
Heparan Sulfate Proteoglycans in Tauopathy
 , , , and
, , , and
Abstract
:1. Introduction
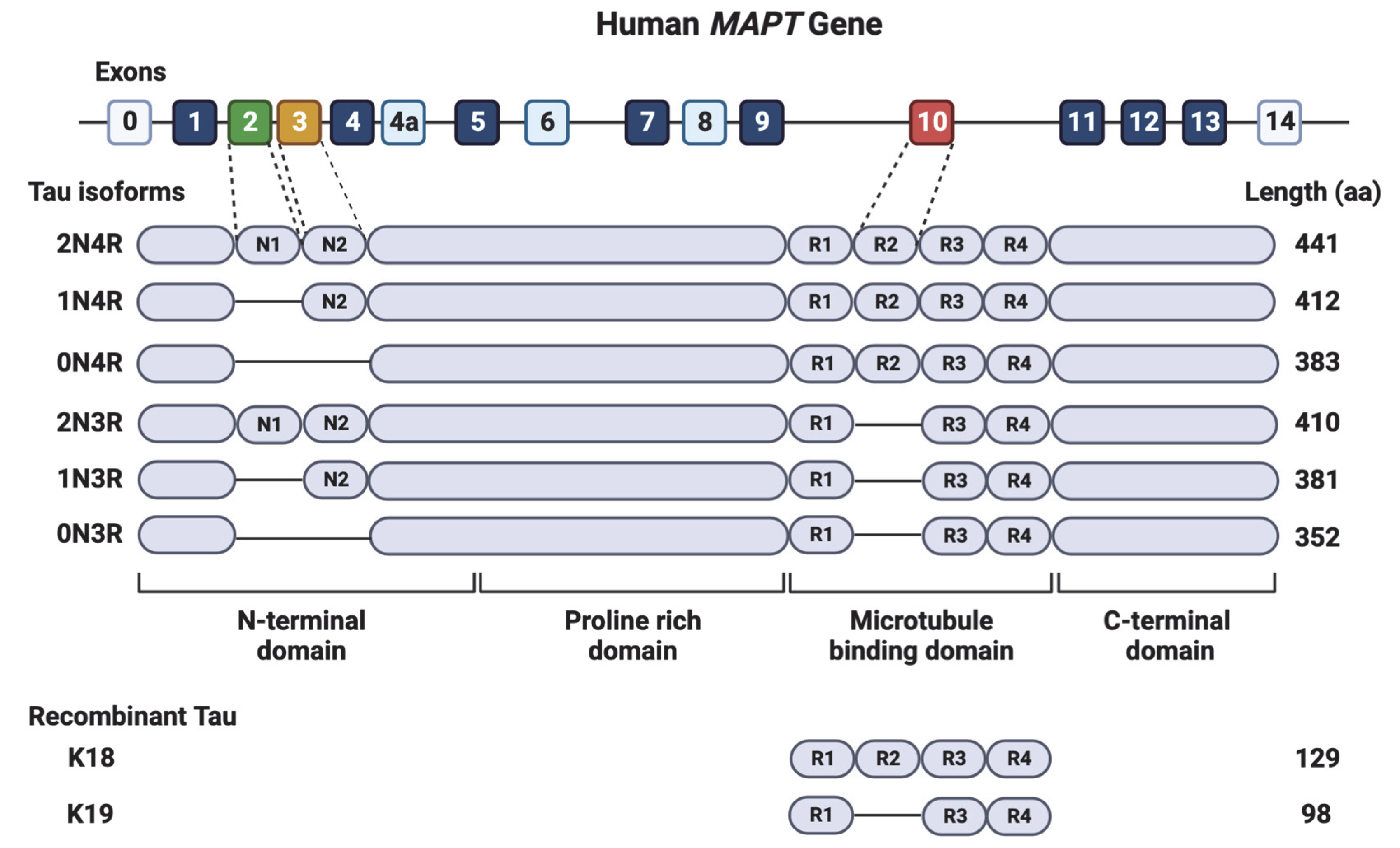
2. The Tau Protein
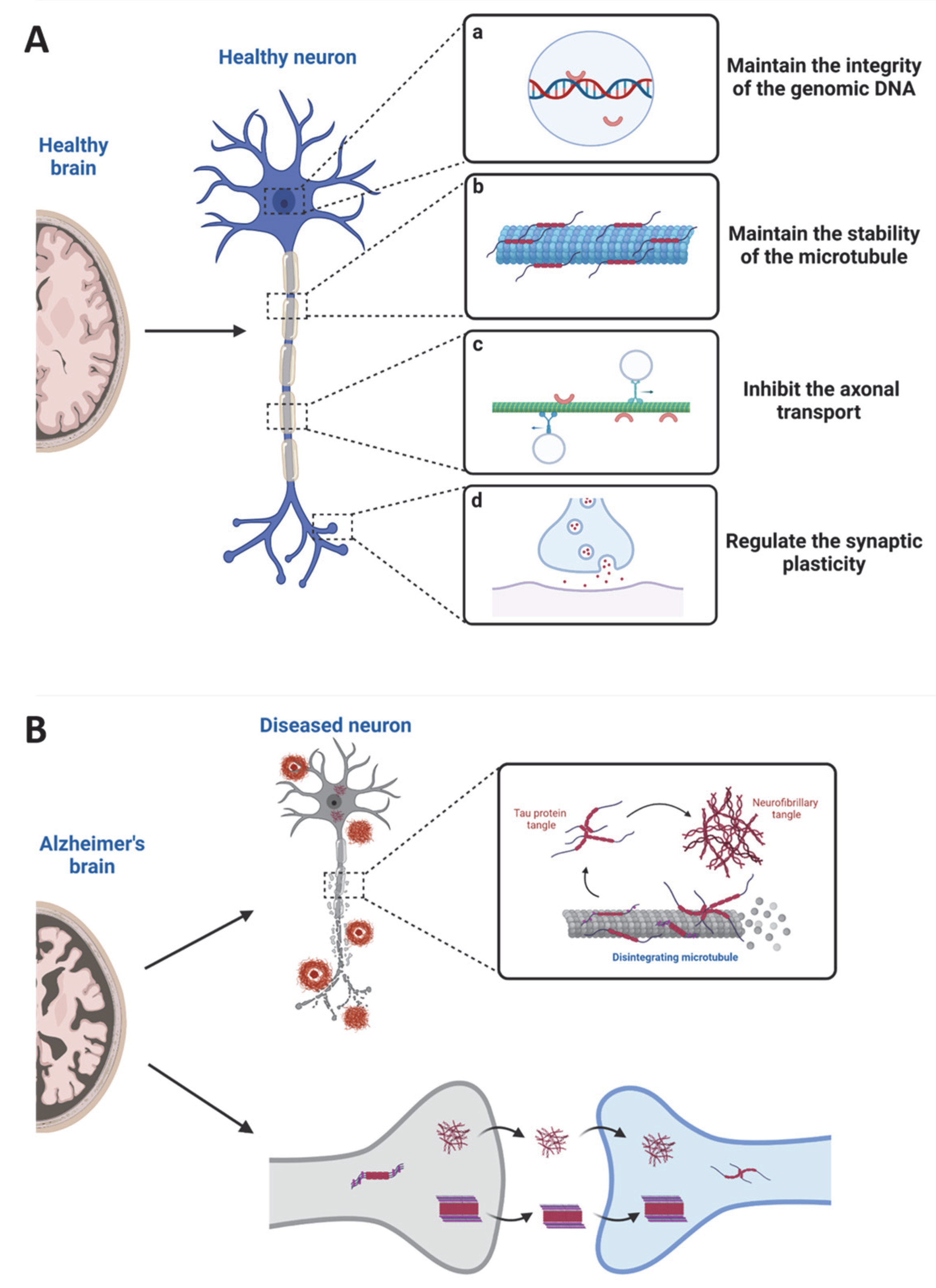
3. Tau in Physiological States
4. Tau in Pathological States
4.1. Tau Mutations
4.2. Tau Post-Translational Modifications
4.3. Tau Seed Propagation
4.4. Tau-Mediated Neurotoxicity
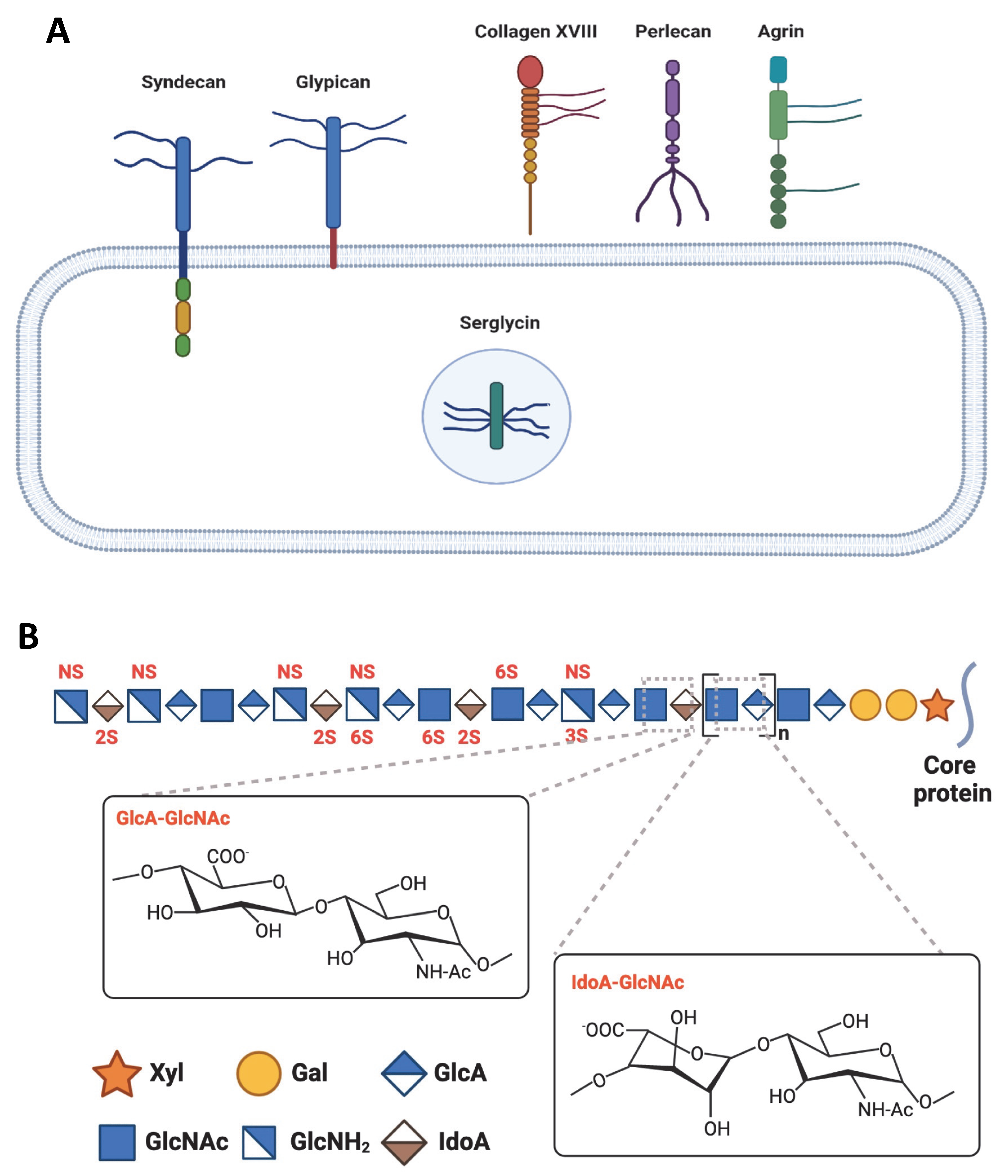
5. Heparan Sulfate Proteoglycans
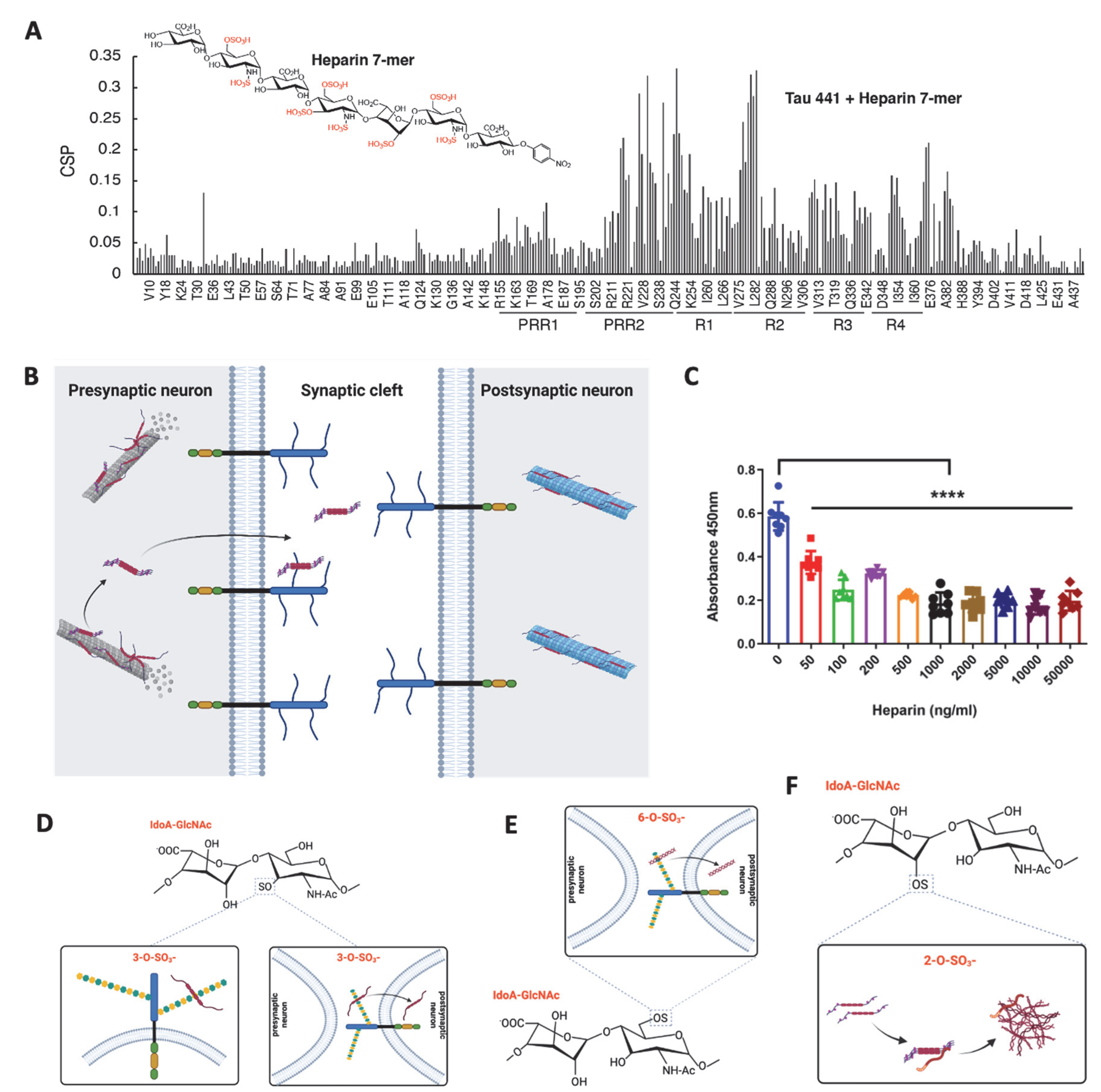
6. Heparan Sulfate-Tau Interaction: The Related Structures
7. The Role of Heparan Sulfate in the Tau-Mediated Pathological Process
7.1. HS in Tau Secretion
7.2. HS in Tau Cell Surface Binding
7.3. HS in Tau Internalization
7.4. HS in Tau Aggregation
8. Aberrant HSPG Expression in AD and Other Tauopathies
9. Future Studies from the HSPG Aspect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Wu, K.M.; Yang, L.; Dong, Q.; Yu, J.T. Tauopathies: New perspectives and challenges. Mol. Neurodegener. 2022, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. 2019, 14, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron. 2018, 99, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drubin, D.G.; Caput, D.; Kirschner, M.W. Studies on the expression of the microtubule-associated protein, tau, during mouse brain development, with newly isolated complementary DNA probes. J. Cell Biol. 1984, 98, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [Green Version]
- Papasozomenos, S.C.; Binder, L.I. Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil. Cytoskeleton 1987, 8, 210–226. [Google Scholar] [CrossRef]
- Liu, C.; Götz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 2021, 371, eabb8255. [Google Scholar] [CrossRef]
- Andreadis, A. Tau gene alternative splicing: Expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2005, 1739, 91–103. [Google Scholar] [CrossRef]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef] [Green Version]
- Panda, D.; Goode, B.L.; Feinstein, S.C.; Wilson, L. Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 1995, 34, 11117–11127. [Google Scholar] [CrossRef]
- Trinczek, B.; Biernat, J.; Baumann, K.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol. Biol. Cell 1995, 6, 1887–1902. [Google Scholar] [CrossRef] [Green Version]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar] [CrossRef]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef]
- Tucker, K.L.; Meyer, M.; Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001, 4, 29–37. [Google Scholar] [CrossRef]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Konzack, S.; Thies, E.; Marx, A.; Mandelkow, E.M.; Mandelkow, E. Swimming against the tide: Mobility of the microtubule-associated protein tau in neurons. J. Neurosci. 2007, 27, 9916–9927. [Google Scholar] [CrossRef] [Green Version]
- Utton, M.A.; Noble, W.J.; Hill, J.E.; Anderton, B.H.; Hanger, D.P. Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J. Cell Sci. 2005, 118, 4645–4654. [Google Scholar] [CrossRef] [Green Version]
- Vershinin, M.; Carter, B.C.; Razafsky, D.S.; King, S.J.; Gross, S.P. Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. USA 2007, 104, 87–92. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Xu, J.C.; Fomenko, V.; Miyamoto, T.; Suberbielle, E.; Knox, J.A.; Ho, K.; Kim, D.H.; Yu, G.Q.; Mucke, L. Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J. Cell Biol. 2015, 209, 419–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau reduction prevents Abeta-induced defects in axonal transport. Science 2010, 330, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.; Kumar, A.; Peterhoff, C.; Duff, K.; Nixon, R.A. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 2008, 28, 1682–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, Y.; Teng, J.; Harada, A.; Hirokawa, N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 2000, 150, 989–1000. [Google Scholar] [CrossRef] [Green Version]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lanté, F.; Buisson, A. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R Soc. Lond B. Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.; Van der Jeugd, A.; Blum, D.; Galas, M.C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Mueller, R.L.; Combs, B.; Alhadidy, M.M.; Brady, S.T.; Morfini, G.A.; Kanaan, N.M. Tau: A Signaling Hub Protein. Front. Mol. Neurosci. 2021, 14, 647054. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Hong, X.P.; Peng, C.X.; Wei, W.; Tian, Q.; Liu, Y.H.; Yao, X.Q.; Zhang, Y.; Cao, F.Y.; Wang, Q.; Wang, J.Z. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 2010, 20, 1339–1349. [Google Scholar] [CrossRef]
- Dioli, C.; Patrício, P.; Trindade, R.; Pinto, L.G.; Silva, J.M.; Morais, M.; Ferreiro, E.; Borges, S.; Mateus-Pinheiro, A.; Rodrigues, A.J.; et al. Tau-dependent suppression of adult neurogenesis in the stressed hippocampus. Mol. Psychiatry 2017, 22, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Criado-Marrero, M.; Sabbagh, J.J.; Jones, M.R.; Chaput, D.; Dickey, C.A.; Blair, L.J. Hippocampal Neurogenesis Is Enhanced in Adult Tau Deficient Mice. Cells 2020, 9, 210. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, S.; Harada, A.; Hirokawa, N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 2000, 279, 129–132. [Google Scholar] [CrossRef]
- Ma, Q.L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Kiosea, N.C.; Nazari, S.; Chen, P.P.; Nothias, F.; et al. Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris Water Maze with aging. J. Neurosci. 2014, 34, 7124–7136. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Caroppo, P.; Prioni, S.; Maderna, E.; Grisoli, M.; Rossi, G. New MAPT variant in a FTD patient with Alzheimer’s disease phenotype at onset. Neurol. Sci. 2021, 42, 2111–2114. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Nasif, L.; Giasson, B.I. Pathogenic MAPT mutations Q336H and Q336R have isoform-dependent differences in aggregation propensity and microtubule dysfunction. J. Neurochem. 2021, 158, 455–466. [Google Scholar] [CrossRef]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front. Cell Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef] [Green Version]
- Vega, I.E.; Cui, L.; Propst, J.A.; Hutton, M.L.; Lee, G.; Yen, S.H. Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Brain Res. Mol. Brain Res. 2005, 138, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Johnson, G.V. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J. Neurochem. 2004, 88, 349–358. [Google Scholar] [CrossRef]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta. Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Han, Y.; Li, P.; Su, Z.; Huang, Y. The Role of Post-Translational Modifications on the Structure and Function of Tau Protein. J. Mol. Neurosci. 2022, 72, 1557–1571. [Google Scholar] [CrossRef]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [Green Version]
- Cantrelle, F.X.; Loyens, A.; Trivelli, X.; Reimann, O.; Despres, C.; Gandhi, N.S.; Hackenberger, C.P.R.; Landrieu, I.; Smet-Nocca, C. Phosphorylation and O-GlcNAcylation of the PHF-1 Epitope of Tau Protein Induce Local Conformational Changes of the C-Terminus and Modulate Tau Self-Assembly Into Fibrillar Aggregates. Front. Mol. Neurosci. 2021, 14, 661368. [Google Scholar] [CrossRef]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Munari, F.; Barracchia, C.G.; Franchin, C.; Parolini, F.; Capaldi, S.; Romeo, A.; Bubacco, L.; Assfalg, M.; Arrigoni, G.; D’Onofrio, M. Semisynthetic and Enzyme-Mediated Conjugate Preparations Illuminate the Ubiquitination-Dependent Aggregation of Tau Protein. Angew. Chem. Int. Ed. Engl. 2020, 59, 6607–6611. [Google Scholar] [CrossRef]
- Luo, H.B.; Xia, Y.Y.; Shu, X.J.; Liu, Z.C.; Feng, Y.; Liu, X.H.; Yu, G.; Yin, G.; Xiong, Y.S.; Zeng, K.; et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2014, 111, 16586–16591. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, J.; Choi, W.H.; Park, S.; Park, S.H.; Lee, J.H.; Lim, S.M.; Mun, J.Y.; Cho, H.S.; Han, D.; et al. CHIP-mediated hyperubiquitylation of tau promotes its self-assembly into the insoluble tau filaments. Chem. Sci. 2021, 12, 5599–5610. [Google Scholar] [CrossRef]
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. 2002, 512, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.P.; Biernat, J.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E.M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef]
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derisbourg, M.; Leghay, C.; Chiappetta, G.; Fernandez-Gomez, F.J.; Laurent, C.; Demeyer, D.; Carrier, S.; Buee-Scherrer, V.; Blum, D.; Vinh, J.; et al. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci. Rep. 2015, 5, 9659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; Weiner, M.; Aisen, P.; Petersen, R.; et al. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta. Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Mahoney, E.R.; Dumitrescu, L.; Moore, A.M.; Cambronero, F.E.; De Jager, P.L.; Koran, M.E.I.; Petyuk, V.A.; Robinson, R.A.S.; Goyal, S.; Schneider, J.A.; et al. Brain expression of the vascular endothelial growth factor gene family in cognitive aging and alzheimer’s disease. Mol. Psychiatry 2021, 26, 888–896. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Rashid, I.; Pathak, A.K.; Kumar, R.; Srivastava, P.; Singh, M.; Murali, S.; Kushwaha, B. Genome-Wide Comparative Analysis of HIF Binding Sites in Cyprinus Carpio for In Silico Identification of Functional Hypoxia Response Elements. Front. Genet. 2019, 10, 659. [Google Scholar] [CrossRef]
- Meisl, G.; Hidari, E.; Allinson, K.; Rittman, T.; DeVos, S.L.; Sanchez, J.S.; Xu, C.K.; Duff, K.E.; Johnson, K.A.; Rowe, J.B.; et al. In vivo rate-determining steps of tau seed accumulation in Alzheimer’s disease. Sci. Adv. 2021, 7, eabh1448. [Google Scholar] [CrossRef]
- DeVos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.P.; Buee, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta. Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [Green Version]
- Brunello, C.A.; Merezhko, M.; Uronen, R.L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell Mol. Life Sci. 2020, 77, 1721–1744. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Eckermann, K.; Mocanu, M.M.; Khlistunova, I.; Biernat, J.; Nissen, A.; Hofmann, A.; Schonig, K.; Bujard, H.; Haemisch, A.; Mandelkow, E.; et al. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J. Biol. Chem. 2007, 282, 31755–31765. [Google Scholar] [CrossRef] [Green Version]
- Chee, F.; Mudher, A.; Newman, T.A.; Cuttle, M.; Lovestone, S.; Shepherd, D. Overexpression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Biochem. Soc. Trans. 2006, 34, 88–90. [Google Scholar] [CrossRef] [Green Version]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [Green Version]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Thies, E.; Mandelkow, E.M. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 2007, 27, 2896–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, M.; Chaudhury, P.; Kabiru, H.; Shea, T.B. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskeleton 2008, 65, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; Leung, K.Y.; Ward, M.A.; Anderton, B.H.; Hanger, D.P. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008, 22, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Samuel, J.C.; Massie, M.; Feinstein, S.C.; Wilson, L. Differential regulation of microtubule dynamics by three- and four-repeat tau: Implications for the onset of neurodegenerative disease. Proc. Natl. Acad. Sci. USA 2003, 100, 9548–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekinci, F.J.; Shea, T.B. Phosphorylation of tau alters its association with the plasma membrane. Cell Mol. Neurobiol. 2000, 20, 497–508. [Google Scholar] [CrossRef]
- Kanki, R.; Nakamizo, T.; Yamashita, H.; Kihara, T.; Sawada, H.; Uemura, K.; Kawamata, J.; Shibasaki, H.; Akaike, A.; Shimohama, S. Effects of mitochondrial dysfunction on glutamate receptor-mediated neurotoxicity in cultured rat spinal motor neurons. Brain Res. 2004, 1015, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Kim, B.T.; Kitagawa, H.; Tanaka, J.; Tamura, J.; Sugahara, K. In vitro heparan sulfate polymerization: Crucial roles of core protein moieties of primer substrates in addition to the EXT1-EXT2 interaction. J. Biol. Chem. 2003, 278, 41618–41623. [Google Scholar] [CrossRef] [Green Version]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. [Google Scholar] [CrossRef]
- De Boer, C.; Armstrong, Z.; Lit, V.A.J.; Barash, U.; Ruijgrok, G.; Boyango, I.; Weitzenberg, M.M.; Schroder, S.P.; Sarris, A.J.C.; Meeuwenoord, N.J.; et al. Mechanism-based heparanase inhibitors reduce cancer metastasis in vivo. Proc. Natl. Acad. Sci. USA 2022, 119, e2203167119. [Google Scholar] [CrossRef]
- Huynh, M.B.; Ouidja, M.O.; Chantepie, S.; Carpentier, G.; Maiza, A.; Zhang, G.; Vilares, J.; Raisman-Vozari, R.; Papy-Garcia, D. Glycosaminoglycans from Alzheimer’s disease hippocampus have altered capacities to bind and regulate growth factors activities and to bind tau. PLoS ONE 2019, 14, e0209573. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, Y.; Song, X.; Xiao, Y.; Su, G.; Liu, X.; Wang, Z.; Xu, Y.; Liu, J.; Eliezer, D.; et al. 3-O-Sulfation of Heparan Sulfate Enhances Tau Interaction and Cellular Uptake. Angew. Chem. Int. Ed. Engl. 2020, 59, 1818–1827. [Google Scholar] [CrossRef]
- Snow, A.D.; Cummings, J.A.; Lake, T. The Unifying Hypothesis of Alzheimer’s Disease: Heparan Sulfate Proteoglycans/Glycosaminoglycans Are Key as First Hypothesized Over 30 Years Ago. Front. Aging Neurosci. 2021, 13, 710683. [Google Scholar] [CrossRef]
- Stopschinski, B.E.; Holmes, B.B.; Miller, G.M.; Manon, V.A.; Vaquer-Alicea, J.; Prueitt, W.L.; Hsieh-Wilson, L.C.; Diamond, M.I. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates. J. Biol. Chem. 2018, 293, 10826–10840. [Google Scholar] [CrossRef] [Green Version]
- Ledin, J.; Staatz, W.; Li, J.P.; Gotte, M.; Selleck, S.; Kjellen, L.; Spillmann, D. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J. Biol. Chem. 2004, 279, 42732–42741. [Google Scholar] [CrossRef] [Green Version]
- Mah, D.; Zhao, J.; Liu, X.; Zhang, F.; Liu, J.; Wang, L.; Linhardt, R.; Wang, C. The Sulfation Code of Tauopathies: Heparan Sulfate Proteoglycans in the Prion Like Spread of Tau Pathology. Front. Mol. Biosci. 2021, 8, 671458. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Heparan Sulfate as a Therapeutic Target in Tauopathies: Insights From Zebrafish. Front. Cell Dev. Biol. 2018, 6, 163. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Cummings, B.J.; Cotman, C.W. Localization of heparan sulfate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer’s disease. Neuroscience 1992, 51, 801–813. [Google Scholar] [CrossRef]
- Snow, A.D.; Mar, H.; Nochlin, D.; Sekiguchi, R.T.; Kimata, K.; Koike, Y.; Wight, T.N. Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am. J. Pathol. 1990, 137, 1253–1270. [Google Scholar]
- Farshi, P.; Ohlig, S.; Pickhinke, U.; Hoing, S.; Jochmann, K.; Lawrence, R.; Dreier, R.; Dierker, T.; Grobe, K. Dual roles of the Cardin-Weintraub motif in multimeric Sonic hedgehog. J. Biol. Chem. 2011, 286, 23608–23619. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Nogues, M.V.; Andreu, D.; Boix, E. The “CPC clip motif”: A conserved structural signature for heparin-binding proteins. PLoS ONE 2012, 7, e42692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Sasaki, H.; Katagiri, T.; Sasaki, H.; Koiwai, K.; Youki, H.; Totsuka, S.; Ishii, T. The binding of basic fibroblast growth factor to Alzheimer’s neurofibrillary tangles and senile plaques. Neurosci. Lett. 1991, 122, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Siedlak, S.L.; Richey, P.; Kawai, M.; Cras, P.; Kalaria, R.N.; Galloway, P.G.; Scardina, J.M.; Cordell, B.; Greenberg, B.D.; et al. Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci. 1991, 11, 3679–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Tolnay, M.; Love, S.; Goedert, M. Microtubule-associated protein tau, heparan sulphate and alpha-synuclein in several neurodegenerative diseases with dementia. Acta Neuropathol. 1999, 97, 585–594. [Google Scholar] [CrossRef]
- Zhao, J.; Huvent, I.; Lippens, G.; Eliezer, D.; Zhang, A.; Li, Q.; Tessier, P.; Linhardt, R.J.; Zhang, F.; Wang, C. Glycan Determinants of Heparin-Tau Interaction. Biophys. J. 2017, 112, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Mukrasch, M.D.; Biernat, J.; von Bergen, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. J. Biol. Chem. 2005, 280, 24978–24986. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.Y.L.; Gibson, J.M.; Liu, J.; Eliezer, D.; Lippens, G.; Zhang, F.; Linhardt, R.J.; Zhao, J.; Wang, C. Proline-Rich Region II (PRR2) Plays an Important Role in Tau-Glycan Interaction: An NMR Study. Biomolecules 2022, 12, 1573. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Crowther, R.A.; Jakes, R.; Goedert, M. Alzheimer-like changes in microtubule-associated protein Tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J. Biol. Chem. 1997, 272, 33118–33124. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda-Diaz, J.E.; Alavi Naini, S.M.; Huynh, M.B.; Ouidja, M.O.; Yanicostas, C.; Chantepie, S.; Villares, J.; Lamari, F.; Jospin, E.; van Kuppevelt, T.H.; et al. HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer’s disease-related tau pathology. Brain 2015, 138, 1339–1354. [Google Scholar] [CrossRef] [Green Version]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Pérez, M.; Avila, J.; Hernández, F. Propagation of Tau via Extracellular Vesicles. Front. Neurosci. 2019, 13, 698. [Google Scholar] [CrossRef]
- Toledo, J.B.; Zetterberg, H.; van Harten, A.C.; Glodzik, L.; Martinez-Lage, P.; Bocchio-Chiavetto, L.; Rami, L.; Hansson, O.; Sperling, R.; Engelborghs, S.; et al. Alzheimer’s disease cerebrospinal fluid biomarker in cognitively normal subjects. Brain 2015, 138, 2701–2715. [Google Scholar] [CrossRef]
- Sutphen, C.L.; Jasielec, M.S.; Shah, A.R.; Macy, E.M.; Xiong, C.; Vlassenko, A.G.; Benzinger, T.L.; Stoops, E.E.; Vanderstichele, H.M.; Brix, B.; et al. Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol. 2015, 72, 1029–1042. [Google Scholar] [CrossRef] [Green Version]
- Pilliod, J.; Desjardins, A.; Pernegre, C.; Jamann, H.; Larochelle, C.; Fon, E.A.; Leclerc, N. Clearance of intracellular tau protein from neuronal cells via VAMP8-induced secretion. J. Biol. Chem. 2020, 295, 17827–17841. [Google Scholar] [CrossRef]
- Merezhko, M.; Uronen, R.L.; Huttunen, H.J. The Cell Biology of Tau Secretion. Front. Mol. Neurosci. 2020, 13, 569818. [Google Scholar] [CrossRef]
- Xu, Y.; Cui, L.; Dibello, A.; Wang, L.; Lee, J.; Saidi, L.; Lee, J.G.; Ye, Y. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov. 2018, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Merezhko, M.; Brunello, C.A.; Yan, X.; Vihinen, H.; Jokitalo, E.; Uronen, R.L.; Huttunen, H.J. Secretion of Tau via an Unconventional Non-vesicular Mechanism. Cell Rep. 2018, 25, 2027–2035.e2024. [Google Scholar] [CrossRef] [Green Version]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Muller, H.M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef]
- Mirbaha, H.; Holmes, B.B.; Sanders, D.W.; Bieschke, J.; Diamond, M.I. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem. 2015, 290, 14893–14903. [Google Scholar] [CrossRef] [Green Version]
- Rauch, J.N.; Chen, J.J.; Sorum, A.W.; Miller, G.M.; Sharf, T.; See, S.K.; Hsieh-Wilson, L.C.; Kampmann, M.; Kosik, K.S. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 2018, 8, 6382. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Shi, S.; Yue, J.; Xin, M.; Nairn, A.V.; Lin, L.; Liu, X.; Li, G.; Archer-Hartmann, S.A.; Dela Rosa, M.; et al. A mutant-cell library for systematic analysis of heparan sulfate structure-function relationships. Nat. Methods 2018, 15, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Lopez, E.; Diez-Ballesteros, J.C.; Avila, J.; Hernandez, F.; Bolos, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrasate, M.; Pérez, M.; Valpuesta, J.M.; Avila, J. Role of glycosaminoglycans in determining the helicity of paired helical filaments. Am. J. Pathol. 1997, 151, 1115–1122. [Google Scholar] [PubMed]
- Perez, M.; Valpuesta, J.M.; Medina, M.; Montejo de Garcini, E.; Avila, J. Polymerization of tau into filaments in the presence of heparin: The minimal sequence required for tau-tau interaction. J. Neurochem. 1996, 67, 1183–1190. [Google Scholar] [CrossRef]
- Fichou, Y.; Lin, Y.; Rauch, J.N.; Vigers, M.; Zeng, Z.; Srivastava, M.; Keller, T.J.; Freed, J.H.; Kosik, K.S.; Han, S. Cofactors are essential constituents of stable and seeding-active tau fibrils. Proc. Natl. Acad. Sci. USA 2018, 115, 13234–13239. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.; Fullwood, N.J.; Yates, E.A.; Middleton, D.A. Aggregation Kinetics and Filament Structure of a Tau Fragment Are Influenced by the Sulfation Pattern of the Cofactor Heparin. Biochemistry 2020, 59, 4003–4014. [Google Scholar] [CrossRef]
- Paudel, H.K.; Li, W. Heparin-induced conformational change in microtubule-associated protein Tau as detected by chemical cross-linking and phosphopeptide mapping. J. Biol. Chem. 1999, 274, 8029–8038. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Maïza, A.; Chantepie, S.; Vera, C.; Fifre, A.; Huynh, M.B.; Stettler, O.; Ouidja, M.O.; Papy-Garcia, D. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 2018, 592, 3806–3818. [Google Scholar] [CrossRef] [Green Version]
- Huynh, M.B.; Rebergue, N.; Merrick, H.; Gomez-Henao, W.; Jospin, E.; Biard, D.S.F.; Papy-Garcia, D. HS3ST2 expression induces the cell autonomous aggregation of tau. Sci. Rep. 2022, 12, 10850. [Google Scholar] [CrossRef]
- Hudak, A.; Kusz, E.; Domonkos, I.; Josvay, K.; Kodamullil, A.T.; Szilak, L.; Hofmann-Apitius, M.; Letoha, T. Contribution of syndecans to cellular uptake and fibrillation of alpha-synuclein and tau. Sci. Rep. 2019, 9, 16543. [Google Scholar] [CrossRef] [Green Version]
- Donahue, J.E.; Berzin, T.M.; Rafii, M.S.; Glass, D.J.; Yancopoulos, G.D.; Fallon, J.R.; Stopa, E.G. Agrin in Alzheimer’s disease: Altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proc. Natl. Acad. Sci. USA 1999, 96, 6468–6472. [Google Scholar] [CrossRef] [Green Version]
- Kolset, S.O.; Pejler, G. Serglycin: A structural and functional chameleon with wide impact on immune cells. J. Immunol. 2011, 187, 4927–4933. [Google Scholar] [CrossRef] [Green Version]
- Lorente-Gea, L.; Garcia, B.; Martin, C.; Ordiales, H.; Garcia-Suarez, O.; Pina-Batista, K.M.; Merayo-Lloves, J.; Quiros, L.M.; Fernandez-Vega, I. Heparan Sulfate Proteoglycans Undergo Differential Expression Alterations in Alzheimer Disease Brains. J. Neuropathol. Exp. Neurol. 2020, 79, 474–483. [Google Scholar] [CrossRef]
- Shimizu, H.; Ghazizadeh, M.; Sato, S.; Oguro, T.; Kawanami, O. Interaction between β-amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer’s disease. J. Clin. Neurosci. 2009, 16, 277–282. [Google Scholar] [CrossRef]
- Wang, Z.; Arnold, K.; Dhurandahare, V.M.; Xu, Y.; Pagadala, V.; Labra, E.; Jeske, W.; Fareed, J.; Gearing, M.; Liu, J. Analysis of 3-O-Sulfated Heparan Sulfate Using Isotopically L.Labeled Oligosaccharide Calibrants. Anal. Chem. 2022, 94, 2950–2957. [Google Scholar] [CrossRef]
- Roberts, R.O.; Kang, Y.N.; Hu, C.; Moser, C.D.; Wang, S.; Moore, M.J.; Graham, R.P.; Lai, J.-P.; Petersen, R.C.; Roberts, L.R. Decreased Expression of Sulfatase 2 in the Brains of Alzheimer’s Disease Patients: Implications for Regulation of Neuronal Cell Signaling. J. Alzheimer’s Dis. Rep. 2017, 1, 115–124. [Google Scholar] [CrossRef]
- Pérez-López, N.; Martín, C.; García, B.; Solís-Hernández, M.P.; Rodríguez, D.; Alcalde, I.; Merayo, J.; Fernández-Vega, I.; Quirós, L.M. Alterations in the Expression of the Genes Responsible for the Synthesis of Heparan Sulfate in Brains With Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2021, 80, 446–456. [Google Scholar] [CrossRef]
- Garcia, B.; Martin, C.; Garcia-Suarez, O.; Muniz-Alonso, B.; Ordiales, H.; Fernandez-Menendez, S.; Santos-Juanes, J.; Lorente-Gea, L.; Castanon, S.; Vicente-Etxenausia, I.; et al. Upregulated Expression of Heparanase and Heparanase 2 in the Brains of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 185–192. [Google Scholar] [CrossRef]
- Lorente-Gea, L.; Garcia, B.; Martin, C.; Quiros, L.M.; Fernandez-Vega, I. Heparan sulfate proteoglycans and heparanases in Alzheimer’s disease: Current outlook and potential therapeutic targets. Neural. Regen. Res. 2017, 12, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, O.; Matsuo, A.; Tooyama, I.; Kimura, H.; McGeer, E.G.; McGeer, P.L. Pick’s disease immunohistochemistry: New alterations and Alzheimer’s disease comparisons. Acta. Neuropathol. 1995, 89, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Dudas, B.; Cornelli, U.; Lee, J.M.; Hejna, M.J.; Walzer, M.; Lorens, S.A.; Mervis, R.F.; Fareed, J.; Hanin, I. Oral and subcutaneous administration of the glycosaminoglycan C3 attenuates Abeta(25-35)-induced abnormal tau protein immunoreactivity in rat brain. Neurobiol. Aging 2002, 23, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Stopschinski, B.E.; Thomas, T.L.; Nadji, S.; Darvish, E.; Fan, L.; Holmes, B.B.; Modi, A.R.; Finnell, J.G.; Kashmer, O.M.; Estill-Terpack, S.; et al. A synthetic heparinoid blocks Tau aggregate cell uptake and amplification. J. Biol. Chem. 2020, 295, 2974–2983. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lo Cascio, F.; Gao, J.; Kayed, R.; Huang, X. Binding and neurotoxicity mitigation of toxic tau oligomers by synthetic heparin like oligosaccharides. Chem. Commun. 2018, 54, 10120–10123. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updates 2016, 29, 54–75. [Google Scholar] [CrossRef] [Green Version]
- Lanzi, C.; Cassinelli, G. Heparan Sulfate Mimetics in Cancer Therapy: The Challenge to Define Structural Determinants and the Relevance of Targets for Optimal Activity. Molecules 2018, 23, 2915. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Kim, H.; Ho, M. Human monoclonal antibody targeting the heparan sulfate chains of Glypican-3 inhibits HGF-mediated migration and motility of hepatocellular carcinoma cells. PLoS ONE 2015, 10, e0137664. [Google Scholar] [CrossRef] [Green Version]
- Rapraeger, A.C. Synstatin: A selective inhibitor of the syndecan-1-coupled IGF1R-alphavbeta3 integrin complex in tumorigenesis and angiogenesis. FEBS J. 2013, 280, 2207–2215. [Google Scholar] [CrossRef]
- Metwaly, H.A.; El-Gayar, A.M.; El-Shishtawy, M.M. Inhibition of the signaling pathway of syndecan-1 by synstatin: A promising anti-integrin inhibitor of angiogenesis and proliferation in HCC in rats. Arch. Biochem. Biophys. 2018, 652, 50–58. [Google Scholar] [CrossRef]
- Schuksz, M.; Fuster, M.M.; Brown, J.R.; Crawford, B.E.; Ditto, D.P.; Lawrence, R.; Glass, C.A.; Wang, L.; Tor, Y.; Esko, J.D. Surfen, a small molecule antagonist of heparan sulfate. Proc. Natl. Acad. Sci. USA 2008, 105, 13075–13080. [Google Scholar] [CrossRef]
- Logun, M.T.; Wynens, K.E.; Simchick, G.; Zhao, W.; Mao, L.; Zhao, Q.; Mukherjee, S.; Brat, D.J.; Karumbaiah, L. Surfen-mediated blockade of extratumoral chondroitin sulfate glycosaminoglycans inhibits glioblastoma invasion. FASEB J. 2019, 33, 11973–11992. [Google Scholar] [CrossRef] [Green Version]
- Vasileva, E.; Warren, M.; Triche, T.J.; Amatruda, J.F. Dysregulated heparan sulfate proteoglycan metabolism promotes Ewing sarcoma tumor growth. eLife 2022, 11, e69734. [Google Scholar] [CrossRef]
- Zhou, H.; Roy, S.; Cochran, E.; Zouaoui, R.; Chu, C.L.; Duffner, J.; Zhao, G.; Smith, S.; Galcheva-Gargova, Z.; Karlgren, J.; et al. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS ONE 2011, 6, e21106. [Google Scholar] [CrossRef] [Green Version]
- Chua, J.S.; Kuberan, B. Synthetic Xylosides: Probing the Glycosaminoglycan Biosynthetic Machinery for Biomedical Applications. Acc. Chem. Res. 2017, 50, 2693–2705. [Google Scholar] [CrossRef]
- Kalita, M.; Villanueva-Meyer, J.; Ohkawa, Y.; Kalyanaraman, C.; Chen, K.; Mohamed, E.; Parker, M.F.L.; Jacobson, M.P.; Phillips, J.J.; Evans, M.J.; et al. Synthesis and Screening of alpha-Xylosides in Human Glioblastoma Cells. Mol. Pharm. 2021, 18, 451–460. [Google Scholar] [CrossRef]
- Raman, K.; Kuberan, B. Click-xylosides mitigate glioma cell invasion in vitro. Mol. Biosyst. 2010, 6, 1800–1802. [Google Scholar] [CrossRef]
- Raman, K.; Ninomiya, M.; Nguyen, T.K.; Tsuzuki, Y.; Koketsu, M.; Kuberan, B. Novel glycosaminoglycan biosynthetic inhibitors affect tumor-associated angiogenesis. Biochem. Biophys. Res. Commun. 2011, 404, 86–89. [Google Scholar] [CrossRef] [Green Version]
- Mani, K.; Belting, M.; Ellervik, U.; Falk, N.; Svensson, G.; Sandgren, S.; Cheng, F.; Fransson, L.A. Tumor attenuation by 2(6-hydroxynaphthyl)-beta-D-xylopyranoside requires priming of heparan sulfate and nuclear targeting of the products. Glycobiology 2004, 14, 387–397. [Google Scholar] [CrossRef]
- Naini, S.M.A.; Yanicostas, C.; Hassan-Abdi, R.; Blondeel, S.; Bennis, M.; Weiss, R.J.; Tor, Y.; Esko, J.D.; Soussi-Yanicostas, N. Surfen and oxalyl surfen decrease tau hyperphosphorylation and mitigate neuron deficits in vivo in a zebrafish model of tauopathy. Transl. Neurodegener. 2018, 7, 6. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Diagnosis | Predominant Tau Isoforms | Human Brain Samples | GAGs/Gene Expression in Disease | GAGs Function in Disease | Reference |
|---|---|---|---|---|---|
| AD | 3R + 4R Tau | 7 AD vs. 4 control | HS ↑ | N/A | [99] |
| AD | 3R + 4R Tau | N/A | N/A | Helicity of PHFs changed (potential) | [124] |
| AD | 3R + 4R Tau | 25 AD vs. 10 control | HS ↑ | N/A | [136] |
| AD | 3R + 4R Tau | 20 AD vs. 20 control | Sulf1 -; Sulf2 ↓ | N/A | [138] |
| AD | 3R + 4R Tau | 5 AD vs. 5 control | HS ↑; Ndst2 ↑; Hs3st2 ↑; Hs3st4 ↑; Glce ↑; HPSE ↑ | HS-tau binding capacity ↑ | [92] |
| AD | 3R + 4R Tau | 18 AD vs. 6 control | Altered expression of multiple HS biosynthesis/remodeling genes | N/A | [139] |
| AD | 3R + 4R Tau | 5 AD vs. 5 control | HS ↑; 3-o-sulfation ↑ | N/A | [137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Gandy, L.; Zhang, F.; Liu, J.; Wang, C.; Blair, L.J.; Linhardt, R.J.; Wang, L. Heparan Sulfate Proteoglycans in Tauopathy. Biomolecules 2022, 12, 1792. https://doi.org/10.3390/biom12121792
Zhu Y, Gandy L, Zhang F, Liu J, Wang C, Blair LJ, Linhardt RJ, Wang L. Heparan Sulfate Proteoglycans in Tauopathy. Biomolecules. 2022; 12(12):1792. https://doi.org/10.3390/biom12121792
Chicago/Turabian StyleZhu, Yanan, Lauren Gandy, Fuming Zhang, Jian Liu, Chunyu Wang, Laura J. Blair, Robert J. Linhardt, and Lianchun Wang. 2022. "Heparan Sulfate Proteoglycans in Tauopathy" Biomolecules 12, no. 12: 1792. https://doi.org/10.3390/biom12121792