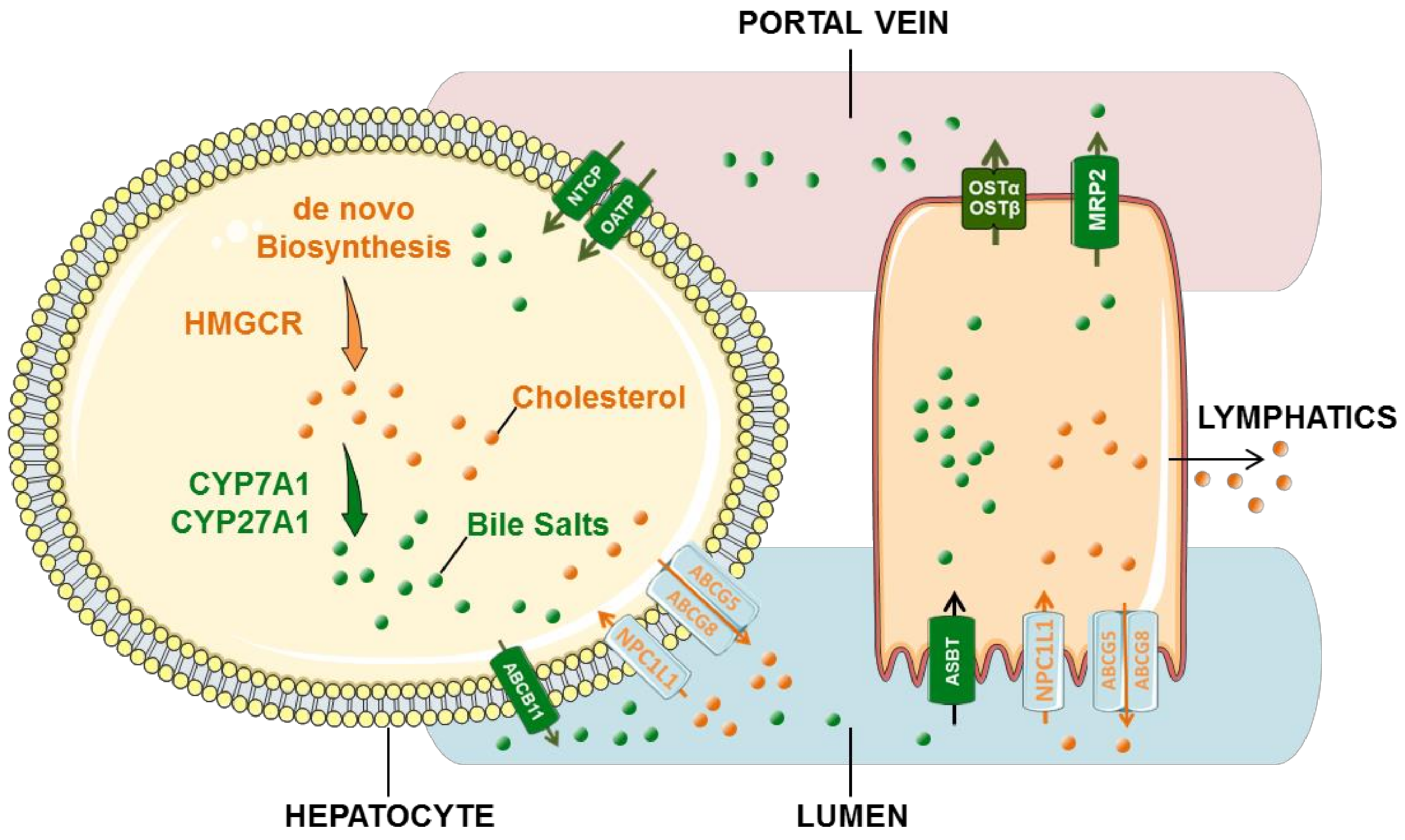
3. Cholesterol and Bile Acid Circulation
3.1. Cholesterol Circulation
3.2. Bile Acid Cycle
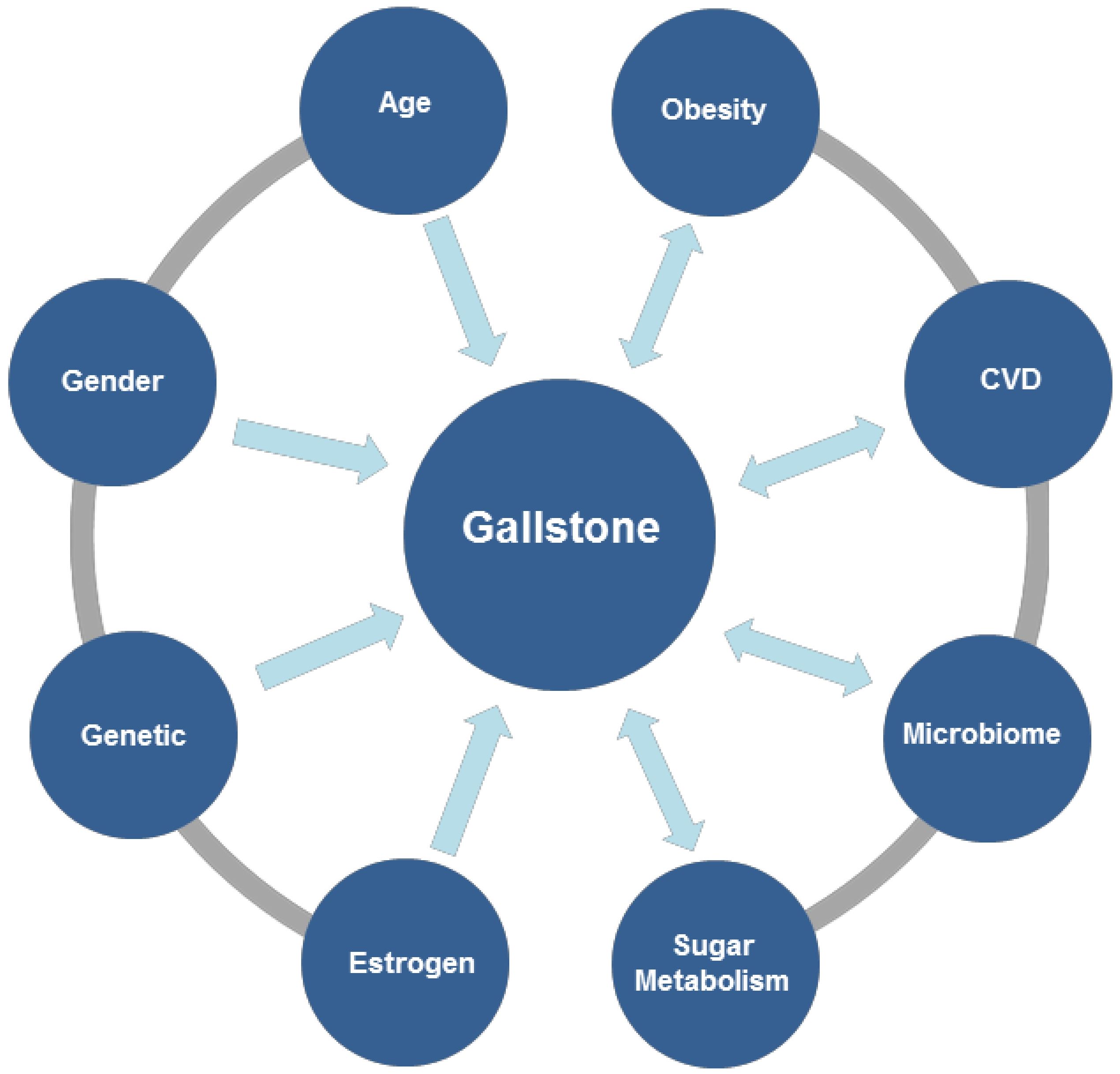
4. Factors Influencing Gallstone Formation
4.1. Genetic Mechanism of Gallstone Formation
4.2. Gallbladder Contraction
4.3. Microbiome
4.4. Effect of Estrogen in Gallstone Formation
4.5. Obesity and Gallstone
4.6. Carbohydrate Metabolism
5. Gallstones, Cardiovascular and Cerebrovascular Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tanaka, H.; Imasato, M.; Yamazaki, Y.; Matsumoto, K.; Kunimoto, K.; Delpierre, J.; Meyer, K.; Zerial, M.; Kitamura, N.; Watanabe, M.; et al. Claudin-3 regulates bile canalicular paracellular barrier and cholesterol gallstone core formation in mice. J. Hepatol. 2018, 69, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Shabanzadeh, D.M.; Sorensen, L.T.; Jorgensen, T. Abdominal Symptoms and Incident Gallstones in a Population Unaware of Gallstone Status. Can. J. Gastroenterol. Hepatol. 2016, 2016, 9730687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barahona Ponce, C.; Scherer, D.; Brinster, R.; Boekstegers, F.; Marcelain, K.; Garate-Calderon, V.; Muller, B.; de Toro, G.; Retamales, J.; Barajas, O.; et al. Gallstones, Body Mass Index, C-Reactive Protein, and Gallbladder Cancer: Mendelian Randomization Analysis of Chilean and European Genotype Data. Hepatology 2021, 73, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Portincasa, P.; Afdhal, N.H.; Wang, D.Q. Lith genes and genetic analysis of cholesterol gallstone formation. Gastroenterol. Clin. 2010, 39, 185–207. [Google Scholar] [CrossRef]
- Idowu, B.M.; Onigbinde, S.O.; Ebie, I.U.; Adeyemi, M.T. Gallbladder diseases in pregnancy: Sonographic findings in an indigenous African population. J. Ultrason. 2019, 19, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Shabanzadeh, D.M.; Holmboe, S.A.; Sorensen, L.T.; Linneberg, A.; Andersson, A.M.; Jorgensen, T. Are incident gallstones associated to sex-dependent changes with age? A cohort Study. Andrology 2017, 5, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Granel-Villach, L.; Gil-Fortuno, M.; Fortea-Sanchis, C.; Gamon-Giner, R.L.; Martinez-Ramos, D.; Escrig-Sos, V.J. Factors that influence bile fluid microbiology in cholecystectomized patients. Rev. Gastroenterol. Mex. 2020, 85, 257–263. [Google Scholar] [CrossRef]
- Nardone, G.; Ferber, I.A.; Miller, L.J. The integrity of the cholecystokinin receptor gene in gallbladder disease and obesity. Hepatology 1995, 22, 1751–1753. [Google Scholar] [PubMed]
- Qiao, T.; Ma, R.H.; Luo, X.B.; Yang, L.Q.; Luo, Z.L.; Zheng, P.M. The systematic classification of gallbladder stones. PLoS ONE 2013, 8, e74887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.R.; Crowther, R.S.; Cozart, J.C.; Sharrock, P.; Wu, J.; Soloway, R.D. Calcium carbonate in cholesterol gallstones: Polymorphism, distribution, and hypotheses about pathogenesis. Hepatology 1995, 22, 488–496. [Google Scholar] [PubMed]
- Tazuma, S.; Kajiyama, G. Carcinogenesis of malignant lesions of the gall bladder. The impact of chronic inflammation and gallstones. Langenbecks Arch. Surg. 2001, 386, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.E.; Boeltz, S.; Bilyy, R.; Schauer, C.; Mahajan, A.; Widulin, N.; Gruneboom, A.; Herrmann, I.; Boada, E.; Rauh, M.; et al. Neutrophil Extracellular Traps Initiate Gallstone Formation. Immunity 2019, 51, 443–450.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.Y.; Shi, X.J.; Hu, A.; Wang, J.Q.; Ding, Y.; Jiang, W.; Sun, M.; Zhao, X.; Luo, J.; Qi, W.; et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature 2020, 588, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Hancock-Cerutti, W.; Rader, D.J. Opposing effects of ABCG5/8 function on myocardial infarction and gallstone disease. J. Am. Coll. Cardiol. 2014, 63, 2129–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghikia, A.; Zimmermann, F.; Schumann, P.; Jasina, A.; Roessler, J.; Schmidt, D.; Heinze, P.; Kaisler, J.; Nageswaran, V.; Aigner, A.; et al. Propionate attenuates atherosclerosis by immune-dependent regulation of intestinal cholesterol metabolism. Eur. Heart J. 2021, 43, 518–533. [Google Scholar] [CrossRef]
- Ticho, A.L.; Calzadilla, N.; Malhotra, P.; Lee, H.; Anbazhagan, A.N.; Saksena, S.; Dudeja, P.K.; Lee, D.; Gill, R.K.; Alrefai, W.A. NPC1L1-dependent transport of 27-alkyne cholesterol in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2021, 320, C916–C925. [Google Scholar] [CrossRef]
- Chambers, K.F.; Day, P.E.; Aboufarrag, H.T.; Kroon, P.A. Polyphenol Effects on Cholesterol Metabolism via Bile Acid Biosynthesis, CYP7A1: A Review. Nutrients 2019, 11, 2588. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.D.; Paumgartner, G.; Wahlstrom, A.; Schwabl, P.; Reiberger, T.; Leditznig, N.; Stojakovic, T.; Rohr-Udilova, N.; Chiba, P.; Marschall, H.U.; et al. Metabolic preconditioning protects BSEP/ABCB11(-/-) mice against cholestatic liver injury. J. Hepatol. 2017, 66, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Deng, F.; Bae, Y.H. Bile acid transporter-mediated oral drug delivery. J. Control. Release 2020, 327, 100–116. [Google Scholar] [CrossRef]
- Gao, T.; Feridooni, H.A.; Howlett, S.E.; Pelis, R.M. Influence of age on intestinal bile acid transport in C57BL/6 mice. Pharmacol. Res. Perspect. 2017, 5, e00287. [Google Scholar] [CrossRef] [Green Version]
- Oswald, S. Organic Anion Transporting Polypeptide (OATP) transporter expression, localization and function in the human intestine. Pharmacol. Ther. 2019, 195, 39–53. [Google Scholar] [CrossRef]
- Li, T.T.; An, J.X.; Xu, J.Y.; Tuo, B.G. Overview of organic anion transporters and organic anion transporter polypeptides and their roles in the liver. World J. Clin. Cases 2019, 7, 3915–3933. [Google Scholar] [CrossRef]
- Jaruvongvanich, V.; Sanguankeo, A.; Upala, S. Significant Association Between Gallstone Disease and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Dig. Dis. Sci. 2016, 61, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Grigor’eva, I.N.; Romanova, T.I. Gallstone Disease and Microbiome. Microorganisms 2020, 8, 835. [Google Scholar] [CrossRef]
- Wang, T.Y.; Portincasa, P.; Liu, M.; Tso, P.; Wang, D.Q. Mouse models of gallstone disease. Curr. Opin. Gastroenterol. 2018, 34, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mitsche, M.A.; Lutjohann, D.; Cohen, J.C.; Xie, X.S.; Hobbs, H.H. Relative roles of ABCG5/ABCG8 in liver and intestine. J. Lipid Res. 2015, 56, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.H.; Liu, M.; Portincasa, P.; Wang, D.Q. Recent Advances in the Critical Role of the Sterol Efflux Transporters ABCG5/G8 in Health and Disease. Adv. Exp. Med. Biol. 2020, 1276, 105–136. [Google Scholar]
- Miettinen, T.A. Phytosterolaemia, xanthomatosis and premature atherosclerotic arterial disease: A case with high plant sterol absorption, impaired sterol elimination and low cholesterol synthesis. Eur. J. Clin. Investig. 1980, 10, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.E.; Tian, H.; Graf, G.A.; Yu, L.; Grishin, N.V.; Schultz, J.; Kwiterovich, P.; Shan, B.; Barnes, R.; Hobbs, H.H. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 2000, 290, 1771–1775. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Lu, K.; Hazard, S.; Yu, H.; Shulenin, S.; Hidaka, H.; Kojima, H.; Allikmets, R.; Sakuma, N.; Pegoraro, R.; et al. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat. Genet. 2001, 27, 79–83. [Google Scholar] [CrossRef]
- Wang, H.H.; Portincasa, P.; Liu, M.; Tso, P.; Wang, D.Q. An Update on the Lithogenic Mechanisms of Cholecystokinin a Receptor (CCKAR), an Important Gallstone Gene for Lith13. Genes 2020, 11, 1438. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Portincasa, P.; Wang, D.Q. Update on the Molecular Mechanisms Underlying the Effect of Cholecystokinin and Cholecystokinin-1 Receptor on the Formation of Cholesterol Gallstones. Curr. Med. Chem. 2019, 26, 3407–3423. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.J.; Harikumar, K.G.; Desai, A.J.; Siddiki, H.; Nguyen, B.D. Kinetics of Gallbladder Emptying During Cholecystokinin Cholescintigraphy as an Indicator of In Vivo Hormonal Sensitivity. J. Nucl. Med. Technol. 2020, 48, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Gether, I.M.; Nexoe-Larsen, C.; Sonne, D.P.; Knop, F.K. Effects of glucagon-like peptides on gallbladder motility. Ugeskr Laeger 2018, 180, V05180386. [Google Scholar] [PubMed]
- Yusta, B.; Matthews, D.; Flock, G.B.; Ussher, J.R.; Lavoie, B.; Mawe, G.M.; Drucker, D.J. Glucagon-like peptide-2 promotes gallbladder refilling via a TGR5-independent, GLP-2R-dependent pathway. Mol. Metab. 2017, 6, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Helaly, G.F.; El-Ghazzawi, E.F.; Kazem, A.H.; Dowidar, N.L.; Anwar, M.M.; Attia, N.M. Detection of Helicobacter pylori infection in Egyptian patients with chronic calcular cholecystitis. Br. J. Biomed. Sci. 2014, 71, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Neri, V.; Margiotta, M.; de Francesco, V.; Ambrosi, A.; Valle, N.D.; Fersini, A.; Tartaglia, N.; Minenna, M.F.; Ricciardelli, C.; Giorgio, F.; et al. DNA sequences and proteic antigens of H. pylori in cholecystic bile and tissue of patients with gallstones. Aliment. Pharmacol. Ther. 2005, 22, 715–720. [Google Scholar] [CrossRef]
- Maurer, K.J.; Ihrig, M.M.; Rogers, A.B.; Ng, V.; Bouchard, G.; Leonard, M.R.; Carey, M.C.; Fox, J.G. Identification of cholelithogenic enterohepatic helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology 2005, 128, 1023–1033. [Google Scholar] [CrossRef]
- Antharam, V.C.; McEwen, D.C.; Garrett, T.J.; Dossey, A.T.; Li, E.C.; Kozlov, A.N.; Mesbah, Z.; Wang, G.P. An Integrated Metabolomic and Microbiome Analysis Identified Specific Gut Microbiota Associated with Fecal Cholesterol and Coprostanol in Clostridium difficile Infection. PLoS ONE 2016, 11, e0148824. [Google Scholar]
- Stewart, L.; Grifiss, J.M.; Jarvis, G.A.; Way, L.W. Biliary bacterial factors determine the path of gallstone formation. Am. J. Surg. 2006, 192, 598–603. [Google Scholar] [CrossRef]
- Tajeddin, E.; Sherafat, S.J.; Majidi, M.R.; Alebouyeh, M.; Alizadeh, A.H.; Zali, M.R. Association of diverse bacterial communities in human bile samples with biliary tract disorders: A survey using culture and polymerase chain reaction-denaturing gradient gel electrophoresis methods. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.; Smith, A.L.; Pellegrini, C.A.; Motson, R.W.; Way, L.W. Pigment gallstones form as a composite of bacterial microcolonies and pigment solids. Ann. Surg. 1987, 206, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kose, S.H.; Grice, K.; Orsi, W.D.; Ballal, M.; Coolen, M.J.L. Author Correction: Metagenomics of pigmented and cholesterol gallstones: The putative role of bacteria. Sci. Rep. 2020, 10, 4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attaallah, W.; Yener, N.; Ugurlu, M.U.; Manukyan, M.; Asmaz, E.; Aktan, A.O. Gallstones and Concomitant Gastric Helicobacter pylori Infection. Gastroenterol. Res. Pract. 2013, 2013, 643109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, M.; Balsari, A.; Rossini, A.; Selleri, S.; Calcaterra, C.; Gariboldi, S.; Zanobbio, L.; Arnaboldi, F.; Shirai, Y.F.; Serrao, G.; et al. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J. Immunol. 2007, 178, 4296–4303. [Google Scholar] [CrossRef] [Green Version]
- Fremont-Rahl, J.J.; Ge, Z.; Umana, C.; Whary, M.T.; Taylor, N.S.; Muthupalani, S.; Carey, M.C.; Fox, J.G.; Maurer, K.J. An analysis of the role of the indigenous microbiota in cholesterol gallstone pathogenesis. PLoS ONE 2013, 8, e70657. [Google Scholar] [CrossRef] [Green Version]
- Maurer, K.J.; Rogers, A.B.; Ge, Z.; Wiese, A.J.; Carey, M.C.; Fox, J.G. Helicobacter pylori and cholesterol gallstone formation in C57L/J mice: A prospective study. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G175–G182. [Google Scholar] [CrossRef] [Green Version]
- Monstein, H.J.; Jonsson, Y.; Zdolsek, J.; Svanvik, J. Identification of Helicobacter pylori DNA in human cholesterol gallstones. Scand. J. Gastroenterol. 2002, 37, 112–119. [Google Scholar] [CrossRef]
- Kaufman, H.S.; Magnuson, T.H.; Lillemoe, K.D.; Frasca, P.; Pitt, H.A. The role of bacteria in gallbladder and common duct stone formation. Ann. Surg. 1989, 209, 584–592. [Google Scholar] [CrossRef]
- Keren, N.; Konikoff, F.M.; Paitan, Y.; Gabay, G.; Reshef, L.; Naftali, T.; Gophna, U. Interactions between the intestinal microbiota and bile acids in gallstones patients. Environ. Microbiol. Rep. 2015, 7, 874–880. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, Z.; Liu, B.; Hou, D.; Liang, Y.; Zhang, J.; Shi, P. Gut microbiota dysbiosis and bacterial community assembly associated with cholesterol gallstones in large-scale study. BMC Genom. 2013, 14, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juran, B.D.; Lazaridis, K.N. Is the FXR the fix for cholesterol gallstone disease? Hepatology 2005, 42, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Molinero, N.; Ruiz, L.; Sanchez, B.; Margolles, A.; Delgado, S. Intestinal Bacteria Interplay with Bile and Cholesterol Metabolism: Implications on Host Physiology. Front. Physiol. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.L.; Takeda, K.; Sundrud, M.S. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. 2019, 12, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Portincasa, P. Recent advances in understanding and managing cholesterol gallstones. F1000Research 2018, 7, 1529. [Google Scholar] [CrossRef]
- Gerard, P.; Lepercq, P.; Leclerc, M.; Gavini, F.; Raibaud, P.; Juste, C. Bacteroides sp. strain D8, the first cholesterol-reducing bacterium isolated from human feces. Appl. Environ. Microbiol. 2007, 73, 5742–5749. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Guo, M.J.; Gao, Q.; Yang, J.F.; Yang, L.; Pang, X.L.; Jiang, X.J. The effects of probiotics on total cholesterol: A meta-analysis of randomized controlled trials. Medicine 2018, 97, e9679. [Google Scholar] [CrossRef]
- Jones, M.L.; Martoni, C.J.; Parent, M.; Prakash, S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase-active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br. J. Nutr. 2012, 107, 1505–1513. [Google Scholar] [CrossRef] [Green Version]
- Celaj, S.; Kourkoumpetis, T. Gallstones in Pregnancy. JAMA 2021, 325, 2410. [Google Scholar] [CrossRef]
- Schwulst, S.J.; Son, M. Nonoperative Management for Pregnant Individuals with Gallstone Disease in the Third Trimester-Reply. JAMA Surg. 2021, 156, 796–797. [Google Scholar] [CrossRef] [PubMed]
- Hossain, G.A.; Islam, S.M.; Mahmood, S.; Chakrabarty, R.K.; Akhter, N. Gall stone in pregnancy. Mymensingh Med. J. 2003, 12, 112–116. [Google Scholar] [PubMed]
- Brown, K.E.; Hirshberg, J.S.; Conner, S.N. Gallbladder and Biliary Disease in Pregnancy. Clin. Obstet. Gynecol. 2020, 63, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, B.S.; Choi, I.; Delage-Mourroux, R.; Ediger, T.R.; Martini, P.G.; Montano, M.; Sun, J.; Weis, K.; Katzenellenbogen, J.A. Molecular mechanisms of estrogen action: Selective ligands and receptor pharmacology. J. Steroid Biochem. Mol. Biol. 2000, 74, 279–285. [Google Scholar] [CrossRef]
- Korstanje, R.; Paigen, B. From QTL to gene: The harvest begins. Nat. Genet. 2002, 31, 235–236. [Google Scholar] [CrossRef]
- Lander, E.S.; Schork, N.J. Genetic dissection of complex traits. Science 1994, 265, 2037–2048. [Google Scholar] [CrossRef] [Green Version]
- Boston Collaborative Drug Surveillance Program, Boston University Medical Center. Surgically confirmed gallbladder disease, venous thromboembolism, and breast tumors in relation to postmenopausal estrogen therapy. N. Engl. J. Med. 1974, 290, 15–19. [Google Scholar] [CrossRef]
- Bennion, L.J.; Ginsberg, R.L.; Gernick, M.B.; Bennett, P.H. Effects of oral contraceptives on the gallbladder bile of normal women. N. Engl. J. Med. 1976, 294, 189–192. [Google Scholar] [CrossRef]
- Grodstein, F.; Colditz, G.A.; Hunter, D.J.; Manson, J.E.; Willett, W.C.; Stampfer, M.J. A prospective study of symptomatic gallstones in women: Relation with oral contraceptives and other risk factors. Obstet. Gynecol. 1994, 84, 207–214. [Google Scholar]
- Petitti, D.B.; Sidney, S.; Perlman, J.A. Increased risk of cholecystectomy in users of supplemental estrogen. Gastroenterology 1988, 94, 91–95. [Google Scholar] [CrossRef]
- Wang, H.H.; Afdhal, N.H.; Wang, D.Q. Overexpression of estrogen receptor alpha increases hepatic cholesterogenesis, leading to biliary hypersecretion in mice. J. Lipid Res. 2006, 47, 778–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, M.A.; Wittenburg, H. Cholesterol gallstone susceptibility loci: A mouse map, candidate gene evaluation, and guide to human LITH genes. Gastroenterology 2006, 131, 1943–1970. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Wang, D.Q.; Portincasa, P.; Lammert, F. Dissecting the genetic heterogeneity of gallbladder stone formation. Semin. Liver Dis. 2011, 31, 157–172. [Google Scholar] [CrossRef]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- De Bari, O.; Wang, T.Y.; Liu, M.; Portincasa, P.; Wang, D.Q. Estrogen induces two distinct cholesterol crystallization pathways by activating ERalpha and GPR30 in female mice. J. Lipid Res. 2015, 56, 1691–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef]
- Waypa, G.B.; Schumacker, P.T. Roles of HIF1 and HIF2 in pulmonary hypertension: It all depends on the context. Eur. Respir. J. 2019, 54, 1901–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, K.; Sugioka, T.; Tsukada, K.; Aizawa, M.; Takizawa, M.; Shimizu, K.; Morimoto, M.; Suematsu, M.; Goda, N. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, improves hepatic microcirculatory patency and oxygen availability in a high-fat-diet-induced fatty liver in mice. Adv. Exp. Med. Biol. 2010, 662, 77–82. [Google Scholar] [PubMed]
- Karimi, S.; Khatami, S.R.; Azarpira, N.; Galehdari, H.; Pakbaz, S. Investigate of AQP gene expression in the liver of mice after ischemia-reperfusion. Mol. Biol. Rep. 2018, 45, 1769–1774. [Google Scholar] [CrossRef]
- Ferri, D.; Mazzone, A.; Liquori, G.E.; Cassano, G.; Svelto, M.; Calamita, G. Ontogeny, distribution, and possible functional implications of an unusual aquaporin, AQP8, in mouse liver. Hepatology 2003, 38, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Calamita, G.; Ferri, D.; Gena, P.; Liquori, G.E.; Marinelli, R.A.; Meyer, G.; Portincasa, P.; Svelto, M. Water transport into bile and role in bile formation. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Cortes, V.; Barrera, F.; Nervi, F. Nonalcoholic fatty liver disease, cholesterol gallstones, and cholecystectomy: New insights on a complex relationship. Curr. Opin. Gastroenterol. 2018, 34, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Lammert, F.; Acalovschi, M.; Ercolani, G.; van Erpecum, K.J.; Gurusamy, K.; van Laarhoven, C.J.; Portincasa, P. EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J. Hepatol. 2016, 65, 146–181. [Google Scholar]
- Nervi, F.; Arrese, M. Cholecystectomy and NAFLD: Does gallbladder removal have metabolic consequences? Am. J. Gastroenterol. 2013, 108, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Stender, S.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjaerg-Hansen, A. The ABCG5/8 cholesterol transporter and myocardial infarction versus gallstone disease. J. Am. Coll. Cardiol. 2014, 63, 2121–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgadottir, A.; Thorleifsson, G.; Alexandersson, K.F.; Tragante, V.; Thorsteinsdottir, M.; Eiriksson, F.F.; Gretarsdottir, S.; Bjornsson, E.; Magnusson, O.; Sveinbjornsson, G.; et al. Genetic variability in the absorption of dietary sterols affects the risk of coronary artery disease. Eur. Heart J. 2020, 41, 2618–2628. [Google Scholar] [CrossRef]
- Zhao, S.F.; Wang, A.M.; Yu, X.J.; Wang, L.L.; Xu, X.N.; Shi, G.J. Association between gallstone and cardio-cerebrovascular disease: Systematic review and meta-analysis. Exp. Ther. Med. 2019, 17, 3092–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabanzadeh, D.M.; Skaaby, T.; Sorensen, L.T.; Jorgensen, T. Screen-detected gallstone disease and cardiovascular disease. Eur. J. Epidemiol. 2017, 32, 501–510. [Google Scholar] [CrossRef]
- Zidi, W.; Allal-Elasmi, M.; Zayani, Y.; Zaroui, A.; Guizani, I.; Feki, M.; Mourali, M.S.; Mechmeche, R.; Kaabachi, N. Metabolic Syndrome, Independent Predictor for Coronary Artery Disease. Clin. Lab. 2015, 61, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Qiao, Q.H.; Zhang, S.C.; Chen, Y.H.; Chao, G.Q.; Fang, L.Z. Metabolic syndrome and gallstone disease. World J. Gastroenterol. 2012, 18, 4215–4220. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D. Gallstone Disease and Increased Risk of Ischemic Heart Disease: Causal Association or Epiphenomenon? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2073–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiuzzi, C.; Sanchis-Gomar, F.; Lippi, G. Worldwide burden of LDL cholesterol: Implications in cardiovascular disease. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Yamanashi, Y.; Takada, T.; Yamamoto, H.; Suzuki, H. NPC1L1 Facilitates Sphingomyelin Absorption and Regulates Diet-Induced Production of VLDL/LDL-associated S1P. Nutrients 2020, 12, 2641. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W.; Davis, H.R., Jr.; Zhu, L.J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanashi, Y.; Takada, T.; Suzuki, H. Niemann-Pick C1-like 1 overexpression facilitates ezetimibe-sensitive cholesterol and beta-sitosterol uptake in CaCo-2 cells. J. Pharmacol. Exp. Ther. 2007, 320, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Calvo, M.; Lisnock, J.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R., Jr.; Dean, D.C.; Detmers, P.A.; et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137. [Google Scholar] [CrossRef] [Green Version]
- Lauridsen, B.K.; Stender, S.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjaerg-Hansen, A. Genetic variation in the cholesterol transporter NPC1L1, ischaemic vascular disease, and gallstone disease. Eur. Heart J. 2015, 36, 1601–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, P.; Grassia, G.; Botha, S.; Sattar, N.; Maffia, P. Targeting inflammation to reduce cardiovascular disease risk: A realistic clinical prospect? Br. J. Pharmacol. 2017, 174, 3898–3913. [Google Scholar] [CrossRef] [Green Version]
- Seaman, C.D.; Yabes, J.; Comer, D.M.; Ragni, M.V. Does deficiency of von Willebrand factor protect against cardiovascular disease? Analysis of a national discharge register. J. Thromb. Haemost. 2015, 13, 1999–2003. [Google Scholar] [CrossRef] [Green Version]
- Taskin, H.E.; Kocael, A.; Kocael, P.; Zengin, K.; Al, M.; Sozer, V.; Buchwald, J.N.; McGlennon, T.W.; Uzun, H. Original contribution: Sleeve gastrectomy reduces soluble lectin-like oxidized low-density lipoprotein receptor-1 (sLOX-1) levels in patients with morbid obesity. Surg. Endosc. 2022, 36, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.C.; Raffield, L.M.; Moxley, A.H.; Miller-Fleming, T.W.; Auer, P.L.; Franceschini, N.; Ngo, D.; Thornton, T.A.; Lange, E.M.; Li, Y.; et al. Soluble Urokinase Plasminogen Activator Receptor: Genetic Variation and Cardiovascular Disease Risk in Black Adults. Circ. Genom. Precis. Med. 2021, 14, e003421. [Google Scholar] [CrossRef] [PubMed]
- Ballout, R.A.; Remaley, A.T. GlycA: A New Biomarker for Systemic Inflammation and Cardiovascular Disease (CVD) Risk Assessment. J. Lab. Precis. Med. 2020, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Mukewar, S.; Mara, K.C.; Dierkhising, R.A.; Kamath, P.S.; Cummins, N. Epidemiologic Factors, Clinical Presentation, Causes, and Outcomes of Liver Abscess: A 35-Year Olmsted County Study. Mayo Clin. Proc. Innov. Qual. Outcomes 2018, 2, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Warren, J.; Yip, J.; Ji, Y.; Hao, S.; Han, W.; Ding, Y. Factors Influencing Gallstone Formation: A Review of the Literature. Biomolecules 2022, 12, 550. https://doi.org/10.3390/biom12040550
Sun H, Warren J, Yip J, Ji Y, Hao S, Han W, Ding Y. Factors Influencing Gallstone Formation: A Review of the Literature. Biomolecules. 2022; 12(4):550. https://doi.org/10.3390/biom12040550
Chicago/Turabian StyleSun, Hao, Jonathan Warren, James Yip, Yu Ji, Shaolong Hao, Wei Han, and Yuchuan Ding. 2022. "Factors Influencing Gallstone Formation: A Review of the Literature" Biomolecules 12, no. 4: 550. https://doi.org/10.3390/biom12040550