
Similarity and Diversity of Presynaptic Molecules at Neuromuscular Junctions and Central Synapses


Abstract
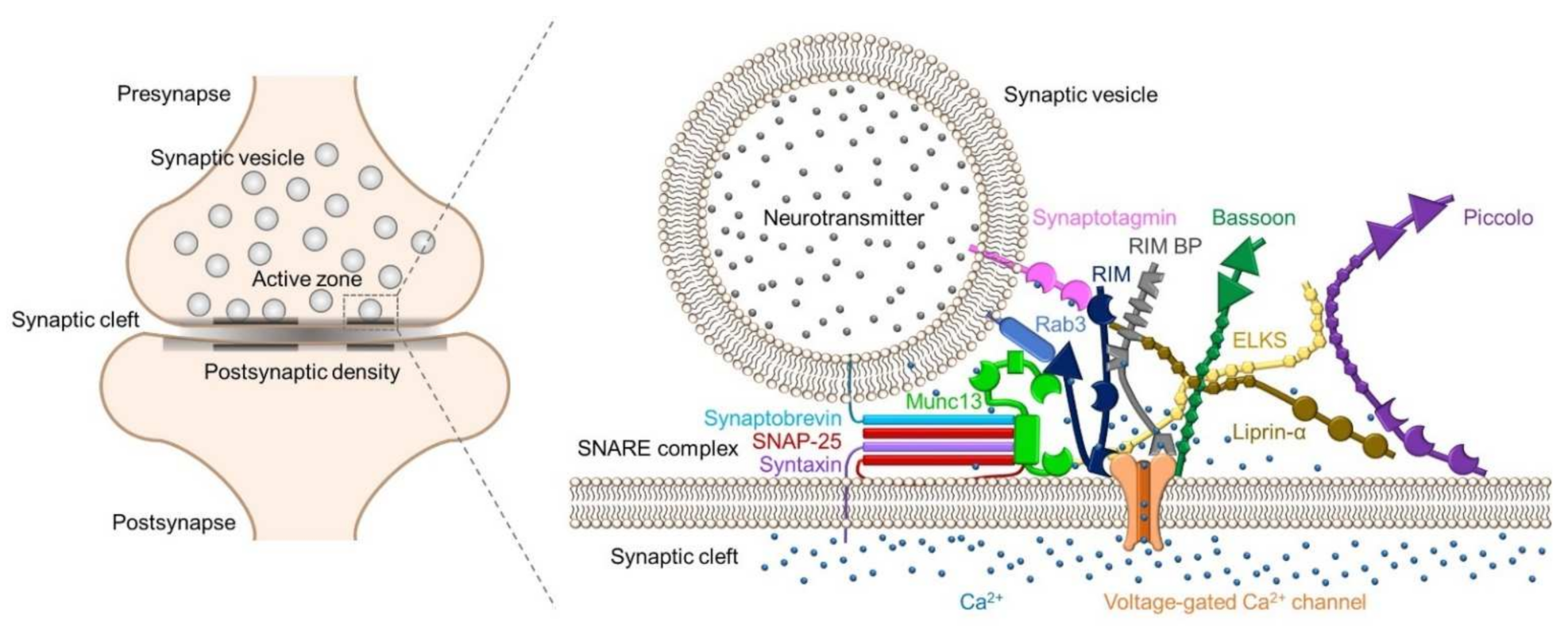
:1. Introduction
2. Similarities and Differences in Presynaptic Structure and Molecules at NMJs and Central Synapses
2.1. Number and Size of Active Zones at NMJs and Central Nervous System Synapses
2.2. Synaptic Proteins in Active Zones
2.3. VGCC Types and Clusters in Active Zones
2.4. Spatial Arrangement of VGCCs and Active Zone Proteins
2.5. Nanocolumn Structures Linking Pre- and Postsynaptic Sites
3. Roles of Exocytosis-Related Proteins during Aging and in Neurological Diseases
3.1. Molecules Inhibiting Exocytosis-Related Proteins and Neurological Diseases of the Peripheral and Central Nervous Systems
3.2. Genetic Alterations in Exocytosis-Related Proteins and Neurological Diseases
3.3. Presynaptic Structure and Aging
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Couteaux, R.; Pecot-Dechavassine, M. Synaptic vesicles and pouches at the level of “active zones” of the neuromuscular junction. C. R. Acad. Hebd. Seances Acad. Sci. D 1970, 271, 2346–2349. [Google Scholar] [PubMed]
- Sudhof, T.C. The presynaptic active zone. Neuron 2012, 75, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Willig, K.I.; Rizzoli, S.O.; Westphal, V.; Jahn, R.; Hell, S.W. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature 2006, 440, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Holderith, N.; Lorincz, A.; Katona, G.; Rozsa, B.; Kulik, A.; Watanabe, M.; Nusser, Z. Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat. Neurosci. 2012, 15, 988–997. [Google Scholar] [CrossRef] [Green Version]
- Nishimune, H.; Badawi, Y.; Mori, S.; Shigemoto, K. Dual-color STED microscopy reveals a sandwich structure of Bassoon and Piccolo in active zones of adult and aged mice. Sci. Rep. 2016, 6, 27935. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.-H.; Chen, H.; Li, T.P.; Metzbower, S.R.; MacGillavry, H.D.; Blanpied, T.A. A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature 2016, 536, 210–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, H.; Ariyoshi, T.; Kimpara, N.; Sugao, K.; Taiko, I.; Takikawa, K.; Asanuma, D.; Namiki, S.; Hirose, K. Synaptic weight set by Munc13-1 supramolecular assemblies. Nat. Neurosci. 2018, 21, 41–49. [Google Scholar] [CrossRef]
- Takikawa, K.; Asanuma, D.; Namiki, S.; Sakamoto, H.; Ariyoshi, T.; Kimpara, N.; Hirose, K. High-throughput development of a hybrid-type fluorescent glutamate sensor for analysis of synaptic transmission. Angew. Chem. Int. Ed. Engl. 2014, 53, 13439–13443. [Google Scholar] [CrossRef]
- Thevenon, J.; Callier, P.; Andrieux, J.; Delobel, B.; David, A.; Sukno, S.; Minot, D.; Mosca Anne, L.; Marle, N.; Sanlaville, D.; et al. 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. Eur. J. Hum. Genet. 2013, 21, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, D.N.; Horvath, R.; Sowden, J.E.; Gonzalez, M.; Sanchez-Mejias, A.; Guan, Z.; Whittaker, R.G.; Almodovar, J.L.; Lane, M.; Bansagi, B.; et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am. J. Hum. Genet. 2014, 95, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipstein, N.; Verhoeven-Duif, N.M.; Michelassi, F.E.; Calloway, N.; van Hasselt, P.M.; Pienkowska, K.; van Haaften, G.; van Haelst, M.M.; van Empelen, R.; Cuppen, I.; et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J. Clin. Investig. 2017, 127, 1005–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salpietro, V.; Lin, W.; Delle Vedove, A.; Storbeck, M.; Liu, Y.; Efthymiou, S.; Manole, A.; Wiethoff, S.; Ye, Q.; Saggar, A.; et al. Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann. Neurol. 2017, 81, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingo, A.P.; Dammer, E.B.; Breen, M.S.; Logsdon, B.A.; Duong, D.M.; Troncosco, J.C.; Thambisetty, M.; Beach, T.G.; Serrano, G.E.; Reiman, E.M.; et al. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat. Commun. 2019, 10, 1619. [Google Scholar] [CrossRef] [Green Version]
- Clarke, G.L.; Chen, J.; Nishimune, H. Presynaptic Active Zone Density during Development and Synaptic Plasticity. Front. Mol. Neurosci. 2012, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, R.; Cano, R.; Casanas, J.J.; Gaffield, M.A.; Betz, W.J.; Tabares, L. Active zones and the readily releasable pool of synaptic vesicles at the neuromuscular junction of the mouse. J. Neurosci. 2011, 31, 2000–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, C.R. The Structure of Human Neuromuscular Junctions: Some Unanswered Molecular Questions. Int. J. Mol. Sci. 2017, 18, 2183. [Google Scholar] [CrossRef]
- Chen, J.; Mizushige, T.; Nishimune, H. Active zone density is conserved during synaptic growth but impaired in aged mice. J. Comp. Neurol. 2012, 520, 434–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, I.; Alhindi, A.; Leite, A.S.; Logie, C.; Gibbs, A.; Murray, O.; Farrukh, R.; Pirie, R.; Proudfoot, C.; Clutton, R.; et al. Comparative anatomy of the mammalian neuromuscular junction. J. Anat. 2020, 237, 827–836. [Google Scholar] [CrossRef]
- Fukunaga, H.; Engel, A.G.; Osame, M.; Lambert, E.H. Paucity and disorganization of presynaptic membrane active zones in the Lambert-Eaton myasthenic syndrome. Muscle Nerve 1982, 5, 686–697. [Google Scholar] [CrossRef]
- Schikorski, T.; Stevens, C.F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 1997, 17, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Schikorski, T.; Stevens, C.F. Quantitative fine-structural analysis of olfactory cortical synapses. Proc. Natl. Acad. Sci. USA 1999, 96, 4107–4112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satzler, K.; Sohl, L.F.; Bollmann, J.H.; Borst, J.G.; Frotscher, M.; Sakmann, B.; Lubke, J.H. Three-dimensional reconstruction of a calyx of Held and its postsynaptic principal neuron in the medial nucleus of the trapezoid body. J. Neurosci. 2002, 22, 10567–10579. [Google Scholar] [CrossRef] [PubMed]
- Rowley, K.L.; Mantilla, C.B.; Ermilov, L.G.; Sieck, G.C. Synaptic vesicle distribution and release at rat diaphragm neuromuscular junctions. J. Neurophysiol. 2007, 98, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Taschenberger, H.; Leao, R.M.; Rowland, K.C.; Spirou, G.A.; von Gersdorff, H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron 2002, 36, 1127–1143. [Google Scholar] [CrossRef] [Green Version]
- Yeow, M.B.; Peterson, E.H. Active zone organization and vesicle content scale with bouton size at a vertebrate central synapse. J. Comp. Neurol. 1991, 307, 475–486. [Google Scholar] [CrossRef]
- Tom Dieck, S.; Sanmarti-Vila, L.; Langnaese, K.; Richter, K.; Kindler, S.; Soyke, A.; Wex, H.; Smalla, K.H.; Kampf, U.; Franzer, J.T.; et al. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J. Cell Biol. 1998, 142, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, T.; Takao-Rikitsu, E.; Inoue, E.; Inoue, M.; Takeuchi, M.; Matsubara, K.; Deguchi-Tawarada, M.; Satoh, K.; Morimoto, K.; Nakanishi, H.; et al. Cast: A novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13-1. J. Cell Biol. 2002, 158, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, X.; Biederer, T.; Sudhof, T.C. A family of RIM-binding proteins regulated by alternative splicing: Implications for the genesis of synaptic active zones. Proc. Natl. Acad. Sci. USA 2002, 99, 14464–14469. [Google Scholar] [CrossRef] [Green Version]
- Brose, N.; Hofmann, K.; Hata, Y.; Sudhof, T.C. Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J. Biol. Chem. 1995, 270, 25273–25280. [Google Scholar] [CrossRef] [Green Version]
- Cases-Langhoff, C.; Voss, B.; Garner, A.M.; Appeltauer, U.; Takei, K.; Kindler, S.; Veh, R.W.; De Camilli, P.; Gundelfinger, E.D.; Garner, C.C. Piccolo, a novel 420 kDa protein associated with the presynaptic cytomatrix. Eur. J. Cell Biol. 1996, 69, 214–223. [Google Scholar] [PubMed]
- Wang, Y.; Okamoto, M.; Schmitz, F.; Hofmann, K.; Sudhof, T.C. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 1997, 388, 593–598. [Google Scholar] [CrossRef]
- Varoqueaux, F.; Sons, M.S.; Plomp, J.J.; Brose, N. Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol. Cell. Biol. 2005, 25, 5973–5984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, S.; Mittelstaedt, T.; Kaeser, P.S.; Padgett, D.; Feldmann, N.; Chevaleyre, V.; Castillo, P.E.; Hammer, R.E.; Han, W.; Schmitz, F.; et al. Redundant functions of RIM1alpha and RIM2alpha in Ca(2+)-triggered neurotransmitter release. EMBO J. 2006, 25, 5852–5863. [Google Scholar] [CrossRef]
- Chen, J.; Billings, S.E.; Nishimune, H. Calcium channels link the muscle-derived synapse organizer laminin beta2 to Bassoon and CAST/Erc2 to organize presynaptic active zones. J. Neurosci. 2011, 31, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, F.; Waites, C.L.; Garner, C.C. Presynaptic active zones in invertebrates and vertebrates. EMBO Rep. 2015, 16, 923–938. [Google Scholar] [CrossRef] [Green Version]
- Badawi, Y.; Nishimune, H. Presynaptic active zones of mammalian neuromuscular junctions: Nanoarchitecture and selective impairments in aging. Neurosci. Res. 2018, 127, 78–88. [Google Scholar] [CrossRef]
- Kaeser, P.S.; Kwon, H.B.; Chiu, C.Q.; Deng, L.; Castillo, P.E.; Sudhof, T.C. RIM1alpha and RIM1beta are synthesized from distinct promoters of the RIM1 gene to mediate differential but overlapping synaptic functions. J. Neurosci. 2008, 28, 13435–13447. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, P.S.; Deng, L.; Fan, M.; Sudhof, T.C. RIM genes differentially contribute to organizing presynaptic release sites. Proc. Natl. Acad. Sci. USA 2012, 109, 11830–11835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Kaeser, P.S.; Sudhof, T.C.; Schneggenburger, R. RIM determines Ca(2)+ channel density and vesicle docking at the presynaptic active zone. Neuron 2011, 69, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Kaeser, P.S.; Xu, W.; Sudhof, T.C. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 2011, 69, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Kaeser, P.S.; Deng, L.; Wang, Y.; Dulubova, I.; Liu, X.; Rizo, J.; Sudhof, T.C. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 2011, 144, 282–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, I.; Rosenmund, C.; Südhof, T.C.; Brose, N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 1999, 400, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Varoqueaux, F.; Sigler, A.; Rhee, J.S.; Brose, N.; Enk, C.; Reim, K.; Rosenmund, C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. USA 2002, 99, 9037–9042. [Google Scholar] [CrossRef] [Green Version]
- Kaeser, P.S.; Deng, L.; Chávez, A.E.; Liu, X.; Castillo, P.E.; Südhof, T.C. ELKS2α/CAST Deletion Selectively Increases Neurotransmitter Release at Inhibitory Synapses. Neuron 2009, 64, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Radulovic, T.; Goral, R.O.; Thomas, C.; Suarez Montesinos, M.; Guerrero-Given, D.; Hagiwara, A.; Putzke, T.; Hida, Y.; Abe, M.; et al. CAST/ELKS Proteins Control Voltage-Gated Ca(2+) Channel Density and Synaptic Release Probability at a Mammalian Central Synapse. Cell Rep. 2018, 24, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Held, R.G.; Liu, C.; Kaeser, P.S. ELKS controls the pool of readily releasable vesicles at excitatory synapses through its N-terminal coiled-coil domains. eLife 2016, 5, e14862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Bickford, L.S.; Held, R.G.; Nyitrai, H.; Sudhof, T.C.; Kaeser, P.S. The active zone protein family ELKS supports Ca2+ influx at nerve terminals of inhibitory hippocampal neurons. J. Neurosci. 2014, 34, 12289–12303. [Google Scholar] [CrossRef] [Green Version]
- Betz, A.; Thakur, P.; Junge, H.J.; Ashery, U.; Rhee, J.S.; Scheuss, V.; Rosenmund, C.; Rettig, J.; Brose, N. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron 2001, 30, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Dulubova, I.; Lou, X.; Lu, J.; Huryeva, I.; Alam, A.; Schneggenburger, R.; Sudhof, T.C.; Rizo, J. A Munc13/RIM/Rab3 tripartite complex: From priming to plasticity? EMBO J. 2005, 24, 2839–2850. [Google Scholar] [CrossRef]
- Andrews-Zwilling, Y.S.; Kawabe, H.; Reim, K.; Varoqueaux, F.; Brose, N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J. Biol. Chem. 2006, 281, 19720–19731. [Google Scholar] [CrossRef] [Green Version]
- Schoch, S.; Castillo, P.E.; Jo, T.; Mukherjee, K.; Geppert, M.; Wang, Y.; Schmitz, F.; Malenka, R.C.; Sudhof, T.C. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 2002, 415, 321–326. [Google Scholar] [CrossRef]
- Wang, Y.; Sugita, S.; Sudhof, T.C. The RIM/NIM family of neuronal C2 domain proteins. Interactions with Rab3 and a new class of Src homology 3 domain proteins. J. Biol. Chem. 2000, 275, 20033–20044. [Google Scholar] [CrossRef] [Green Version]
- Hibino, H.; Pironkova, R.; Onwumere, O.; Vologodskaia, M.; Hudspeth, A.J.; Lesage, F. RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca(2+) channels. Neuron 2002, 34, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Kiyonaka, S.; Wakamori, M.; Miki, T.; Uriu, Y.; Nonaka, M.; Bito, H.; Beedle, A.M.; Mori, E.; Hara, Y.; De Waard, M.; et al. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat. Neurosci. 2007, 10, 691–701. [Google Scholar] [CrossRef] [Green Version]
- Brose, N.; Rosenmund, C.; Rettig, J. Regulation of transmitter release by Unc-13 and its homologues. Curr. Opin. Neurobiol. 2000, 10, 303–311. [Google Scholar] [CrossRef]
- Augustin, I.; Betz, A.; Herrmann, C.; Jo, T.; Brose, N. Differential expression of two novel Munc13 proteins in rat brain. Biochem. J. 1999, 337 Pt 3, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, H.; Mitkovski, M.; Kaeser, P.S.; Hirrlinger, J.; Opazo, F.; Nestvogel, D.; Kalla, S.; Fejtova, A.; Verrier, S.E.; Bungers, S.R.; et al. ELKS1 localizes the synaptic vesicle priming protein bMunc13-2 to a specific subset of active zones. J. Cell Biol. 2017, 216, 1143–1161. [Google Scholar] [CrossRef] [Green Version]
- Rosenmund, C.; Sigler, A.; Augustin, I.; Reim, K.; Brose, N.; Rhee, J.S. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 2002, 33, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Siksou, L.; Varoqueaux, F.; Pascual, O.; Triller, A.; Brose, N.; Marty, S. A common molecular basis for membrane docking and functional priming of synaptic vesicles. Eur. J. Neurosci. 2009, 30, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Camacho, M.; Basu, J.; Trimbuch, T.; Chang, S.; Pulido-Lozano, C.; Chang, S.S.; Duluvova, I.; Abo-Rady, M.; Rizo, J.; Rosenmund, C. Heterodimerization of Munc13 C2A domain with RIM regulates synaptic vesicle docking and priming. Nat. Commun. 2017, 8, 15293. [Google Scholar] [CrossRef] [Green Version]
- Pulido, C.; Trigo, F.F.; Llano, I.; Marty, A. Vesicular release statistics and unitary postsynaptic current at single GABAergic synapses. Neuron 2015, 85, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Augustin, I.; Korte, S.; Rickmann, M.; Kretzschmar, H.A.; Südhof, T.C.; Herms, J.W.; Brose, N. The cerebellum-specific Munc13 isoform Munc13-3 regulates cerebellar synaptic transmission and motor learning in mice. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Deguchi-Tawarada, M.; Inoue, E.; Takao-Rikitsu, E.; Inoue, M.; Ohtsuka, T.; Takai, Y. CAST2: Identification and characterization of a protein structurally related to the presynaptic cytomatrix protein CAST. Genes Cells 2004, 9, 15–23. [Google Scholar] [CrossRef]
- Tokoro, T.; Higa, S.; Deguchi-Tawarada, M.; Inoue, E.; Kitajima, I.; Ohtsuka, T. Localization of the active zone proteins CAST, ELKS, and Piccolo at neuromuscular junctions. Neuroreport 2007, 18, 313–316. [Google Scholar] [CrossRef]
- Ohara-Imaizumi, M.; Aoyagi, K.; Yamauchi, H.; Yoshida, M.; Mori, M.X.; Hida, Y.; Tran, H.N.; Ohkura, M.; Abe, M.; Akimoto, Y.; et al. ELKS/Voltage-Dependent Ca(2+) Channel-beta Subunit Module Regulates Polarized Ca(2+) Influx in Pancreatic beta Cells. Cell Rep. 2019, 26, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Hida, Y.; Ishizaki, H.; Inoue, E.; Tanaka-Okamoto, M.; Yamasaki, M.; Miyazaki, T.; Fukaya, M.; Kitajima, I.; Takai, Y.; et al. The active zone protein CAST regulates synaptic vesicle recycling and quantal size in the mouse hippocampus. Eur. J. Neurosci. 2016, 44, 2272–2284. [Google Scholar] [CrossRef] [PubMed]
- Kittel, R.J.; Wichmann, C.; Rasse, T.M.; Fouquet, W.; Schmidt, M.; Schmid, A.; Wagh, D.A.; Pawlu, C.; Kellner, R.R.; Willig, K.I.; et al. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 2006, 312, 1051–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, S.E.; Clarke, G.L.; Nishimune, H. ELKS1 and Ca(2+) channel subunit beta4 interact and colocalize at cerebellar synapses. Neuroreport 2012, 23, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, S.; Takahashi, T. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J. Physiol. 1998, 509 Pt 2, 419–423. [Google Scholar] [CrossRef]
- Iwasaki, S.; Momiyama, A.; Uchitel, O.D.; Takahashi, T. Developmental changes in calcium channel types mediating central synaptic transmission. J. Neurosci. 2000, 20, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Bischofberger, J.; Jonas, P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J. Neurosci. 2007, 27, 13420–13429. [Google Scholar] [CrossRef] [PubMed]
- Rollenhagen, A.; Satzler, K.; Rodriguez, E.P.; Jonas, P.; Frotscher, M.; Lubke, J.H. Structural determinants of transmission at large hippocampal mossy fiber synapses. J. Neurosci. 2007, 27, 10434–10444. [Google Scholar] [CrossRef] [Green Version]
- Stephens, G.J.; Morris, N.P.; Fyffe, R.E.; Robertson, B. The Cav2.1/alpha1A (P/Q-type) voltage-dependent calcium channel mediates inhibitory neurotransmission onto mouse cerebellar Purkinje cells. Eur. J. Neurosci. 2001, 13, 1902–1912. [Google Scholar] [CrossRef]
- Hefft, S.; Jonas, P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat. Neurosci. 2005, 8, 1319–1328. [Google Scholar] [CrossRef]
- Rosato Siri, M.D.; Uchitel, O.D. Calcium channels coupled to neurotransmitter release at neonatal rat neuromuscular junctions. J. Physiol. 1999, 514 Pt 2, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Protti, D.A.; Reisin, R.; Mackinley, T.A.; Uchitel, O.D. Calcium channel blockers and transmitter release at the normal human neuromuscular junction. Neurology 1996, 46, 1391–1396. [Google Scholar] [CrossRef]
- Indriati, D.W.; Kamasawa, N.; Matsui, K.; Meredith, A.L.; Watanabe, M.; Shigemoto, R. Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: Somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J. Neurosci. 2013, 33, 3668–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Harada, H.; Kamasawa, N.; Matsui, K.; Rothman, J.S.; Shigemoto, R.; Silver, R.A.; DiGregorio, D.A.; Takahashi, T. Nanoscale distribution of presynaptic Ca(2+) channels and its impact on vesicular release during development. Neuron 2015, 85, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Miki, T.; Kaufmann, W.A.; Malagon, G.; Gomez, L.; Tabuchi, K.; Watanabe, M.; Shigemoto, R.; Marty, A. Numbers of presynaptic Ca(2+) channel clusters match those of functionally defined vesicular docking sites in single central synapses. Proc. Natl. Acad. Sci. USA 2017, 114, E5246–E5255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucurenciu, I.; Kulik, A.; Schwaller, B.; Frotscher, M.; Jonas, P. Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron 2008, 57, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Rebola, N.; Reva, M.; Kirizs, T.; Szoboszlay, M.; Lőrincz, A.; Moneron, G.; Nusser, Z.; DiGregorio, D.A. Distinct Nanoscale Calcium Channel and Synaptic Vesicle Topographies Contribute to the Diversity of Synaptic Function. Neuron 2019, 104, 693–710.e699. [Google Scholar] [CrossRef] [PubMed]
- Althof, D.; Baehrens, D.; Watanabe, M.; Suzuki, N.; Fakler, B.; Kulik, Á. Inhibitory and excitatory axon terminals share a common nano-architecture of their Cav2.1 (P/Q-type) Ca(2+) channels. Front. Cell. Neurosci. 2015, 9, 315. [Google Scholar] [CrossRef] [Green Version]
- Pulido, C.; Marty, A. Quantal Fluctuations in Central Mammalian Synapses: Functional Role of Vesicular Docking Sites. Physiol. Rev. 2017, 97, 1403–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littleton, J.T.; Stern, M.; Schulze, K.; Perin, M.; Bellen, H.J. Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell 1993, 74, 1125–1134. [Google Scholar] [CrossRef]
- Geppert, M.; Goda, Y.; Hammer, R.E.; Li, C.; Rosahl, T.W.; Stevens, C.F.; Sudhof, T.C. Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell 1994, 79, 717–727. [Google Scholar] [CrossRef]
- Fernandez-Chacon, R.; Konigstorfer, A.; Gerber, S.H.; Garcia, J.; Matos, M.F.; Stevens, C.F.; Brose, N.; Rizo, J.; Rosenmund, C.; Sudhof, T.C. Synaptotagmin I functions as a calcium regulator of release probability. Nature 2001, 410, 41–49. [Google Scholar] [CrossRef]
- Tucker, W.C.; Weber, T.; Chapman, E.R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 2004, 304, 435–438. [Google Scholar] [CrossRef] [Green Version]
- Naraghi, M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium 1997, 22, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Vyleta, N.P.; Jonas, P. Loose coupling between Ca2+ channels and release sensors at a plastic hippocampal synapse. Science 2014, 343, 665–670. [Google Scholar] [CrossRef]
- Urbano, F.J.; Uchitel, O.D. L-type calcium channels unmasked by cell-permeant Ca2+ buffer at mouse motor nerve terminals. Pflug. Arch. 1999, 437, 523–528. [Google Scholar] [CrossRef]
- Rosato-Siri, M.D.; Piriz, J.; Tropper, B.A.; Uchitel, O.D. Differential Ca2+-dependence of transmitter release mediated by P/Q- and N-type calcium channels at neonatal rat neuromuscular junctions. Eur. J. Neurosci. 2002, 15, 1874–1880. [Google Scholar] [CrossRef]
- Eggermann, E.; Bucurenciu, I.; Goswami, S.P.; Jonas, P. Nanodomain coupling between Ca(2)(+) channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 2011, 13, 7–21. [Google Scholar] [CrossRef]
- Fujimoto, K. Freeze-fracture replica electron microscopy combined with SDS digestion for cytochemical labeling of integral membrane proteins. Application to the immunogold labeling of intercellular junctional complexes. J. Cell Sci. 1995, 108 Pt 11, 3443–3449. [Google Scholar] [CrossRef] [PubMed]
- Nyitrai, H.; Wang, S.S.H.; Kaeser, P.S. ELKS1 Captures Rab6-Marked Vesicular Cargo in Presynaptic Nerve Terminals. Cell Rep. 2020, 31, 107712. [Google Scholar] [CrossRef]
- Valera, A.M.; Doussau, F.; Poulain, B.; Barbour, B.; Isope, P. Adaptation of granule cell to Purkinje cell synapses to high-frequency transmission. J. Neurosci. 2012, 32, 3267–3280. [Google Scholar] [CrossRef] [Green Version]
- Baur, D.; Bornschein, G.; Althof, D.; Watanabe, M.; Kulik, A.; Eilers, J.; Schmidt, H. Developmental tightening of cerebellar cortical synaptic influx-release coupling. J. Neurosci. 2015, 35, 1858–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, I.; Jonas, P. Nanodomain coupling explains Ca(2)(+) independence of transmitter release time course at a fast central synapse. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Kaeser, P.S. Nanoscale Location Matters: Emerging Principles of Ca(2+) Channel Organization at the Presynaptic Active Zone. Neuron 2019, 104, 627–629. [Google Scholar] [CrossRef]
- Davydova, D.; Marini, C.; King, C.; Klueva, J.; Bischof, F.; Romorini, S.; Montenegro-Venegas, C.; Heine, M.; Schneider, R.; Schroder, M.S.; et al. Bassoon specifically controls presynaptic P/Q-type Ca(2+) channels via RIM-binding protein. Neuron 2014, 82, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagwaney, S.; Harlow, M.L.; Jung, J.H.; Szule, J.A.; Ress, D.; Xu, J.; Marshall, R.M.; McMahan, U.J. Macromolecular connections of active zone material to docked synaptic vesicles and presynaptic membrane at neuromuscular junctions of mouse. J. Comp. Neurol. 2009, 513, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.G.; Betz, W.J. Spatial variability in release at the frog neuromuscular junction measured with FM1-43. Can. J. Physiol. Pharm. 1999, 77, 672–678. [Google Scholar] [CrossRef]
- Wyatt, R.M.; Balice-Gordon, R.J. Heterogeneity in synaptic vesicle release at neuromuscular synapses of mice expressing synaptopHluorin. J. Neurosci. 2008, 28, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Dittrich, M.; Stiles, J.R.; Meriney, S.D. Single-pixel optical fluctuation analysis of calcium channel function in active zones of motor nerve terminals. J. Neurosci. 2011, 31, 11268–11281. [Google Scholar] [CrossRef]
- Peled, E.S.; Isacoff, E.Y. Optical quantal analysis of synaptic transmission in wild-type and rab3-mutant Drosophila motor axons. Nat. Neurosci. 2011, 14, 519–526. [Google Scholar] [CrossRef] [PubMed]
- York, A.L.; Zheng, J.Q. Super-Resolution Microscopy Reveals a Nanoscale Organization of Acetylcholine Receptors for Trans-Synaptic Alignment at Neuromuscular Synapses. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voglmaier, S.M.; Kam, K.; Yang, H.; Fortin, D.L.; Hua, Z.; Nicoll, R.A.; Edwards, R.H. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 2006, 51, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Ryan, T.A. CDK5 serves as a major control point in neurotransmitter release. Neuron 2010, 67, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Hruska, M.; Henderson, N.; Le Marchand, S.J.; Jafri, H.; Dalva, M.B. Synaptic nanomodules underlie the organization and plasticity of spine synapses. Nat. Neurosci. 2018, 21, 671–682. [Google Scholar] [CrossRef]
- Haas, K.T.; Compans, B.; Letellier, M.; Bartol, T.M.; Grillo-Bosch, D.; Sejnowski, T.J.; Sainlos, M.; Choquet, D.; Thoumine, O.; Hosy, E. Pre-post synaptic alignment through neuroligin-1 tunes synaptic transmission efficiency. eLife 2018, 7, e31755. [Google Scholar] [CrossRef]
- Crosby, K.C.; Gookin, S.E.; Garcia, J.D.; Hahm, K.M.; Dell’Acqua, M.L.; Smith, K.R. Nanoscale Subsynaptic Domains Underlie the Organization of the Inhibitory Synapse. Cell Rep. 2019, 26, 3284–3297.e3. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Le Corronc, H.; Legendre, P.; Triller, A.; Specht, C.G. Differential regulation of glycinergic and GABAergic nanocolumns at mixed inhibitory synapses. EMBO Rep. 2021, 22, e52154. [Google Scholar] [CrossRef]
- Patton, B.L.; Cunningham, J.M.; Thyboll, J.; Kortesmaa, J.; Westerblad, H.; Edstrom, L.; Tryggvason, K.; Sanes, J.R. Properly formed but improperly localized synaptic specializations in the absence of laminin alpha4. Nat. Neurosci. 2001, 4, 597–604. [Google Scholar] [CrossRef]
- Schiavo, G.; Benfenati, F.; Poulain, B.; Rossetto, O.; Polverino de Laureto, P.; DasGupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Rossetto, O.; Catsicas, S.; Polverino de Laureto, P.; DasGupta, B.R.; Benfenati, F.; Montecucco, C. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A., D, and E. J. Biol. Chem. 1993, 268, 23784–23787. [Google Scholar] [CrossRef]
- Fukunaga, H.; Engel, A.G.; Lang, B.; Newsom-Davis, J.; Vincent, A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc. Natl. Acad. Sci. USA 1983, 80, 7636–7640. [Google Scholar] [CrossRef] [Green Version]
- Lennon, V.A.; Kryzer, T.J.; Griesmann, G.E.; O’Suilleabhain, P.E.; Windebank, A.J.; Woppmann, A.; Miljanich, G.P.; Lambert, E.H. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N. Engl. J. Med. 1995, 332, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, C.; McElroy, O.E.; Eklund, H.W. The Purification and Crystallization of Clostridium botulinum Type A Toxin. Science 1946, 103, 613–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantha, S.S. A centennial review; the 1890 tetanus antitoxin paper of von Behring and Kitasato and the related developments. Keio J. Med. 1991, 40, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Masuyer, G.; Stenmark, P. Botulinum and Tetanus Neurotoxins. Annu. Rev. Biochem. 2019, 88, 811–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fu, Z.; Kim, J.J.; Barbieri, J.T.; Baldwin, M.R. Gangliosides as high affinity receptors for tetanus neurotoxin. J. Biol. Chem. 2009, 284, 26569–26577. [Google Scholar] [CrossRef] [Green Version]
- Blum, F.C.; Tepp, W.H.; Johnson, E.A.; Barbieri, J.T. Multiple domains of tetanus toxin direct entry into primary neurons. Traffic 2014, 15, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Koriazova, L.K.; Montal, M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol. 2003, 10, 13–18. [Google Scholar] [CrossRef]
- Fischer, A.; Montal, M. Crucial role of the disulfide bridge between botulinum neurotoxin light and heavy chains in protease translocation across membranes. J. Biol. Chem. 2007, 282, 29604–29611. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Montal, M. Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 10447–10452. [Google Scholar] [CrossRef] [Green Version]
- Pirazzini, M.; Azarnia Tehran, D.; Zanetti, G.; Megighian, A.; Scorzeto, M.; Fillo, S.; Shone, C.C.; Binz, T.; Rossetto, O.; Lista, F.; et al. Thioredoxin and its reductase are present on synaptic vesicles, and their inhibition prevents the paralysis induced by botulinum neurotoxins. Cell Rep. 2014, 8, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Azarnia Tehran, D.; Zanetti, G.; Lista, F.; Binz, T.; Shone, C.C.; Rossetto, O.; Montecucco, C. The thioredoxin reductase--Thioredoxin redox system cleaves the interchain disulphide bond of botulinum neurotoxins on the cytosolic surface of synaptic vesicles. Toxicon 2015, 107, 32–36. [Google Scholar] [CrossRef]
- Verderio, C.; Rossetto, O.; Grumelli, C.; Frassoni, C.; Montecucco, C.; Matteoli, M. Entering neurons: Botulinum toxins and synaptic vesicle recycling. EMBO Rep. 2006, 7, 995–999. [Google Scholar] [CrossRef] [Green Version]
- Lambert, E.H.; Elmqvist, D. Quantal components of end-plate potentials in the myasthenic syndrome. Ann. N. Y. Acad. Sci. 1971, 183, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Newsom-Davis, J.; Prior, C.; Wray, D. Antibodies to motor nerve terminals: An electrophysiological study of a human myasthenic syndrome transferred to mouse. J. Physiol. 1983, 344, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Waterman, S.A. Autonomic dysfunction in Lambert-Eaton myasthenic syndrome. Clin. Auton. Res. 2001, 11, 145–154. [Google Scholar] [CrossRef]
- Fukuda, T.; Motomura, M.; Nakao, Y.; Shiraishi, H.; Yoshimura, T.; Iwanaga, K.; Tsujihata, M.; Eguchi, K. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann. Neurol. 2003, 53, 21–28. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; Veldink, J.H.; Saris, C.G.; Blauw, H.M.; van Vught, P.W.; Birve, A.; Lemmens, R.; Schelhaas, H.J.; Groen, E.J.; Huisman, M.H.; et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 2009, 41, 1083–1087. [Google Scholar] [CrossRef]
- Glessner, J.T.; Reilly, M.P.; Kim, C.E.; Takahashi, N.; Albano, A.; Hou, C.; Bradfield, J.P.; Zhang, H.; Sleiman, P.M.; Flory, J.H.; et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 10584–10589. [Google Scholar] [CrossRef] [Green Version]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourassa, C.V.; Meijer, I.A.; Merner, N.D.; Grewal, K.K.; Stefanelli, M.G.; Hodgkinson, K.; Ives, E.J.; Pryse-Phillips, W.; Jog, M.; Boycott, K.; et al. VAMP1 mutation causes dominant hereditary spastic ataxia in Newfoundland families. Am. J. Hum. Genet. 2012, 91, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.M.; Scola, R.H.; Lorenzoni, P.J.; Kay, C.S.; Werneck, L.C.; Brengman, J.; Selcen, D.; Engel, A.G. Novel synaptobrevin-1 mutation causes fatal congenital myasthenic syndrome. Ann. Clin. Transl. Neurol. 2017, 4, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Malintan, N.T.; Llano-Rivas, I.; Spaeth, C.G.; Efthymiou, S.; Striano, P.; Vandrovcova, J.; Cutrupi, M.C.; Chimenz, R.; David, E.; et al. Mutations in the Neuronal Vesicular SNARE VAMP2 Affect Synaptic Membrane Fusion and Impair Human Neurodevelopment. Am. J. Hum. Genet. 2019, 104, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Sunaga, Y.; Muramatsu, K.; Kosaki, K.; Sugai, K.; Mizuno, T.; Kouno, M.; Tashiro, M. Variant in the neuronal vesicular SNARE VAMP2 (synaptobrevin-2): First report in Japan. Brain Dev. 2020, 42, 529–533. [Google Scholar] [CrossRef]
- Baker, K.; Gordon, S.L.; Grozeva, D.; van Kogelenberg, M.; Roberts, N.Y.; Pike, M.; Blair, E.; Hurles, M.E.; Chong, W.K.; Baldeweg, T.; et al. Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J. Clin. Investig. 2015, 125, 1670–1678. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.; Gordon, S.L.; Melland, H.; Bumbak, F.; Scott, D.J.; Jiang, T.J.; Owen, D.; Turner, B.J.; Boyd, S.G.; Rossi, M.; et al. SYT1-associated neurodevelopmental disorder: A case series. Brain 2018, 141, 2576–2591. [Google Scholar] [CrossRef] [Green Version]
- Bradberry, M.M.; Courtney, N.A.; Dominguez, M.J.; Lofquist, S.M.; Knox, A.T.; Sutton, R.B.; Chapman, E.R. Molecular Basis for Synaptotagmin-1-Associated Neurodevelopmental Disorder. Neuron 2020, 107, 52–64.e7. [Google Scholar] [CrossRef]
- Whittaker, R.G.; Herrmann, D.N.; Bansagi, B.; Hasan, B.A.; Lofra, R.M.; Logigian, E.L.; Sowden, J.E.; Almodovar, J.L.; Littleton, J.T.; Zuchner, S.; et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology 2015, 85, 1964–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes-Chinea, N.I.; Guan, Z.; Coutts, M.; Vidal, C.; Courel, S.; Rebelo, A.P.; Abreu, L.; Zuchner, S.; Littleton, J.T.; Saporta, M.A. Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype. Neurol. Genet. 2018, 4, e282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkervoort, S.; Mohassel, P.; Laugwitz, L.; Zaki, M.S.; Kamsteeg, E.J.; Maroofian, R.; Chao, K.R.; Verschuuren-Bemelmans, C.C.; Horber, V.; Fock, A.J.M.; et al. Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome. Am. J. Med. Genet. A 2020, 182, 2272–2283. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; van der Linden, H., Jr.; Ferns, M. Recessive congenital myasthenic syndrome caused by a homozygous mutation in SYT2 altering a highly conserved C-terminal amino acid sequence. Am. J. Med. Genet. A 2020, 182, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Selcen, D.; Shen, X.M.; Milone, M.; Harper, C.M. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol. Genet. 2016, 2, e105. [Google Scholar] [CrossRef] [Green Version]
- Elferink, L.A.; Trimble, W.S.; Scheller, R.H. Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J. Biol. Chem. 1989, 264, 11061–11064. [Google Scholar] [CrossRef]
- Nystuen, A.M.; Schwendinger, J.K.; Sachs, A.J.; Yang, A.W.; Haider, N.B. A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics 2007, 8, 1–10. [Google Scholar] [CrossRef]
- Li, J.Y.; Edelmann, L.; Jahn, R.; Dahlstrom, A. Axonal transport and distribution of synaptobrevin I and II in the rat peripheral nervous system. J. Neurosci. 1996, 16, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sugiura, Y.; Lin, W. The role of synaptobrevin1/VAMP1 in Ca2+-triggered neurotransmitter release at the mouse neuromuscular junction. J. Physiol. 2011, 589, 1603–1618. [Google Scholar] [CrossRef]
- Trimble, W.S.; Gray, T.S.; Elferink, L.A.; Wilson, M.C.; Scheller, R.H. Distinct patterns of expression of two VAMP genes within the rat brain. J. Neurosci. 1990, 10, 1380–1387. [Google Scholar] [CrossRef] [Green Version]
- Ferecsko, A.S.; Jiruska, P.; Foss, L.; Powell, A.D.; Chang, W.C.; Sik, A.; Jefferys, J.G. Structural and functional substrates of tetanus toxin in an animal model of temporal lobe epilepsy. Brain Struct. Funct. 2015, 220, 1013–1029. [Google Scholar] [CrossRef] [Green Version]
- Vuong, C.K.; Wei, W.; Lee, J.A.; Lin, C.H.; Damianov, A.; de la Torre-Ubieta, L.; Halabi, R.; Otis, K.O.; Martin, K.C.; O’Dell, T.J.; et al. Rbfox1 Regulates Synaptic Transmission through the Inhibitory Neuron-Specific vSNARE Vamp1. Neuron 2018, 98, 127–141 e127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Adler, M.; Demogines, A.; Borrell, A.; Liu, H.; Tao, L.; Tepp, W.H.; Zhang, S.C.; Johnson, E.A.; Sawyer, S.L.; et al. Widespread sequence variations in VAMP1 across vertebrates suggest a potential selective pressure from botulinum neurotoxins. PLoS Pathog. 2014, 10, e1004177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Y.; Jahn, R.; Dahlstrom, A. Synaptotagmin I is present mainly in autonomic and sensory neurons of the rat peripheral nervous system. Neuroscience 1994, 63, 837–850. [Google Scholar] [CrossRef]
- Marqueze, B.; Boudier, J.A.; Mizuta, M.; Inagaki, N.; Seino, S.; Seagar, M. Cellular localization of synaptotagmin I, II, and III mRNAs in the central nervous system and pituitary and adrenal glands of the rat. J. Neurosci. 1995, 15, 4906–4917. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Lee, J.; Nemcova, P.; Liu, C.; Kaeser, P.S. Synaptotagmin-1 is the Ca(2+) sensor for fast striatal dopamine release. eLife 2020, 9. [Google Scholar] [CrossRef]
- Pang, Z.P.; Melicoff, E.; Padgett, D.; Liu, Y.; Teich, A.F.; Dickey, B.F.; Lin, W.; Adachi, R.; Sudhof, T.C. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J. Neurosci. 2006, 26, 13493–13504. [Google Scholar] [CrossRef]
- Chen, C.; Arai, I.; Satterfield, R.; Young, S.M.; Jonas, P. Synaptotagmin 2 Is the Fast Ca 2+ Sensor at a Central Inhibitory Synapse. Cell Rep. 2017, 18, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Schoch, S.; Deak, F.; Konigstorfer, A.; Mozhayeva, M.; Sara, Y.; Sudhof, T.C.; Kavalali, E.T. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 2001, 294, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.; Schinder, A.F.; Wong, S.T.; Poo, M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 2001, 105, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Peters, A.; Sethares, C.; Luebke, J.I. Synapses are lost during aging in the primate prefrontal cortex. Neuroscience 2008, 152, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, R.J.; Vukovic, J.; Dunlop, S.; Grounds, M.D.; Shavlakadze, T. Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS ONE 2011, 6, e28090. [Google Scholar] [CrossRef] [Green Version]
- Badawi, Y.; Nishimune, H. Impairment Mechanisms and Intervention Approaches for Aged Human Neuromuscular Junctions. Front. Mol. Neurosci. 2020, 13, 568426. [Google Scholar] [CrossRef] [PubMed]
- Geinisman, Y.; deToledo-Morrell, L.; Morrell, F.; Persina, I.S.; Rossi, M. Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus 1992, 2, 437–444. [Google Scholar] [CrossRef]
- Adams, M.M.; Donohue, H.S.; Linville, M.C.; Iversen, E.A.; Newton, I.G.; Brunso-Bechtold, J.K. Age-related synapse loss in hippocampal CA3 is not reversed by caloric restriction. Neuroscience 2010, 171, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Geinisman, Y.; Ganeshina, O.; Yoshida, R.; Berry, R.W.; Disterhoft, J.F.; Gallagher, M. Aging, spatial learning, and total synapse number in the rat CA1 stratum radiatum. Neurobiol. Aging 2004, 25, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Park, C.S.; Janssen, W.G.; Punsoni, M.; Rapp, P.R.; Morrison, J.H. Synaptic characteristics of dentate gyrus axonal boutons and their relationships with aging, menopause, and memory in female rhesus monkeys. J. Neurosci. 2011, 31, 7737–7744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglione, M.; Kochlamazashvili, G.; Eisenberg, T.; Racz, B.; Michael, E.; Toppe, D.; Stumpf, A.; Wirth, A.; Zeug, A.; Muller, F.E.; et al. Spermidine protects from age-related synaptic alterations at hippocampal mossy fiber-CA3 synapses. Sci. Rep. 2019, 9, 19616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Applegate, M.D.; Landfield, P.W. Synaptic vesicle redistribution during hippocampal frequency potentiation and depression in young and aged rats. J. Neurosci. 1988, 8, 1096–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanGuilder, H.D.; Yan, H.; Farley, J.A.; Sonntag, W.E.; Freeman, W.M. Aging alters the expression of neurotransmission-regulating proteins in the hippocampal synaptoproteome. J. Neurochem. 2010, 113, 1577–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: Key actors of synapse function and plasticity. Prog Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef]
- Li, L.; Chin, L.S.; Shupliakov, O.; Brodin, L.; Sihra, T.S.; Hvalby, O.; Jensen, V.; Zheng, D.; McNamara, J.O.; Greengard, P.; et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc. Natl. Acad. Sci. USA 1995, 92, 9235–9239. [Google Scholar] [CrossRef] [Green Version]
- Takei, Y.; Harada, A.; Takeda, S.; Kobayashi, K.; Terada, S.; Noda, T.; Takahashi, T.; Hirokawa, N. Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J. Cell Biol. 1995, 131, 1789–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.A.; Li, L.; Chin, L.S.; Greengard, P.; Smith, S.J. Synaptic vesicle recycling in synapsin I knock-out mice. J. Cell Biol. 1996, 134, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Rosahl, T.W.; Spillane, D.; Missler, M.; Herz, J.; Selig, D.K.; Wolff, J.R.; Hammer, R.E.; Malenka, R.C.; Sudhof, T.C. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 1995, 375, 488–493. [Google Scholar] [CrossRef]
- Baldelli, P.; Fassio, A.; Valtorta, F.; Benfenati, F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J. Neurosci. 2007, 27, 13520–13531. [Google Scholar] [CrossRef] [Green Version]
- Johansson, J.U.; Ericsson, J.; Janson, J.; Beraki, S.; Stanic, D.; Mandic, S.A.; Wikstrom, M.A.; Hokfelt, T.; Ogren, S.O.; Rozell, B.; et al. An ancient duplication of exon 5 in the Snap25 gene is required for complex neuronal development/function. PLoS Genet. 2008, 4, e1000278. [Google Scholar] [CrossRef] [Green Version]
- Mishima, T.; Fujiwara, T.; Kofuji, T.; Saito, A.; Terao, Y.; Akagawa, K. Syntaxin 1B regulates synaptic GABA release and extracellular GABA concentration, and is associated with temperature-dependent seizures. J. Neurochem. 2021, 156, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.C.; Barnes, C.A.; Rao, G.; McNaughton, B.L. Increase in perforant path quantal size in aged F-344 rats. Neurobiol. Aging 1991, 12, 441–448. [Google Scholar] [CrossRef]
- Soghomonian, J.J.; Sethares, C.; Peters, A. Effects of age on axon terminals forming axosomatic and axodendritic inhibitory synapses in prefrontal cortex. Neuroscience 2010, 168, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synapse | Species | Age | Active Zone Number Per Presynaptic Terminal | Presynaptic Terminal Size | Active Zone Density | Active Zone Size | Reference |
|---|---|---|---|---|---|---|---|
| NMJ | Mouse | Day 54 | 780 | 295 μm2 | 2.6/μm2 | 0.082 μm2 | [17] |
| NMJ | Mouse | Adult | 850 | 335.9 μm2 | 2.5/μm2 * | N.P. | [15] |
| NMJ | Human | 67 years | N.P. | 122.7 μm2 | N.P. | N.P. | [18] |
| NMJ | Human | Adult | N.P. | N.P. | 2.6/μm2 | N.P. | [19] |
| Stratum radiatum in CA1 hippocampus | Mouse | Adult | one for more than 90% of synapses | 0.086 μm3 | 13.4/μm3 * | 0.039 μm2 | [20] |
| Piriform cortex layer 1a | Mouse | 7 months | one for more than 90% of synapses | 0.367 μm3 | 3.1/μm3 * | 0.095 μm2 | [21] |
| Piriform cortex layer 1b | Mouse | 7 months | one for more than 90% of synapses | 0.208 μm3 | 5.5/μm3 * | 0.097 μm2 | [21] |
| Calyx of Held | Rats | Day 9 | 554 | 1022 μm2 | 0.54/μm2 * | N.P. | [22] |
| Calyx of Held | Rats | Day 14 | 678 | N.P. | N.P. | 0.089 μm2 | [24] |
| Synapse | Gene Knockout | Excitatory/Inhibitory | Evoked EPSC/IPSC/EPP Amplitude | Miniature EPSC/IPSC/EPP Amplitude | Miniature EPSC/IPSC/EPP Frequency | Release Probability | RRP Size | Reference |
|---|---|---|---|---|---|---|---|---|
| Schaffer collateral to hippocampal CA1 pyramidal cell synapses (3- to 7-week) and cultured hippocampal synapses | Rim1a and Rim1b | Excitatory | N.P. | No change | Decrease | Decrease | N.P. | [37,38] |
| Hippocampal CA1 synapses (3- to 7-week) and cultured hippocampal synapses | Rim1a and Rim1b | Inhibitory | Decrease | No change | Decrease | Decrease | Decrease | [37,38] |
| Calyx of Held synapses in brainstem slices (Postnatal day 9–11) | Rim1 and Rim2 | Excitatory | 1/5-fold decrease | No change | N.P. | ~25% decrease | 75% decrease | [39] |
| Cultured hippocampal synapses | Rim1 and Rim2 | Excitatory | 1/10-fold decrease | No change | 1/10-fold decrease | N.P. | 1/3- to 1/4-fold decrease | [40,41] |
| Cultured hippocampal synapses | Rim1 and Rim2 | Inhibitory | 1/10-fold decrease | No change | 1/3-fold decrease | Decrease | 1/3- to 1/4-fold decrease | [40,41] |
| Diaphragm NMJs (Embryonic day 18.5) | Rim1a and Rim2a | Excitatory | 1/10-fold decrease | No change | No change (decrease in 40 mM KCl) | Decrease | N.P. | [33] |
| Cultured hippocampal synapses | Munc13-1 | Excitatory | Markedly decrease | No change | Decrease | No change | Markedly decrease | [42] |
| Cultured hippocampal synapses | Munc13-1 | Inhibitory | No change | N.P. | N.P. | N.P. | No change | [42,43] |
| Cultured hippocampal synapses | Munc13-2 | Excitatory | No change | No change | No change | N.P. | No change | [43] |
| Cultured hippocampal synapses | Munc13-2 | Inhibitory | No change | No change | No change | N.P. | N.P. | [43] |
| Cultured hippocampal synapses | Munc13-1 and Munc13-2 | Excitatory | Completely abolish | Completely abolish | Completely abolish | N.P. | Completely abolish | [43] |
| Cultured hippocampal synapses | Munc13-1 and Munc13-2 | Inhibitory | Completely abolish | Completely abolish | Completely abolish | N.P. | N.P. | [43] |
| Diaphragm NMJs (Embryonic day 18.5) | Munc13-1 and Munc13-2 | Excitatory | 1/16-fold decrease | No change | More than 2-fold increase | Decrease | Decrease | [32] |
| Schaffer collateral to hippocampal CA1 pyramidal cell synapses (4- to 6-week) and cultured hippocampal synapses | ELKS2a | Excitatory | N.P. | No change | No change | No change | No change | [44] |
| Hippocampal CA1 synapses (4- to 6-week) and cultured hippocampal synapses | ELKS2a | Inhibitory | Increase | No change | No change | Increase | Increase | [44] |
| Calyx of Held synapses in brainstem slices (Postnatal day 16–21) | ELKS1 and ELKS2 | Excitatory | No change | No change | Increase | Increase | Decrease | [45] |
| Cultured hippocampal synapses | ELKS1a and ELKS2a | Excitatory | 1/2-fold decrease | No change | 1/2-fold decrease | No change | Decrease | [46] |
| Cultured hippocampal synapses | ELKS1a and ELKS2a | Inhibitory | 1/2-fold decrease | No change | 1/2-fold decrease | Decrease | No change | [46,47] |
| Gene | Mutation | Disease Symptoms | Reference |
|---|---|---|---|
| Synaptobrevin 1 | G18Wfs*5 R49P chr12:g. 6,574,054 T > G; disruption of mRNA splicing and generation of S114R variant in synaptobrevin 1 isoform D | Congenital myasthenic syndrome, motor retardation, muscle weakness, spastic ataxia, gait disturbance | [12,135,136] |
| Synaptobrevin 2 | V43del I45del A67P S75P P77S E78A | Autistic features, developmental delay, moderate to severe intellectual disability, poor visual fixation, absent purposeful hand movements | [137,138] |
| Synaptotagmin 1 | M303K D304G D366E I368T N371K | Profound cognitive impairment, lack of eye contact, severe motor delay, developmental delay varying in severity from moderate to profound | [139,140,141] |
| Synaptotagmin 2 | Exon3-9del V243Gfs*13 E269 * D307A P308L Y309 * I371K R397Sfs*37 | Distal hereditary motor neuropathy, congenital myasthenic syndrome, foot deformity since childhood, distal limb weakness, areflexia, gait abnormality | [10,142,143,144,145] |
| Munc13-1 | Q102 * | Microcephaly, cortical hyperexcitability, fatal myasthenia | [146] |
| P814L | Delayed neurological development, dyskinesia, autism spectrum disorder, comorbid attention-deficit hyperactivity disorder | [11] | |
| ELKS | Microdeletions of chromosome 12p13.33 including ELKS gene | Autism spectrum disorder, childhood apraxia of speech, deficits in gross motor, fine motor, and oral motor function | [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takikawa, K.; Nishimune, H. Similarity and Diversity of Presynaptic Molecules at Neuromuscular Junctions and Central Synapses. Biomolecules 2022, 12, 179. https://doi.org/10.3390/biom12020179
Takikawa K, Nishimune H. Similarity and Diversity of Presynaptic Molecules at Neuromuscular Junctions and Central Synapses. Biomolecules. 2022; 12(2):179. https://doi.org/10.3390/biom12020179
Chicago/Turabian StyleTakikawa, Kenji, and Hiroshi Nishimune. 2022. "Similarity and Diversity of Presynaptic Molecules at Neuromuscular Junctions and Central Synapses" Biomolecules 12, no. 2: 179. https://doi.org/10.3390/biom12020179