
Melanogenesis and the Targeted Therapy of Melanoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pigment Production
2.1. Melanocytes and Melanin
2.2. Melanosomes
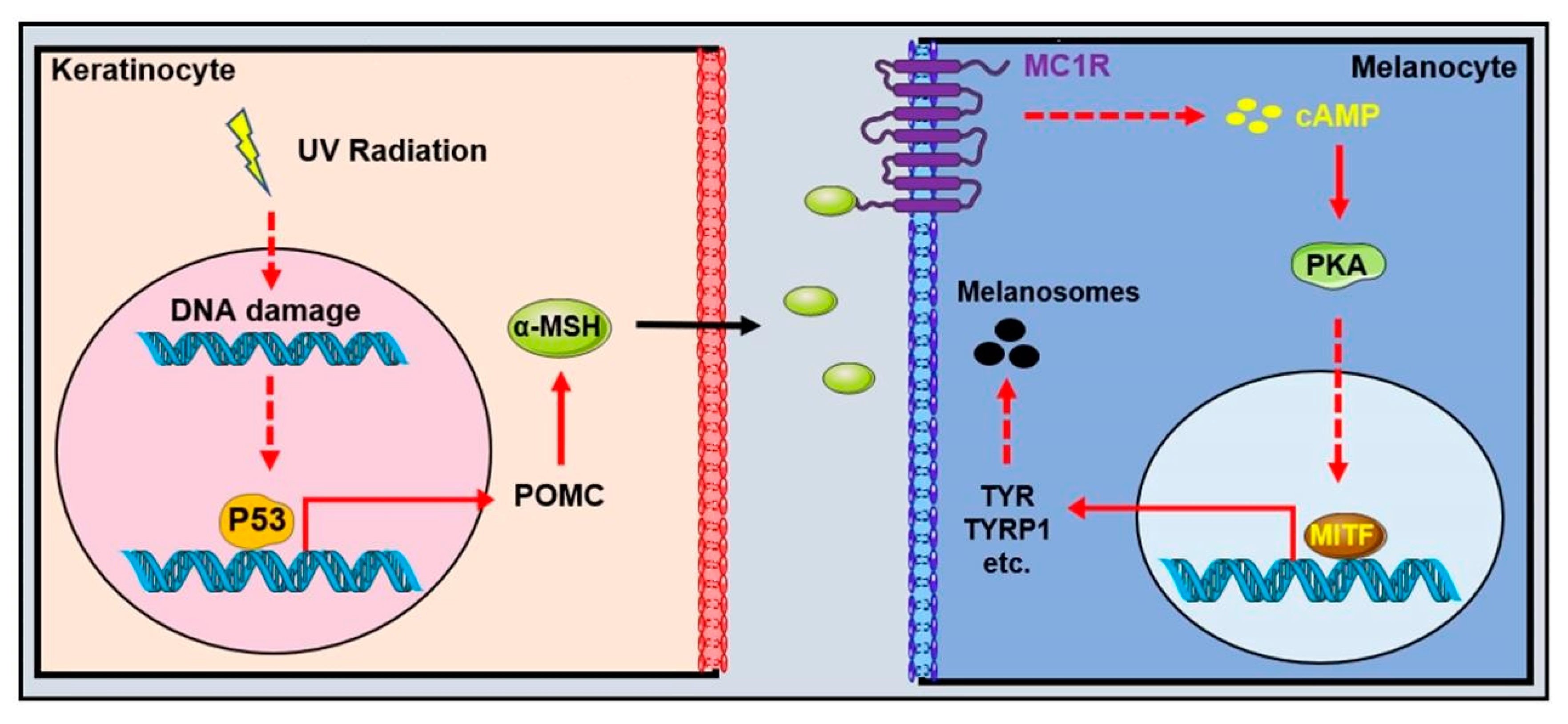
2.3. UVR-Extrinsic Activators and Regulators of Pigmentation
2.4. MC1R, the Central Determinant of Pigmentation
2.5. MITF, the Master Transcriptional Regulator of Pigmentation
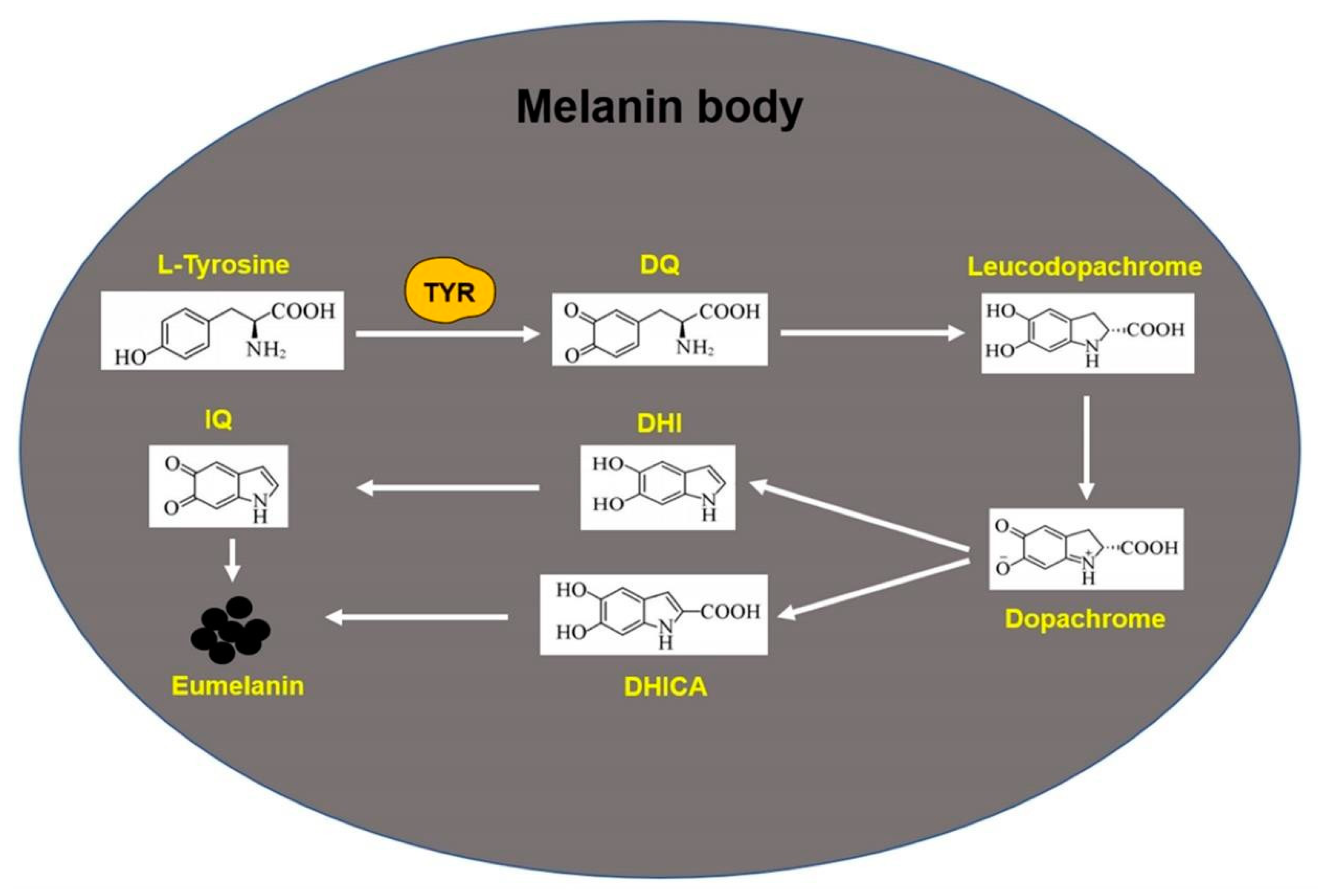
2.6. Melanin Synthesis Process
3. The Targeted Therapeutics of Melanoma
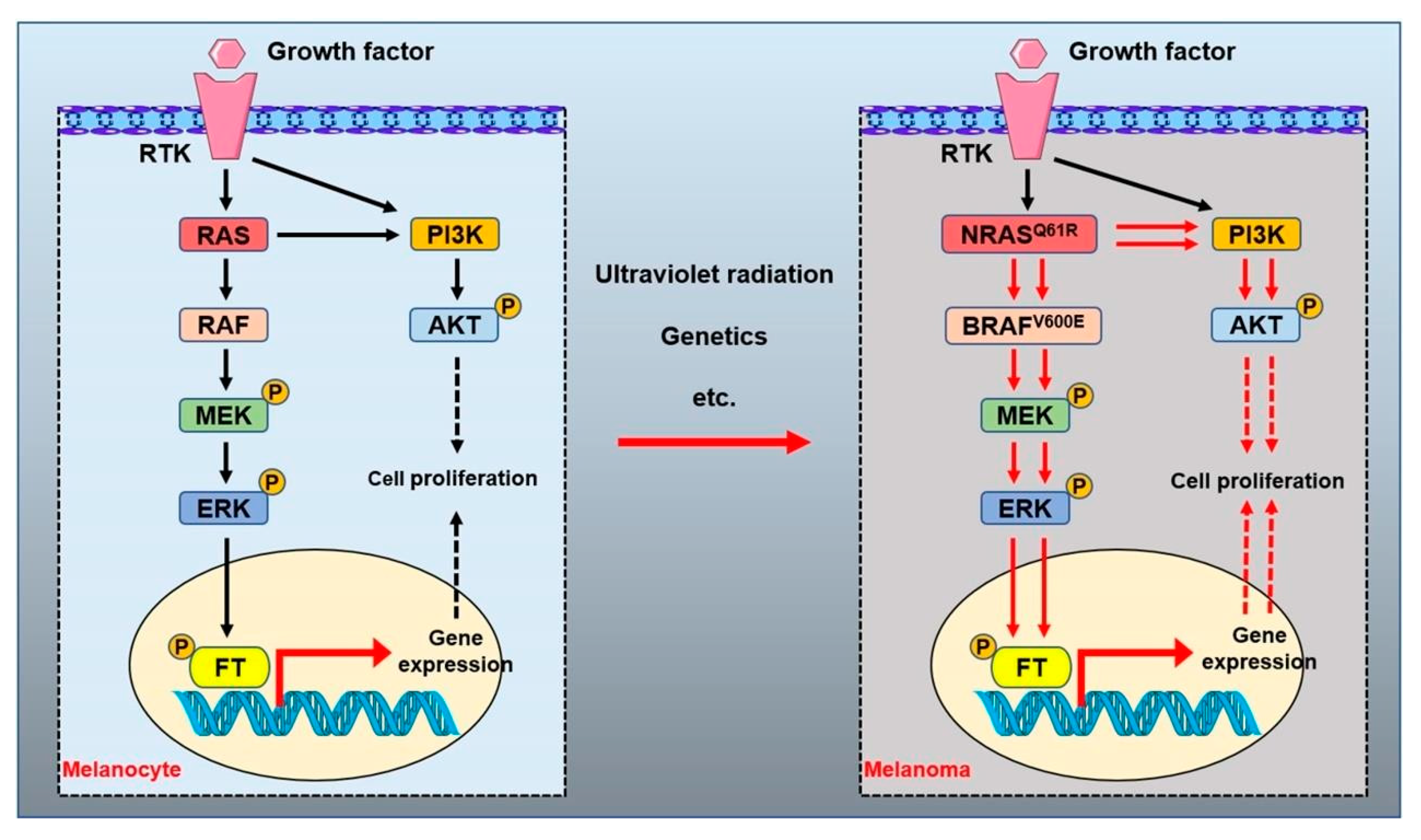
3.1. BRAFV600E Mutation
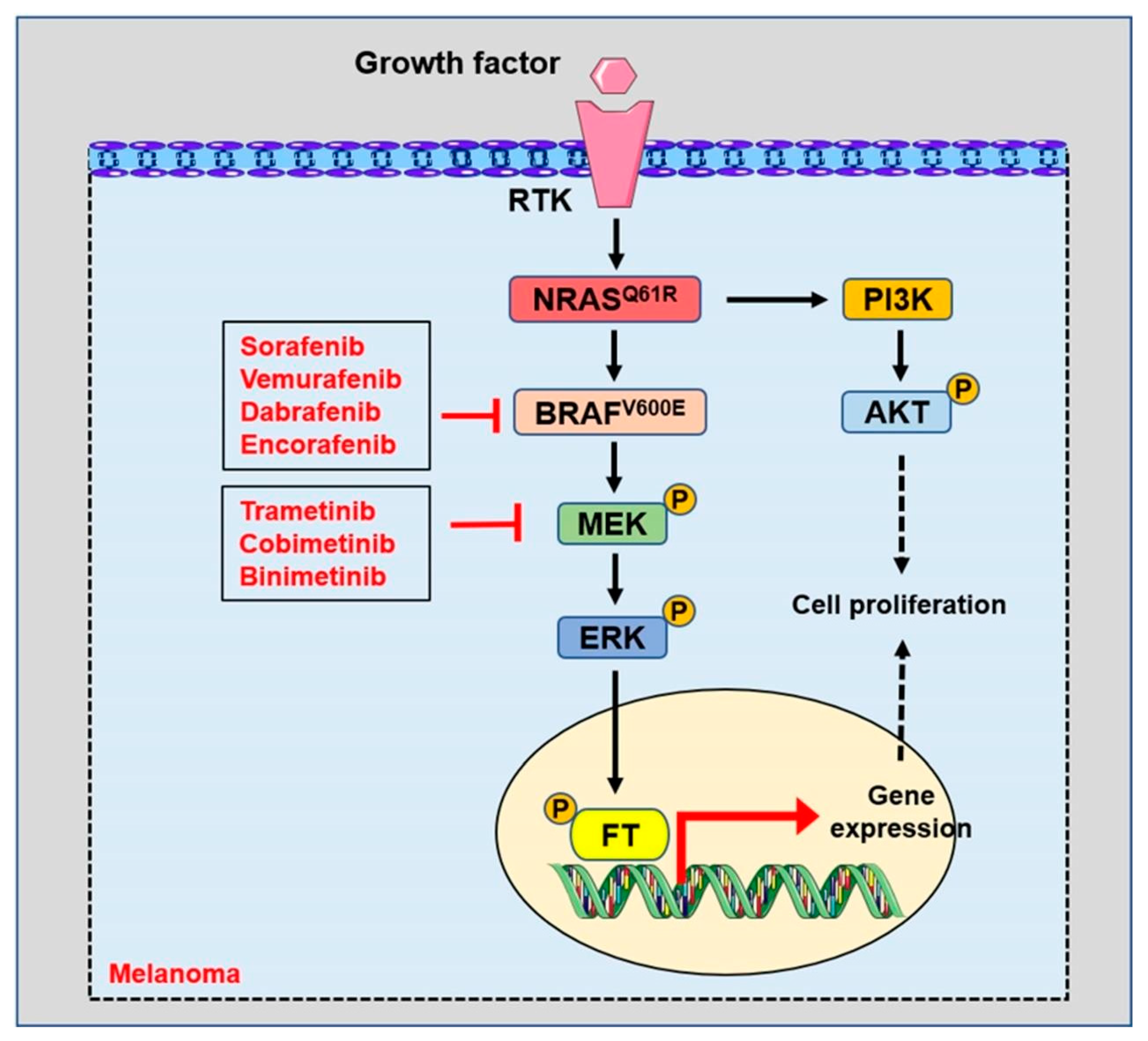
3.1.1. The BRAFV600E-Targeted Therapy in Melanoma
3.1.2. Combined Therapy of BRAF and MEK Inhibitors in Melanoma
3.1.3. The Combined Therapy of BRAF V600E Inhibitor and Immune Checkpoint Inhibitors in Melanoma
3.2. NRAS and Melanoma
3.2.1. The Targeted Therapy in Melanoma with NRASmut
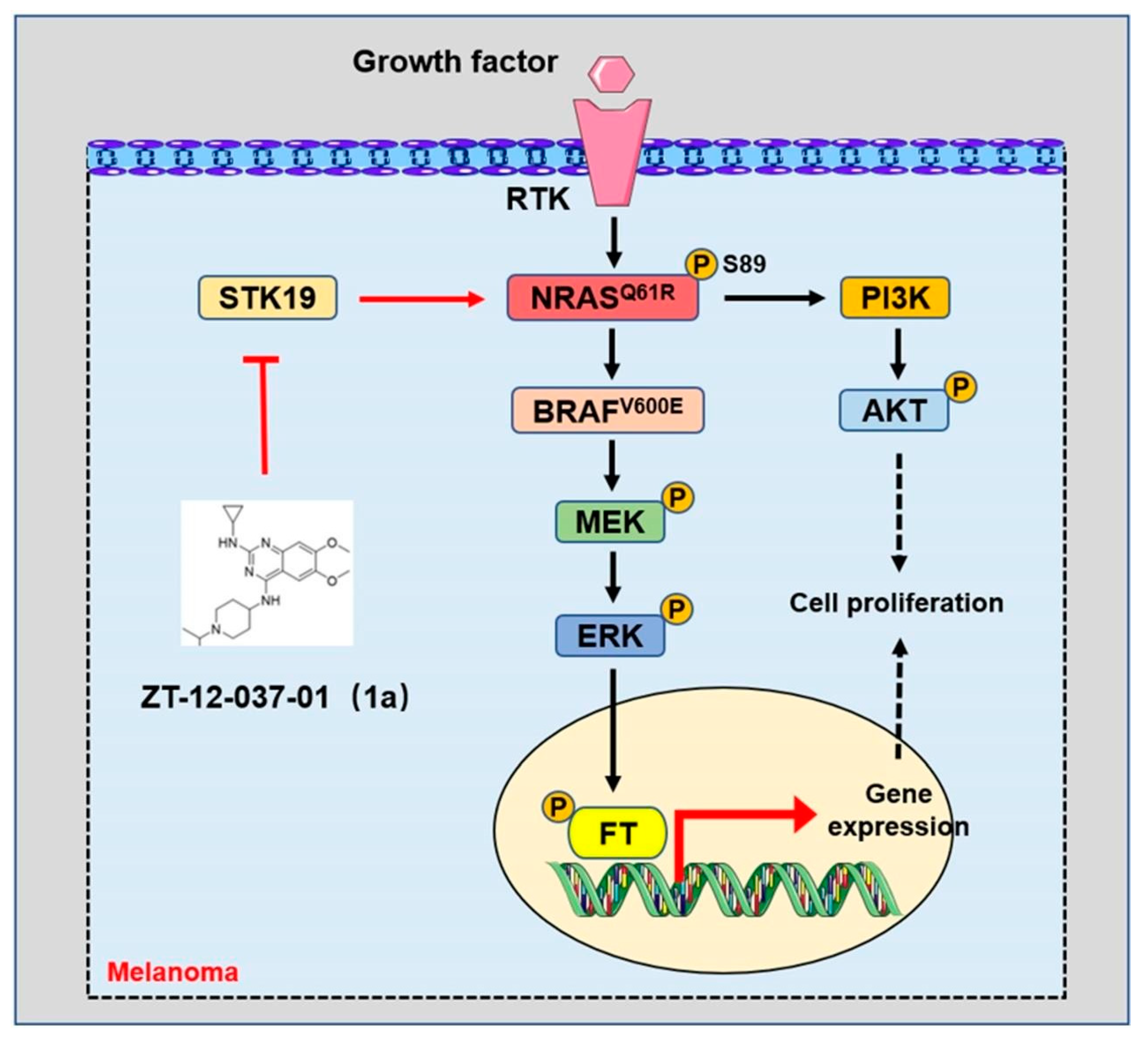
3.2.2. The Targeted Inhibition of NRAS Signaling in Melanoma
3.3. MC1R and Melanoma
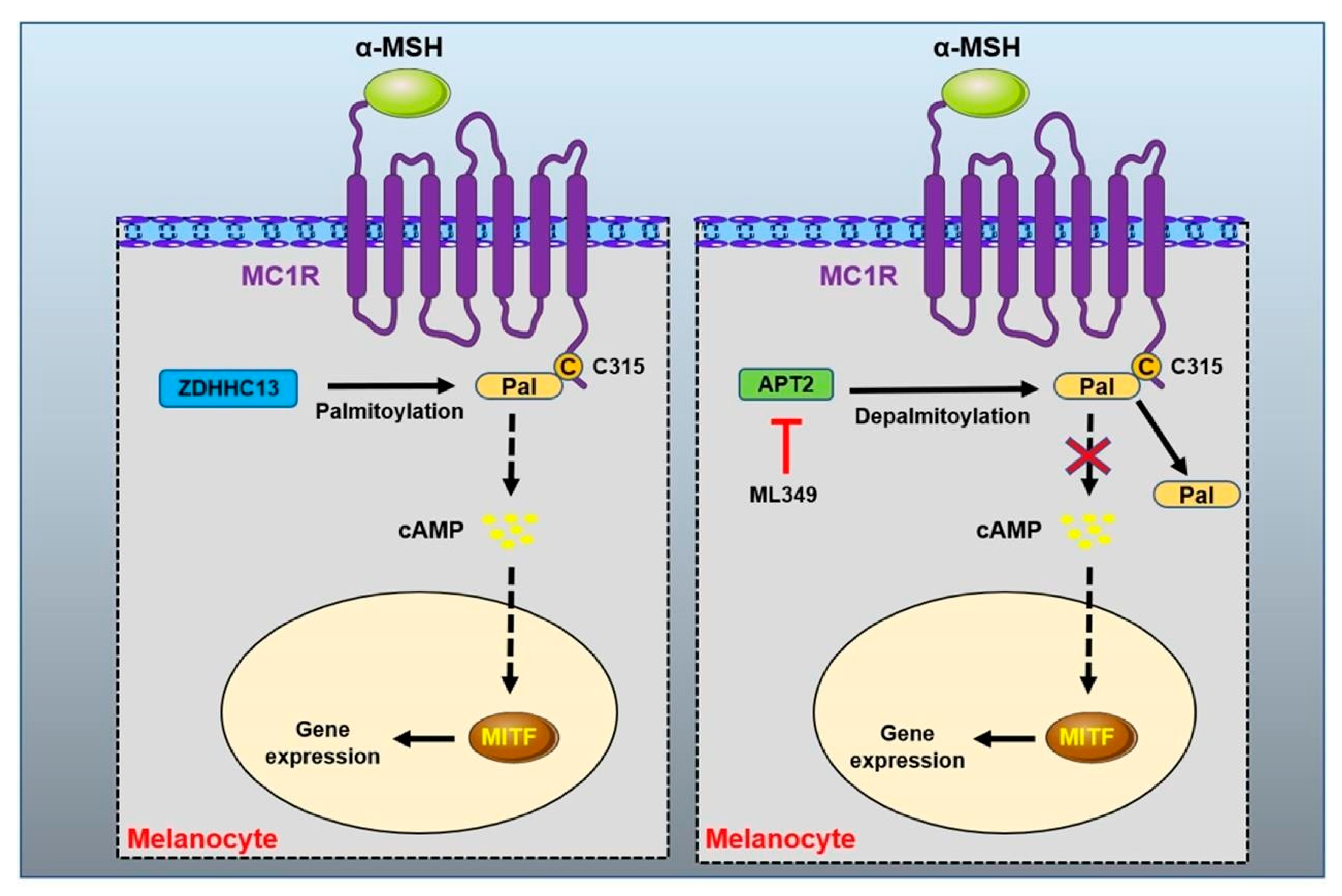
MC1R Protein Palmitoylation Modification
3.4. Other Therapeutical Target in Melanoma
4. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Tian, X.; Cui, Z.; Liu, S.; Zhou, J.; Cui, R. Melanosome transport and regulation in development and disease. Pharmacol. Ther. 2021, 219, 107707. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Videira, I.F.D.S.; Moura, D.F.L.; Magina, S. Mechanisms regulating melanogenesis. An. Bras. De Dermatol. 2013, 88, 76–83. [Google Scholar] [CrossRef] [Green Version]
- D’Mello, S.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling pathways in melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, S.M.; Fisher, D.E. Biology of melanoma. Hematol./Oncol. Clin. N. Am. 2021, 35, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Adameyko, I.; Lallemend, F.; Aquino, J.B.; Pereira, J.A.; Topilko, P.; Müller, T.; Fritz, N.; Beljajeva, A.; Mochii, M.; Liste, I.; et al. Schwann Cell Precursors from Nerve Innervation Are a Cellular Origin of Melanocytes in Skin. Cell 2009, 139, 366–379. [Google Scholar] [CrossRef] [Green Version]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin melanocytes: Biology and development. Adv. Dermatol. Allergol. 2013, 1, 30–41. [Google Scholar] [CrossRef]
- Thody, A.J.; Higgins, E.M.; Wakamatsu, K.; Ito, S.; Burchill, S.A.; Marks, J.M. Pheomelanin as well as Eumelanin Is Present in Human Epidermis. J. Investig. Dermatol. 1991, 97, 340–344. [Google Scholar] [CrossRef] [Green Version]
- Del Bino, S.; Ito, S.; Sok, J.; Nakanishi, Y.; Bastien, P.; Wakamatsu, K.; Bernerd, F. Chemical analysis of constitutive pigmentation of human epidermis reveals constant eumelanin to pheomelanin ratio. Pigment. Cell Melanoma Res. 2015, 28, 707–717. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K.; Sarna, T. Photodegradation of Eumelanin and Pheomelanin and Its Pathophysiological Implications. Photochem. Photobiol. 2018, 94, 409–420. [Google Scholar] [CrossRef]
- Nasti, T.H.; Timares, L. MC1R, Eumelanin and Pheomelanin: Their Role in Determining the Susceptibility to Skin Cancer. Photochem. Photobiol. 2014, 91, 188–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Marmol, V.; Beermann, F.F. Tyrosinase and related proteins in mammalian pigmentation. FEBS Lett. 1996, 381, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Quantitative Analysis of Eumelanin and Pheomelanin in Humans, Mice, and Other Animals: A Comparative Review. Pigment. Cell Res. 2003, 16, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Urabe, K.; Winder, A.; Jiménez-Cervantes, C.; Imokawa, G.; Brewington, T.; Solano, F.; Garcia-Borron, J.C.; Hearing, V. Tyrosinase related protein 1 (TRP1) functions as a DHICA oxidase in melanin biosynthesis. EMBO J. 1994, 13, 5818–5825. [Google Scholar] [CrossRef]
- Seruggia, D.; Josa, S.; Fernández, A.; Montoliu, L. The structure and function of the mouse tyrosinase locus. Pigment. Cell Melanoma Res. 2020, 34, 212–221. [Google Scholar] [CrossRef]
- Boissy, R.E.; Zhao, H.; Oetting, W.S.; Austin, L.M.; Wildenberg, S.C.; Boissy, Y.L.; Zhao, Y.; Sturm, R.A.; Hearing, V.J.; King, R.A.; et al. Mutation in and lack of expression of tyrosinase-related protein-1 (TRP-1) in melanocytes from an individual with brown oculocutaneous albinism: A new subtype of albinism classified as “OCA3”. Am. J. Hum. Genet. 1996, 58, 1145–1156. [Google Scholar]
- Vaish, U.; Kumar, A.A.; Varshney, S.; Ghosh, S.; Sengupta, S.; Sood, C.; Kar, H.K.; Sharma, P.; Natarajan, V.T.; Gokhale, R.S.; et al. Micro RNAs upregulated in Vitiligo skin play an important role in its aetiopathogenesis by altering TRP1 expression and keratinocyte-melanocytes cross-talk. Sci. Rep. 2019, 9, 10079. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Li, Y.; Zhao, B.; Chen, T.; Dong, Y.; Fan, R.; Li, J.; Wang, H.; He, X. TRP-2 mediates coat color pigmentation in sheep skin. Mol. Med. Rep. 2018, 17, 5869–5877. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Raposo, G.; Marks, M.S. Melanosomes—Dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Hoashi, T.; Watabe, H.; Muller, J.; Yamaguchi, Y.; Vieira, W.D.; Hearing, V.J. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J. Biol. Chem. 2005, 280, 14006–14016. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Muller, J.; Vieira, W.D.; Rouzaud, F.; Kikuchi, K.; Tamaki, K.; Hearing, V.J. The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers. J. Biol. Chem. 2006, 281, 21198–21208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theos, A.C.; Berson, J.F.; Theos, S.C.; Herman, K.E.; Harper, D.C.; Tenza, D.; Sviderskaya, E.V.; Lamoreux, M.L.; Bennett, D.C.; Raposo, G.; et al. Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell 2006, 17, 3598–3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaluf, S.; Atkins, D.; Barrett, K.; Blount, M.; Carter, N.; Heath, A. Ethnic Variation in Melanin Content and Composition in Photoexposed and Photoprotected Human Skin. Pigment. Cell Res. 2002, 15, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Murase, D.; Hachiya, A.; Takano, K.; Hicks, R.; Visscher, M.O.; Kitahara, T.; Hase, T.; Takema, Y.; Yoshimori, T. Autophagy Has a Significant Role in Determining Skin Color by Regulating Melanosome Degradation in Keratinocytes. J. Investig. Dermatol. 2013, 133, 2416–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebanks, J.P.; Koshoffer, A.; Wickett, R.R.; Schwemberger, S.; Babcock, G.; Hakozaki, T.; Boissy, R.E. Epidermal Keratinocytes from Light vs. Dark Skin Exhibit Differential Degradation of Melanosomes. J. Investig. Dermatol. 2011, 131, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, J.; Huflejt, M.E.; Rothfuss, L.M.; Wilson, D.S.; Carcamo, G.; Packer, L. Acute effects of near ultraviolet and visible light on the cutaneous antioxidant defense system. Photochem. Photobiol. 1989, 50, 739–744. [Google Scholar] [CrossRef]
- Jablonski, N.G.; Chaplin, G. The colours of humanity: The evolution of pigmentation in the human lineage. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160349. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, T.; Yamaguchi, Y.; Batzer, J.; Coelho, S.G.; Zmudzka, B.Z.; Miller, S.A.; Wolber, R.; Beer, J.Z.; Hearing, V.J. Mechanisms of Skin Tanning in Different Racial/Ethnic Groups in Response to Ultraviolet Radiation. J. Investig. Dermatol. 2005, 124, 1326–1332. [Google Scholar] [CrossRef] [Green Version]
- Friedmann, P.S.; Gilchrest, B.A. Ultraviolet radiation directly induces pigment production by cultured human melanocytes. J. Cell. Physiol. 1987, 133, 88–94. [Google Scholar] [CrossRef]
- Hu, J.; Adar, S. The Cartography of UV-induced DNA Damage Formation and DNA Repair. Photochem. Photobiol. 2017, 93, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Heenen, M.; Giacomoni, P.; Golstein, P. Individual variations in the correlation between erythemal threshold, UV-induced DNA damage and sun-burn cell formation. J. Photochem. Photobiol. B Biol. 2001, 63, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Garmyn, M.; Young, A.R.; Miller, S.A. Mechanisms of and variables affecting UVR photoadaptation in human skin. Photochem. Photobiol. Sci. 2018, 17, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Valencia, J.C.; Namiki, T.; Suzuki, T.; Hearing, V.J. Diacylglycerol Kinase Regulates Tyrosinase Expression and Function in Human Melanocytes. J. Investig. Dermatol. 2012, 132, 2791–2799. [Google Scholar] [CrossRef] [Green Version]
- Friedmann, P.S.; Wren, F.E.; Matthews, J.N.S. Ultraviolet stimulated melanogenesis by human melanocytes is augmented by di-acyl glycerol but not TPA. J. Cell. Physiol. 1990, 142, 334–341. [Google Scholar] [CrossRef]
- Eller, M.S.; Ostrom, K.; Gilchrest, B.A. DNA damage enhances melanogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Khlgatian, M.K.; Hadshiew, I.M.; Asawanonda, P.; Yaar, M.; Eller, M.S.; Gilchrest, B.A.; Fujita, M.; Norris, D.A. Tyrosinase Gene Expression is Regulated by p53. J. Investig. Dermatol. 2002, 118, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central Role of p53 in the Suntan Response and Pathologic Hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Goding, C.R.; Arnheiter, H. MITF—The first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Jin, Z. Paracrine regulation of melanogenesis. Br. J. Dermatol. 2018, 178, 632–639. [Google Scholar] [CrossRef]
- Li, W.; Wang, Z.; Xiao, M.; Miyoshi, T.; Yang, X.; Hu, Z.; Liu, C.; Chuang, S.S.C.; Shawkey, M.D.; Gianneschi, N.C.; et al. Mechanism of UVA Degradation of Synthetic Eumelanin. Biomacromolecules 2019, 20, 4593–4601. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Kobayashi, N.; Zmudzka, B.Z.; Ito, S.; Wakamatsu, K.; Yamaguchi, Y.; Korossy, K.S.; Miller, S.A.; Beer, J.Z.; Hearing, V.J. UV-induced DNA damage and melanin content in human skin differing in racial/ethnic origin. FASEB J. 2003, 17, 1177–1179. [Google Scholar] [CrossRef] [Green Version]
- Hennessy, A.; Oh, C.; Diffey, B.; Wakamatsu, K.; Ito, S.; Rees, J. Eumelanin and pheomelanin concentrations in human epidermis before and after UVB irradiation. Pigment Cell Res. 2005, 18, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Gantz, I.; Konda, Y.; Tashiro, T.; Shimoto, Y.; Miwa, H.; Munzert, G.; Watson, S.; DelValle, J.; Yamada, T. Molecular cloning of a novel melanocortin receptor. J. Biol. Chem. 1993, 268, 8246–8250. [Google Scholar] [CrossRef] [PubMed]
- Guida, S.; Guida, G.; Goding, C.R. MC1R functions, expression, and implications for targeted therapy. J. Investig. Dermatol. 2022, 142, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G.; Robbins, L.S.; Mortrud, M.T.; Cone, R.D. The Cloning of a Family of Genes That Encode the Melanocortin Receptors. Science 1992, 257, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Horrell, E.M.W.; Boulanger, M.C.; D’Orazio, J.A. Melanocortin 1 Receptor: Structure, Function, and Regulation. Front. Genet. 2016, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Ji, R.-L.; Tao, Y.-X. Melanocortin-1 receptor mutations and pigmentation: Insights from large animals. Prog. Mol. Biol. Transl. Sci. 2022, 189, 179–213. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, B.; Yin, C.; Liu, W.; Han, C.; Chen, B.; Liu, T.; Li, X.; Chen, X.; Li, C.; et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 2017, 549, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Rees, J.L. Genetics of Hair and Skin Color. Annu. Rev. Genet. 2003, 37, 67–90. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Sera, F.; Gandini, S.; Iodice, S.; Caini, S.; Maisonneuve, P.; Fargnoli, M.C. MC1R variants, melanoma and red hair color phenotype: A meta-analysis. Int. J. Cancer 2008, 122, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.F.; Olsen, C.M.; Hayward, N.K.; Whiteman, D. Melanocortin 1 receptor and risk of cutaneous melanoma: A meta-analysis and estimates of population burden. Int. J. Cancer 2011, 129, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, C.D.R.; Roberts, N.; Chen, S.; Leacy, F.P.; Alexandrov, L.B.; Pornputtapong, N.; Halaban, R.; Krauthammer, M.; Cui, R.; Bishop, D.T.; et al. Germline MC1R status influences somatic mutation burden in melanoma. Nat. Commun. 2016, 7, 12064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, I.; Tada, A.; Ollmann, M.M.; Barsh, G.S.; Im, S.; Lamoreux, M.L.; Hearing, V.J.; Nordlund, J.J.; Abdel-Malek, Z.A. Agouti Signaling Protein Inhibits Melanogenesis and the Response of Human Melanocytes to α-Melanotropin. J. Investig. Dermatol. 1997, 108, 838–842. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.D.; Ollmann, M.M.; Kang, L.; Stoffel, M.; Bell, G.I.; Barsh, G.S. Structure and function of ASP, the human homolog of the mouseagouti gene. Hum. Mol. Genet. 1995, 4, 223–230. [Google Scholar] [CrossRef]
- Slominski, A.; Płonka, P.; Pisarchik, A.; Smart, J.L.; Tolle, V.; Wortsman, J.; Low, M.J. Preservation of Eumelanin Hair Pigmentation in Proopiomelanocortin-Deficient Mice on a Nonagouti (a/a) Genetic Background. Endocrinology 2005, 146, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Goding, C.R. Mitf from neural crest to melanoma: Signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000, 14, 1712–1728. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Ploper, D.; De Robertis, E.M. The MITF family of transcription factors: Role in endolysosomal biogenesis, Wnt signaling, and oncogenesis. Pharmacol. Res. 2015, 99, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Hertwig, P. Neue mutationen und koppelungsgruppen bei der hausmaus. Z. Für Indukt. Abstamm.-Und Vererb. 1942, 80, 220–246. [Google Scholar] [CrossRef]
- Hodgkinson, C.A.; Moore, K.J.; Nakayama, A.; Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A.; Arnheiter, H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 1993, 74, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Seberg, H.E.; Van Otterloo, E.; Cornell, R.A. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res. 2017, 30, 454–466. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Miller, A.J.; Widlund, H.R.; Horstmann, M.A.; Ramaswamy, S.; Fisher, D.E. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am. J. Pathol. 2003, 163, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Goshima, Y.; Masukawa, D.; Kasahara, Y.; Hashimoto, T.; Aladeokin, A.C. l-DOPA and Its Receptor GPR143: Implications for Pathogenesis and Therapy in Parkinson’s Disease. Front. Pharmacol. 2019, 10, 1119. [Google Scholar] [CrossRef] [Green Version]
- Chiaverini, C.; Beuret, L.; Flori, E.; Busca, R.; Abbe, P.; Bille, K.; Bahadoran, P.; Ortonne, J.-P.; Bertolotto, C.; Ballotti, R. Microphthalmia-associated Transcription Factor Regulates RAB27A Gene Expression and Controls Melanosome Transport. J. Biol. Chem. 2008, 283, 12635–12642. [Google Scholar] [CrossRef] [Green Version]
- Carreira, S.; Liu, B.; Goding, C.R. The Gene Encoding the T-box Factor Tbx2 Is a Target for the Microphthalmia-associated Transcription Factor in Melanocytes. J. Biol. Chem. 2000, 275, 21920–21927. [Google Scholar] [CrossRef] [Green Version]
- McGill, G.G.; Horstmann, M.; Widlund, H.; Du, J.; Motyckova, G.; Nishimura, E.K.; Lin, Y.-L.; Ramaswamy, S.; Avery, W.; Ding, H.-F.; et al. Bcl2 Regulation by the Melanocyte Master Regulator Mitf Modulates Lineage Survival and Melanoma Cell Viability. Cell 2002, 109, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Carreira, S.; Goodall, J.; Aksan, I.; La Rocca, S.A.; Galibert, M.-D.; Denat, L.; Larue, L.; Goding, C.R. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 2005, 433, 764–769. [Google Scholar] [CrossRef]
- Tripathi, R.; Hearing, V.; Urabe, K.; Aroca, P.; Spritz, R. Mutational mapping of the catalytic activities of human tyrosinase. J. Biol. Chem. 1992, 267, 23707–23712. [Google Scholar] [CrossRef]
- Kishida, R.; Ushijima, Y.; Saputro, A.G.; Kasai, H. Effect of pH on elementary steps of dopachrome conversion from first-principles calculation. Pigment Cell Melanoma Res. 2014, 27, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Kishida, R.; Ito, S.; Sugumaran, M.; Arevalo, R.; Nakanishi, H.; Kasai, H. Density Functional Theory-Based Calculation Shed New Light on the Bizarre Addition of Cysteine Thiol to Dopaquinone. Int. J. Mol. Sci. 2021, 22, 1373. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA A Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and lpilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef]
- Kalaora, S.; Nagler, A.; Wargo, J.A.; Samuels, Y. Mechanisms of immune activation and regulation: Lessons from melanoma. Nat. Rev. Cancer 2022, 22, 195–207. [Google Scholar] [CrossRef]
- Jansen, H.W.; Lurz, R.; Bister, K.; Bonner, T.I.; Mark, G.E.; Rapp, U.R. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature 1984, 307, 281–284. [Google Scholar] [CrossRef]
- Sutrave, P.; Bonner, T.I.; Rapp, U.R.; Jansen, H.W.; Patschinsky, T.; Bister, K. Nucleotide sequence of avian retroviral oncogene v-mil: Homologue of murine retroviral oncogene v-raf. Nature 1984, 309, 85–88. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; van Akkooi, A.C.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Ito, T.; Tanaka, Y.; Murata, M.; Kaku-Ito, Y.; Furue, K.; Furue, M. BRAF heterogeneity in melanoma. Curr. Treat. Options Oncol. 2021, 22, 20. [Google Scholar] [CrossRef]
- Strickler, J.H.; Wu, C.; Bekaii-Saab, T. Targeting BRAF in metastatic colorectal cancer: Maximizing molecular approaches. Cancer Treat. Rev. 2017, 60, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Eisen, T.; Ahmad, T.; Flaherty, K.T.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L.M.; et al. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer 2006, 95, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Agarwala, S.S.; Trefzer, U.; Hogg, D.; Robert, C.; Hersey, P.; Eggermont, A.; Grabbe, S.; Gonzalez, R.; Gille, J.; et al. Results of a Phase III, Randomized, Placebo-Controlled Study of Sorafenib in Combination With Carboplatin and Paclitaxel As Second-Line Treatment in Patients With Unresectable Stage III or Stage IV Melanoma. J. Clin. Oncol. 2009, 27, 2823–2830. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600Emutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. OncoTargets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef]
- Long, G.V.; Weber, J.S.; Infante, J.R.; Kim, K.B.; Daud, A.; Gonzalez, R.; Sosman, J.A.; Hamid, O.; Schuchter, L.; Cebon, J.; et al. Overall Survival and Durable Responses in Patients With BRAF V600–Mutant Metastatic Melanoma Receiving Dabrafenib Combined With Trametinib. J. Clin. Oncol. 2016, 34, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Sandhu, S.; Hospers, G.; Long, G.V.; Aglietta, M.; Ferrucci, P.F.; Tulyte, S.; Cappellini, G.C.A.; Soriano, V.; Ali, S.; et al. Dabrafenib plus trametinib is effective in the treatment of BRAF V600-mutated metastatic melanoma patients: Analysis of patients from the dabrafenib plus trametinib Named Patient Program (DESCRIBE II). Melanoma Res. 2020, 30, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Gonzalez, R.; Pavlick, A.; Hamid, O.; Gajewski, T.F.; Daud, A.; Flaherty, L.; Logan, T.; Chmielowski, B.; Lewis, K.; et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAFV600-mutated melanoma: A phase 1b study. Lancet Oncol. 2014, 15, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour–host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.K.M.; Leprivier, G. The impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death Dis. 2019, 10, 955–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Randic, T.; Kozar, I.; Margue, C.; Utikal, J.; Kreis, S. NRAS mutant melanoma: Towards better therapies. Cancer Treat. Rev. 2021, 99, 102238. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Posch, C.; Vujic, I.; Monshi, B.; Sanlorenzo, M.; Weihsengruber, F.; Rappersberger, K.; Ortiz-Urda, S. Searching for the Chokehold of NRAS Mutant Melanoma. J. Investig. Dermatol. 2016, 136, 1330–1336. [Google Scholar] [CrossRef] [Green Version]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, K.E.; McArthur, G.A. The Cell-Cycle Regulator CDK4: An Emerging Therapeutic Target in Melanoma. Clin. Cancer Res. 2013, 19, 5320–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, J.A.; Bassett, R.L.; Ng, C.S.; Curry, J.L.; Joseph, R.; Alvarado, G.C.; Apn, M.L.R.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2011, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Thumar, J.; Shahbazian, D.; Aziz, S.A.; Jilaveanu, L.B.; Kluger, H.M. MEK targeting in N-RAS mutated metastatic melanoma. Mol. Cancer 2014, 13, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Michielin, O.; Van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U.; ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef] [Green Version]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncol. 2015, 1, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, S.; Zhang, G.; Wu, B.; Zhu, Y.; Frederick, D.T.; Hu, Y.; Zhong, W.; Randell, S.; Sadek, N.; et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017, 550, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Fusi, L.; Weiss, N.; Goller, S.G.; Meder, K.; Frings, V.G.; Kneitz, H.; Goebeler, M.; Houben, R.; Schrama, D.; et al. Efficient Suppression of NRAS-Driven Melanoma by Co-Inhibition of ERK1/2 and ERK5 MAPK Pathways. J. Investig. Dermatol. 2020, 140, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef] [Green Version]
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In Melanoma, RAS Mutations Are Accompanied by Switching Signaling from BRAF to CRAF and Disrupted Cyclic AMP Signaling. Cancer Res 2006, 66, 9483–9491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-Mediated MEK Activation Is Required for Effective MEK Inhibition in KRAS Mutant Tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Atefi, M.; Titz, B.; Avramis, E.; Ng, C.; Wong, D.J.; Lassen, A.; Cerniglia, M.; Escuin-Ordinas, H.; Foulad, D.; Comin-Anduix, B.; et al. Combination of pan-RAF and MEK inhibitors in NRAS mutant melanoma. Mol. Cancer 2015, 14, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorard, C.; Estrada, C.; Barbotin, C.; Larcher, M.; Garancher, A.; Leloup, J.; Beermann, F.; Baccarini, M.; Pouponnot, C.; Larue, L.; et al. RAF proteins exert both specific and compensatory functions during tumour progression of NRAS-driven melanoma. Nat. Commun. 2017, 8, 15262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posch, C.; Moslehi, H.; Feeney, L.; Green, G.A.; Ebaee, A.; Feichtenschlager, V.; Chong, K.; Peng, L.; Dimon, M.T.; Phillips, T.; et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4015–4020. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.-Z.; Wang, H.-B.; Chen, X.-L.; Cao, W.-T.; Fu, L.; Li, Y.; Quan, H.-T.; Xie, C.-Y.; Lou, L.-G. Aberrant modulation of ribosomal protein S6 phosphorylation confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma. Acta Pharmacol. Sin. 2018, 40, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Teh, J.L.F.; Cheng, P.F.; Purwin, T.J.; Nikbakht, N.; Patel, P.; Chervoneva, I.; Ertel, A.; Fortina, P.M.; Kleiber, I.; HooKim, K.; et al. In vivo E2F reporting reveals efficacious schedules of MEK1/2-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer Discov. 2018, 8, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Appleton, K.; Palsuledesai, C.; Misek, S.; Blake, M.; Zagorski, J.; Gallo, K.; Dexheimer, T.; Neubig, R. Inhibition of the Myocardin-Related Transcription Factor Pathway Increases Efficacy of Trametinib in NRAS-Mutant Melanoma Cell Lines. Cancers 2021, 13, 2012. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Lakhter, A.J.; Sahu, R.P.; Sun, Y.; Kaufmann, W.K.; Androphy, E.J.; Travers, J.B.; Naidu, S.R. Chloroquine Promotes Apoptosis in Melanoma Cells by Inhibiting BH3 Domain–Mediated PUMA Degradation. J. Investig. Dermatol. 2013, 133, 2247–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posch, C.; Cholewa, B.D.; Vujic, I.; Sanlorenzo, M.; Ma, J.; Kim, S.T.; Kleffel, S.; Schatton, T.; Rappersberger, K.; Gutteridge, R.; et al. Combined Inhibition of MEK and Plk1 Has Synergistic Antitumor Activity in NRAS Mutant Melanoma. J. Investig. Dermatol. 2015, 135, 2475–2483. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.Q.; Teh, J.L.; Purwin, T.J.; Chervoneva, I.; Davies, M.A.; Nathanson, K.L.; Cheng, P.F.; Levesque, M.P.; Dummer, R.; Aplin, A.E. Targeting PHGDH Upregulation Reduces Glutathione Levels and Resensitizes Resistant NRAS-Mutant Melanoma to MAPK Kinase Inhibition. J. Investig. Dermatol. 2020, 140, 2242–2252.e7. [Google Scholar] [CrossRef]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Zhu, B.; Zhang, T.; Liu, T.; Chen, S.; Liu, Y.; Li, X.; Miao, X.; Li, S.; Mi, X.; et al. Pharmacological Targeting of STK19 Inhibits Oncogenic NRAS-Driven Melanomagenesis. Cell 2019, 176, 1113–1127.e16. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.S.; Duffy, D.L.; Box, N.F.; Aitken, J.F.; O’Gorman, L.E.; Green, A.C.; Hayward, N.K.; Martin, N.G.; Sturm, R.A. Melanocortin-1 receptor polymorphisms and risk of melanoma: Is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet. 2000, 66, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.T.; Demenais, F.; Iles, M.; Harland, M.; Taylor, J.C.; Corda, E.; Randerson-Moor, J.; Aitken, J.; Avril, M.-F.; Azizi, E.; et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 2009, 41, 920–925. [Google Scholar] [CrossRef]
- Miao, X.; Chen, S.; Zhu, B.; Yin, C.; Li, X.; Han, C.; Cui, R.; Li, B. Are redheads at an increased risk of melanoma? Future Oncol. 2018, 14, 413–416. [Google Scholar] [CrossRef]
- Kennedy, C.; ter Huurne, J.; Berkhout, M.; Gruis, N.; Bastiaens, M.; Bergman, W.; Willemze, R.; Bavinck, J.N.B. Melanocortin 1 Receptor (MC1R) Gene Variants are Associated with an Increased Risk for Cutaneous Melanoma Which is Largely Independent of Skin Type and Hair Color. J. Investig. Dermatol. 2001, 117, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Bauer, J.; Pfeiffer, R.M.; Elder, D.E.; Hulley, B.; Minghetti, P.; Calista, D.; Kanetsky, P.A.; Pinkel, D.; Bastian, B.C. MC1R germline variants confer risk for BRAF-mutant melanoma. Science 2006, 313, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Han, C.; Miao, X.; Li, X.; Yin, C.; Zou, J.; Liu, M.; Li, S.; Stawski, L.; Zhu, B.; et al. Targeting MC1R depalmitoylation to prevent melanomagenesis in redheads. Nat. Commun. 2019, 10, 877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwardhan, A.; Cheng, N.; Trejo, J. Post-Translational Modifications of G Protein–Coupled Receptors Control Cellular Signaling Dynamics in Space and Time. Pharmacol. Rev. 2020, 73, 120–151. [Google Scholar] [CrossRef] [PubMed]
- González, F.E.; Ramírez, M.; Allerbring, E.B.; Fasching, N.; Lundqvist, A.; Poschke, I.; Achour, A.; Salazar-Onfray, F. Melanocortin 1 Receptor-derived peptides are efficiently recognized by cytotoxic T lymphocytes from melanoma patients. Immunobiology 2014, 219, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Wankowicz-Kalinska, A.; Mailliard, R.B.; Olson, K.; Graham, F.; Edington, H.; Kirkwood, J.M.; Martinek, S.; Das, P.K.; Storkus, W.J. Accumulation of low-avidity anti-melanocortin receptor 1 (anti-MC1R) CD8+ T cells in the lesional skin of a patient with melanoma-related depigmentation. Melanoma Res. 2006, 16, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Mascitti, M.; Santarelli, A.; Sartini, D.; Rubini, C.; Colella, G.; Salvolini, E.; Ganzetti, G.; Offidani, A.; Emanuelli, M. Analysis of nicotinamide N-methyltransferase in oral malignant melanoma and potential prognostic significance. Melanoma Res. 2019, 29, 151–156. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef]
- Ganzetti, G.; Sartini, D.; Campanati, A.; Rubini, C.; Molinelli, E.; Brisigotti, V.; Cecati, M.; Pozzi, V.; Campagna, R.; Offidani, A.; et al. Nicotinamide N-methyltransferase: Potential involvement in cutaneous malignant melanoma. Melanoma Res. 2018, 28, 82–88. [Google Scholar] [CrossRef]
- Campagna, R.; Salvolini, E.; Pompei, V.; Pozzi, V.; Salvucci, A.; Molinelli, E.; Brisigotti, V.; Sartini, D.; Campanati, A.; Offidani, A.; et al. Nicotinamide N-methyltransferase gene silencing enhances chemosensitivity of melanoma cell lines. Pigment. Cell Melanoma Res. 2021, 34, 1039–1048. [Google Scholar] [CrossRef]
- van Haren, M.J.; Zhang, Y.; Thijssen, V.; Buijs, N.; Gao, Y.; Mateuszuk, L.; Fedak, F.A.; Kij, A.; Campagna, R.; Sartini, D.; et al. Macrocyclic peptides as allosteric inhibitors of nicotinamide N-methyltransferase (NNMT). RSC Chem. Biol. 2021, 2, 1546–1555. [Google Scholar] [CrossRef]
- Barrows, R.D.; Jeffries, D.E.; Vishe, M.; Tukachinsky, H.; Zheng, S.-L.; Li, F.; Ma, Z.; Li, X.; Jin, S.; Song, H.; et al. Potent Uncompetitive Inhibitors of Nicotinamide N-Methyltransferase (NNMT) as In Vivo Chemical Probes. J. Med. Chem. 2022, 65, 14642–14654. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Kuai, L.; Cui, R.; Miao, X. Melanogenesis and the Targeted Therapy of Melanoma. Biomolecules 2022, 12, 1874. https://doi.org/10.3390/biom12121874
Li C, Kuai L, Cui R, Miao X. Melanogenesis and the Targeted Therapy of Melanoma. Biomolecules. 2022; 12(12):1874. https://doi.org/10.3390/biom12121874
Chicago/Turabian StyleLi, Cang, Le Kuai, Rutao Cui, and Xiao Miao. 2022. "Melanogenesis and the Targeted Therapy of Melanoma" Biomolecules 12, no. 12: 1874. https://doi.org/10.3390/biom12121874