



A Novel Tandem-Tag Purification Strategy for Challenging Disordered Proteins

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of pSUMO Plasmid
2.2. Generation of pSUMO Expression Constructs
2.2.1. Generating pSUMO Constructs by HiFi Cloning (pSUMO-AF1, pSUMO-Tau-441)
2.2.2. Generating pSUMO Constructs by Site-Directed Mutagenesis (pSUMO-Tau-NTMT, pSUMO-Tau-MTBR and pSUMO-AF1 (Only N-tag))
2.3. Expression and Purification of pSUMO Constructs
2.3.1. Expression and Purification of pSUMO-AF1(Only N-tag)
2.3.2. Expression and Purification of pSUMO-AF1
2.3.3. Expression and Purification of pSUMO-Tau-441
2.3.4. Expression and Purification of pSUMO-Tau-NTMT
2.3.5. Expression and Purification of pSUMO-Tau-MTBR
2.4. LC-MS/MS Analysis
3. Results
3.1. Generation of pSUMO Plasmid with Tandem-Tags
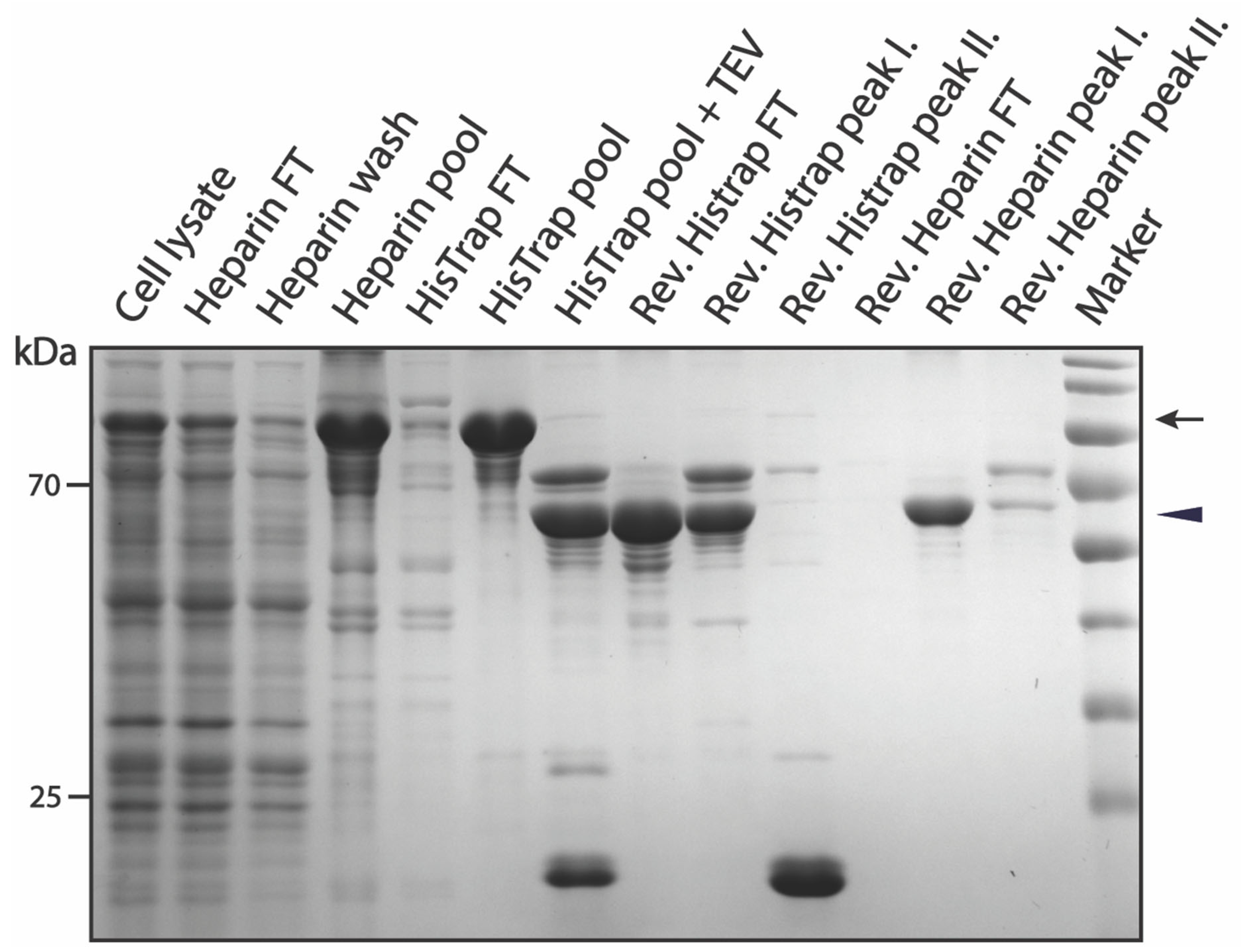
3.2. Purification of pSUMO-AF1 and pSUMO-AF1(Only N-tag): Comparison of the Two Methods
3.3. Purification of pSUMO-Tau Constructs
3.3.1. Purification of pSUMO-Tau-441
3.3.2. Purification of pSUMO-Tau-NTMT
3.3.3. Purification of pSUMO-Tau-MTBR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wingfield, P.T. Overview of the Purification of Recombinant Proteins. Curr. Protoc. Protein Sci. 2015, 80, 6.1.1–6.1.35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, G. Biopharmaceutical Benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Bigelow, L.; Borovilos, M.; Dementieva, I.; Duggan, E.; Eschenfeldt, W.; Hatzos, C.; Joachimiak, G.; Li, H.; Maltseva, N.; et al. Chapter 3. High-Throughput Protein Purification for X-Ray Crystallography and NMR. Adv. Protein Chem. Struct. Biol. 2008, 75, 85–105. [Google Scholar] [PubMed] [Green Version]
- Edwards, A.M.; Arrowsmith, C.H.; Christendat, D.; Dharamsi, A.; Friesen, J.D.; Greenblatt, J.F.; Vedadi, M. Protein Production: Feeding the Crystallographers and NMR Spectroscopists. Nat. Struct. Biol. 2000, 7, 970–972. [Google Scholar] [CrossRef] [PubMed]
- Hura, G.L.; Menon, A.L.; Hammel, M.; Rambo, R.P.; Poole, F.L., 2nd; Tsutakawa, S.E.; Jenney, F.E., Jr.; Classen, S.; Frankel, K.A.; Hopkins, R.C.; et al. Robust, High-Throughput Solution Structural Analyses by Small Angle X-Ray Scattering (SAXS). Nat. Methods 2009, 6, 606–612. [Google Scholar] [CrossRef]
- Stark, H.; Chari, A. Sample Preparation of Biological Macromolecular Assemblies for the Determination of High-Resolution Structures by Cryo-Electron Microscopy. Microscopy 2016, 65, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Li, Y. Commonly Used Tag Combinations for Tandem Affinity Purification. Biotechnol. Appl. Biochem. 2010, 55, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.G.M.; Skerra, A. The Strep-Tag System for One-Step Purification and High-Affinity Detection or Capturing of Proteins. Nat. Protoc. 2007, 2, 1528–1535. [Google Scholar] [CrossRef]
- Kronqvist, N.; Sarr, M.; Lindqvist, A.; Nordling, K.; Otikovs, M.; Venturi, L.; Pioselli, B.; Purhonen, P.; Landreh, M.; Biverstål, H.; et al. Efficient Protein Production Inspired by How Spiders Make Silk. Nat. Commun. 2017, 8, 15504. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef]
- Demain, A.L.; Vaishnav, P. Production of Recombinant Proteins by Microbes and Higher Organisms. Biotechnol. Adv. 2009, 27, 297–306. [Google Scholar] [CrossRef]
- Adrio, J.-L.; Demain, A.L. Recombinant Organisms for Production of Industrial Products. Bioeng. Bugs 2010, 1, 116–131. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Shukla, P. Advanced Technologies for Improved Expression of Recombinant Proteins in Bacteria: Perspectives and Applications. Crit. Rev. Biotechnol. 2016, 36, 1089–1098. [Google Scholar] [CrossRef]
- Ferrer-Miralles, N.; Domingo-Espín, J.; Corchero, J.L.; Vázquez, E.; Villaverde, A. Microbial Factories for Recombinant Pharmaceuticals. Microb. Cell Fact. 2009, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Mamat, U.; Wilke, K.; Bramhill, D.; Schromm, A.B.; Lindner, B.; Kohl, T.A.; Corchero, J.L.; Villaverde, A.; Schaffer, L.; Head, S.R.; et al. Detoxifying Escherichia coli for Endotoxin-Free Production of Recombinant Proteins. Microb. Cell Fact. 2015, 14, 57. [Google Scholar] [CrossRef] [Green Version]
- Carrió, M.M.; Villaverde, A. Protein Aggregation as Bacterial Inclusion Bodies Is Reversible. FEBS Lett. 2001, 489, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Carrió, M.M.; Villaverde, A. Construction and Deconstruction of Bacterial Inclusion Bodies. J. Biotechnol. 2002, 96, 3–12. [Google Scholar] [CrossRef]
- Owczarek, B.; Gerszberg, A.; Hnatuszko-Konka, K. A Brief Reminder of Systems of Production and Chromatography-Based Recovery of Recombinant Protein Biopharmaceuticals. Biomed Res. Int. 2019, 2019, 4216060. [Google Scholar] [CrossRef]
- Fletcher, E.; Krivoruchko, A.; Nielsen, J. Industrial Systems Biology and Its Impact on Synthetic Biology of Yeast Cell Factories. Biotechnol. Bioeng. 2016, 113, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, E.A.; Abbott, W.M. Expression of Recombinant Proteins in Insect and Mammalian Cells. Methods 2018, 147, 40–49. [Google Scholar] [CrossRef]
- Puetz, J.; Wurm, F.M. Recombinant Proteins for Industrial versus Pharmaceutical Purposes: A Review of Process and Pricing. Processes 2019, 7, 476. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, F.; Tozawa, Y. Cell-Free Expression--Making a Mark. Curr. Opin. Struct. Biol. 2013, 23, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Silverman, A.D.; Karim, A.S.; Jewett, M.C. Cell-Free Gene Expression: An Expanded Repertoire of Applications. Nat. Rev. Genet. 2020, 21, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant Protein Expression in Escherichia coli: Advances and Challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezonov, G.; Joseleau-Petit, D.; D’Ari, R. Escherichia coli Physiology in Luria-Bertani Broth. J. Bacteriol. 2007, 189, 8746–8749. [Google Scholar] [CrossRef] [Green Version]
- Sahdev, S.; Khattar, S.K.; Saini, K.S. Production of Active Eukaryotic Proteins through Bacterial Expression Systems: A Review of the Existing Biotechnology Strategies. Mol. Cell. Biochem. 2008, 307, 249–264. [Google Scholar] [CrossRef]
- Gopal, G.J.; Kumar, A. Strategies for the Production of Recombinant Protein in Escherichia coli. Protein J. 2013, 32, 419–425. [Google Scholar] [CrossRef]
- Rosano, G.L.; Morales, E.S.; Ceccarelli, E.A. New Tools for Recombinant Protein Production in Escherichia coli: A 5-Year Update. Protein Sci. 2019, 28, 1412–1422. [Google Scholar] [CrossRef]
- Liu, M.; Wang, B.; Wang, F.; Yang, Z.; Gao, D.; Zhang, C.; Ma, L.; Yu, X. Soluble Expression of Single-Chain Variable Fragment (scFv) in Escherichia coli Using Superfolder Green Fluorescent Protein as Fusion Partner. Appl. Microbiol. Biotechnol. 2019, 103, 6071–6079. [Google Scholar] [CrossRef]
- Paraskevopoulou, V.; Falcone, F.H. Polyionic Tags as Enhancers of Protein Solubility in Recombinant Protein Expression. Microorganisms 2018, 6, 47. [Google Scholar] [CrossRef]
- De Marco, A. Protocol for Preparing Proteins with Improved Solubility by Co-Expressing with Molecular Chaperones in Escherichia coli. Nat. Protoc. 2007, 2, 2632–2639. [Google Scholar] [CrossRef]
- Jo, B.H. An Intrinsically Disordered Peptide Tag That Confers an Unusual Solubility to Aggregation-Prone Proteins. Appl. Environ. Microbiol. 2022, 88, e0009722. [Google Scholar] [CrossRef]
- Gupta, S.K.; Dangi, A.K.; Smita, M.; Dwivedi, S.; Shukla, P. Effectual Bioprocess Development for Protein Production. In Applied Microbiology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2019; pp. 203–227. ISBN 9780128154076. [Google Scholar]
- Wacker, M.; Linton, D.; Hitchen, P.G.; Nita-Lazar, M.; Haslam, S.M.; North, S.J.; Panico, M.; Morris, H.R.; Dell, A.; Wren, B.W.; et al. N-Linked Glycosylation in Campylobacter Jejuni and Its Functional Transfer into E. coli. Science 2002, 298, 1790–1793. [Google Scholar] [CrossRef]
- Valderrama-Rincon, J.D.; Fisher, A.C.; Merritt, J.H.; Fan, Y.-Y.; Reading, C.A.; Chhiba, K.; Heiss, C.; Azadi, P.; Aebi, M.; DeLisa, M.P. An Engineered Eukaryotic Protein Glycosylation Pathway in Escherichia coli. Nat. Chem. Biol. 2012, 8, 434–436. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically Disordered Proteins: A 10-Year Recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Uversky, V.N. Introduction to Intrinsically Disordered Proteins (IDPs). Chem. Rev. 2014, 114, 6557–6560. [Google Scholar] [CrossRef]
- Suskiewicz, M.J.; Sussman, J.L.; Silman, I.; Shaul, Y. Context-Dependent Resistance to Proteolysis of Intrinsically Disordered Proteins. Protein Sci. 2011, 20, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Hammarberg, B.; Nygren, P.A.; Holmgren, E.; Elmblad, A.; Tally, M.; Hellman, U.; Moks, T.; Uhlén, M. Dual Affinity Fusion Approach and Its Use to Express Recombinant Human Insulin-like Growth Factor II. Proc. Natl. Acad. Sci. USA 1989, 86, 4367–4371. [Google Scholar] [CrossRef] [Green Version]
- McInnes, J.; Zhou, L.; Verstreken, A.P. Purification of Soluble Recombinant Human Tau Protein from Bacteria Using Double-Tag Affinity Purification. Bio. Protoc. 2018, 8, e3043. [Google Scholar] [CrossRef]
- Ortega, C.; Prieto, D.; Abreu, C.; Oppezzo, P.; Correa, A. Multi-Compartment and Multi-Host Vector Suite for Recombinant Protein Expression and Purification. Front. Microbiol. 2018, 9, 1384. [Google Scholar] [CrossRef]
- Bekesi, A.; Abdellaoui, S.; Holroyd, N.; Van Delm, W.; Pardon, E.; Pauwels, J.; Gevaert, K.; Steyaert, J.; Derveaux, S.; Borysik, A.; et al. Challenges in the Structural-Functional Characterization of Multidomain, Partially Disordered Proteins CBP and p300: Preparing Native Proteins and Developing Nanobody Tools. Methods Enzymol. 2018, 611, 607–675. [Google Scholar] [PubMed]
- Huang, L.; Qu, X.; Chen, Y.; Xu, W.; Huang, C. Sandwiched-Fusion Strategy Facilitates Recombinant Production of Small Labile Proteins. Protein Sci. 2021, 30, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Einhauer, A.; Jungbauer, A. The FLAG Peptide, a Versatile Fusion Tag for the Purification of Recombinant Proteins. J. Biochem. Biophys. Methods 2001, 49, 455–465. [Google Scholar] [CrossRef]
- Guerrero, F.; Ciragan, A.; Iwaï, H. Tandem SUMO Fusion Vectors for Improving Soluble Protein Expression and Purification. Protein Expr. Purif. 2015, 116, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Young, C.L.; Britton, Z.T.; Robinson, A.S. Recombinant Protein Expression and Purification: A Comprehensive Review of Affinity Tags and Microbial Applications. Biotechnol. J. 2012, 7, 620–634. [Google Scholar] [CrossRef]
- Siri, A.; Balza, E.; Carnemolla, B.; Castellani, P.; Borsi, L.; Zardi, L. DNA-Binding Domains of Human Plasma Fibronectin. pH and Calcium Ion Modulation of Fibronectin Binding to DNA and Heparin. Eur. J. Biochem. 1986, 154, 533–538. [Google Scholar] [CrossRef]
- Bolten, S.N.; Rinas, U.; Scheper, T. Heparin: Role in Protein Purification and Substitution with Animal-Component Free Material. Appl. Microbiol. Biotechnol. 2018, 102, 8647–8660. [Google Scholar] [CrossRef] [Green Version]
- Staby, A.; Sand, M.-B.; Hansen, R.G.; Jacobsen, J.H.; Andersen, L.A.; Gerstenberg, M.; Bruus, U.K.; Jensen, I.H. Comparison of Chromatographic Ion-Exchange Resins IV. Strong and Weak Cation-Exchange Resins and Heparin Resins. J. Chromatogr. A 2005, 1069, 65–77. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An Illustrator for the Presentation and Visualization of Biological Sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Monaghan, A.E.; McEwan, I.J. A Sting in the Tail: The N-Terminal Domain of the Androgen Receptor as a Drug Target. Asian J. Androl. 2016, 18, 687–694. [Google Scholar]
- Reid, J.; Kelly, S.M.; Watt, K.; Price, N.C.; McEwan, I.J. Conformational Analysis of the Androgen Receptor Amino-Terminal Domain Involved in Transactivation. Influence of Structure-Stabilizing Solutes and Protein-Protein Interactions. J. Biol. Chem. 2002, 277, 20079–20086. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X. Tau and Neurodegenerative Disease: The Story so Far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural Polymorphism of 441-Residue Tau at Single Residue Resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-Liquid Phase Separation Induces Pathogenic Tau Conformations in vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau Protein Liquid-Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Hochmair, J.; Exner, C.; Franck, M.; Dominguez-Baquero, A.; Diez, L.; Brognaro, H.; Kraushar, M.L.; Mielke, T.; Radbruch, H.; Kaniyappan, S.; et al. Molecular Crowding and RNA Synergize to Promote Phase Separation, Microtubule Interaction, and Seeding of Tau Condensates. EMBO J. 2022, 41, e108882. [Google Scholar] [CrossRef]
- Soeda, Y.; Yoshikawa, M.; Almeida, O.F.X.; Sumioka, A.; Maeda, S.; Osada, H.; Kondoh, Y.; Saito, A.; Miyasaka, T.; Kimura, T.; et al. Toxic Tau Oligomer Formation Blocked by Capping of Cysteine Residues with 1,2-Dihydroxybenzene Groups. Nat. Commun. 2015, 6, 10216. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.L.; Tsai, M.-Y.; Aloe, S.; Bechberger, K.; König, S.; Morfini, G.; Brady, S.T. Defined Tau Phosphospecies Differentially Inhibit Fast Axonal Transport Through Activation of Two Independent Signaling Pathways. Front. Mol. Neurosci. 2020, 13, 610037. [Google Scholar] [CrossRef]
- Venkatramani, A.; Panda, D. Regulation of Neuronal Microtubule Dynamics by Tau: Implications for Tauopathies. Int. J. Biol. Macromol. 2019, 133, 473–483. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau Stabilizes Microtubules by Binding at the Interface between Tubulin Heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed]
- Frappier, T.F.; Georgieff, I.S.; Brown, K.; Shelanski, M.L. Tau Regulation of Microtubule-Microtubule Spacing and Bundling. J. Neurochem. 1994, 63, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimers’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local Beta-Structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-C.; Nana, A.L.; Hepker, M.; Hwang, J.-H.L.; Gaus, S.E.; Spina, S.; Cosme, C.G.; Gan, L.; Grinberg, L.T.; Geschwind, D.H.; et al. Preferential Tau Aggregation in von Economo Neurons and Fork Cells in Frontotemporal Lobar Degeneration with Specific MAPT Variants. Acta Neuropathol. Commun. 2019, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Parolini, F.; Tira, R.; Barracchia, C.G.; Munari, F.; Capaldi, S.; D’Onofrio, M.; Assfalg, M. Ubiquitination of Alzheimer’s-Related Tau Protein Affects Liquid-Liquid Phase Separation in a Site- and Cofactor-Dependent Manner. Int. J. Biol. Macromol. 2022, 201, 173–181. [Google Scholar] [CrossRef]
- Ferrari, L.; Rüdiger, S.G.D. Recombinant Production and Purification of the Human Protein Tau. Protein Eng. Des. Sel. 2018, 31, 447–455. [Google Scholar] [CrossRef]
- Barghorn, S.; Biernat, J.; Mandelkow, E. Purification of Recombinant Tau Protein and Preparation of Alzheimer-Paired Helical Filaments in Vitro. Methods Mol. Biol. 2005, 299, 35–51. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mészáros, A.; Muwonge, K.; Janvier, S.; Ahmed, J.; Tompa, P. A Novel Tandem-Tag Purification Strategy for Challenging Disordered Proteins. Biomolecules 2022, 12, 1566. https://doi.org/10.3390/biom12111566
Mészáros A, Muwonge K, Janvier S, Ahmed J, Tompa P. A Novel Tandem-Tag Purification Strategy for Challenging Disordered Proteins. Biomolecules. 2022; 12(11):1566. https://doi.org/10.3390/biom12111566
Chicago/Turabian StyleMészáros, Attila, Kevin Muwonge, Steven Janvier, Junaid Ahmed, and Peter Tompa. 2022. "A Novel Tandem-Tag Purification Strategy for Challenging Disordered Proteins" Biomolecules 12, no. 11: 1566. https://doi.org/10.3390/biom12111566