

New Betulin Derivatives with Nitrogen Heterocyclic Moiety—Synthesis and Anticancer Activity In Vitro

Abstract
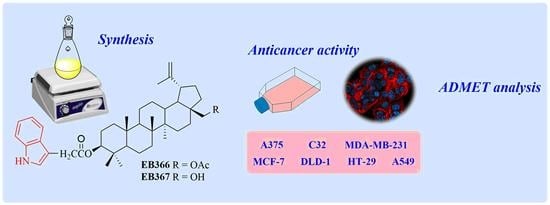
:
1. Introduction
2. Materials and Methods
2.1. General Procedures
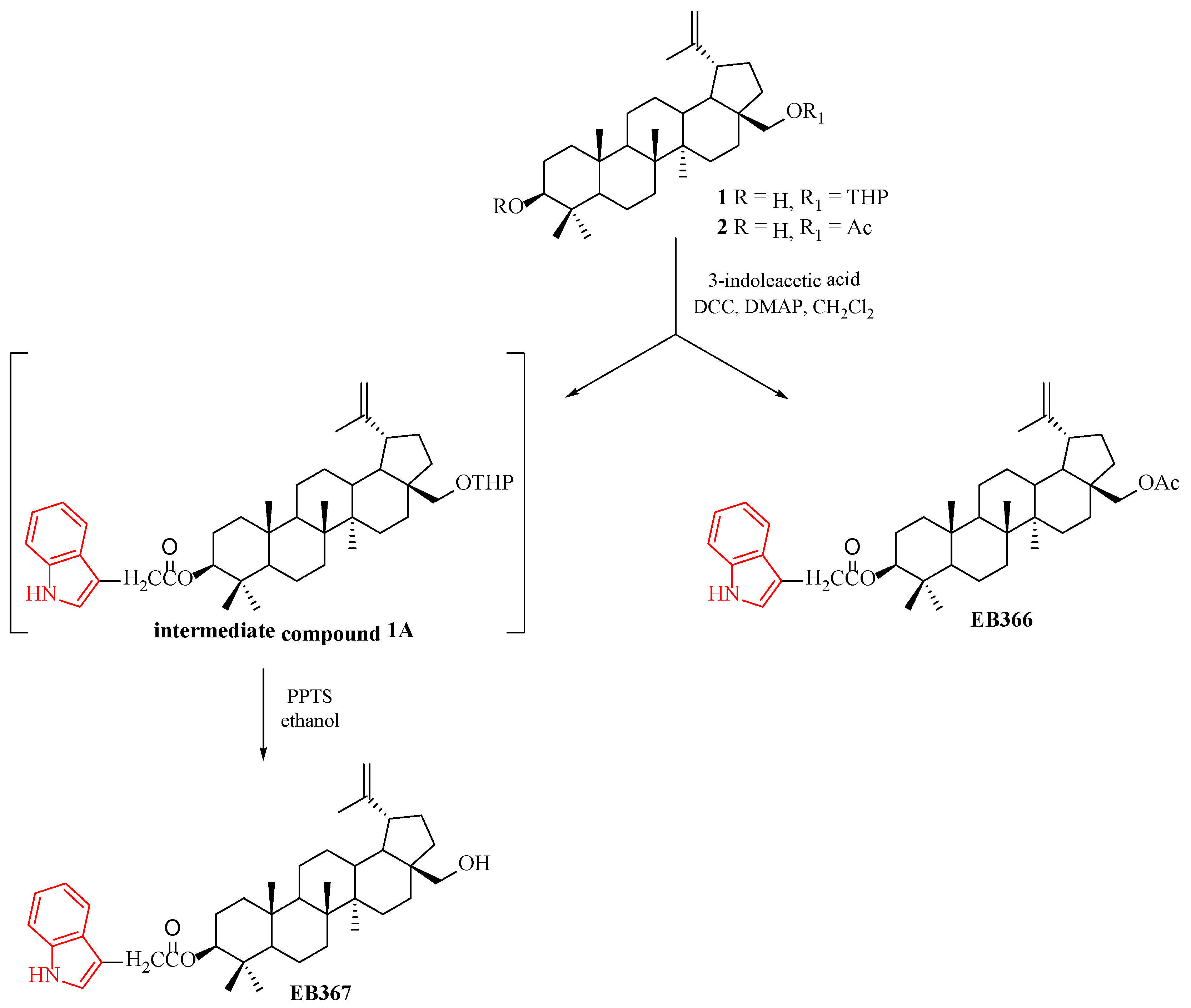
2.2. Synthesis of 3-Indolyl Betulin Derivatives EB366 and EB367
2.2.1. General Grocedure for the Steglich Esterification of 28-Tetrahydropyranyl ether of Betulin 1 and 28-Acetylbetulin 2
2.2.2. General Procedure for the Synthesis of EB367
2.3. Cell Culture
2.4. WST-1 Assay
2.5. Fluorescent Cytometry Analysis
2.6. Microscopic Assessment of Cell Culture
2.7. In Silico Analysis
2.8. Statistical Analysis
3. Results and Discussions
3.1. Chemistry
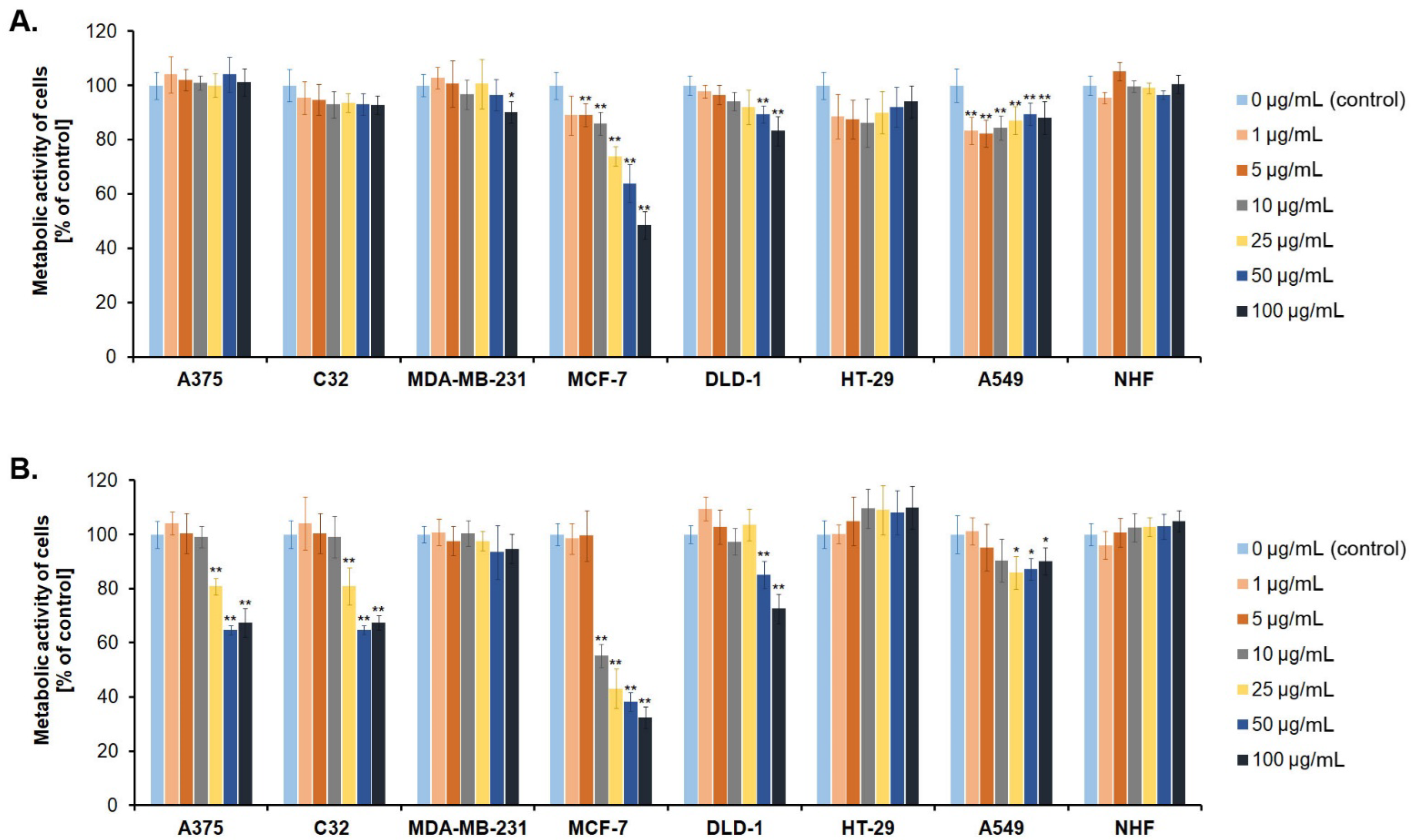
3.2. Screening Cancer Cell Lines for Sensitivity to Novel Betulin Derivatives
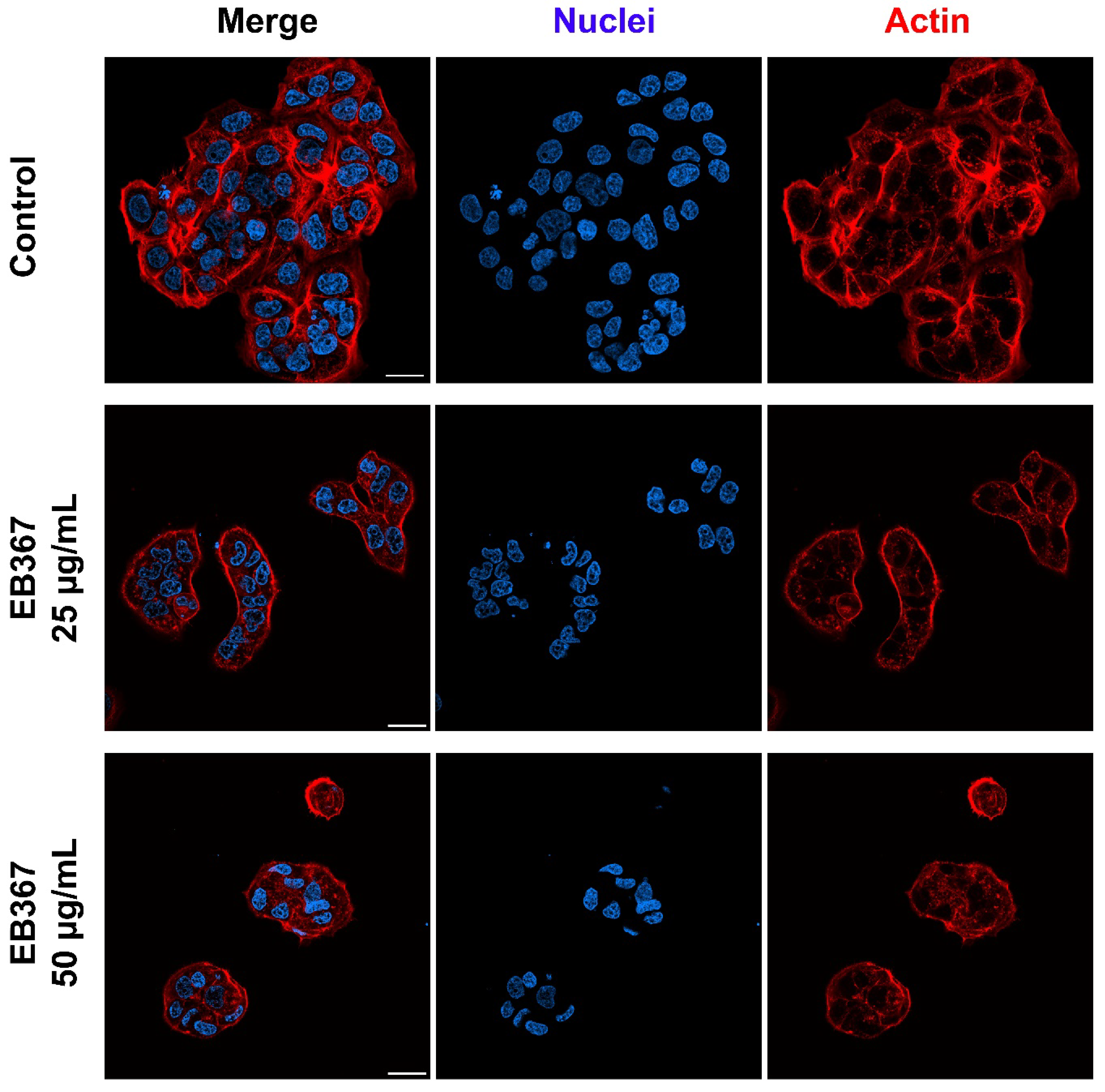
3.3. Analysis of Proliferation Rate and Survival of Breast Cancer Cells Treated with EB367
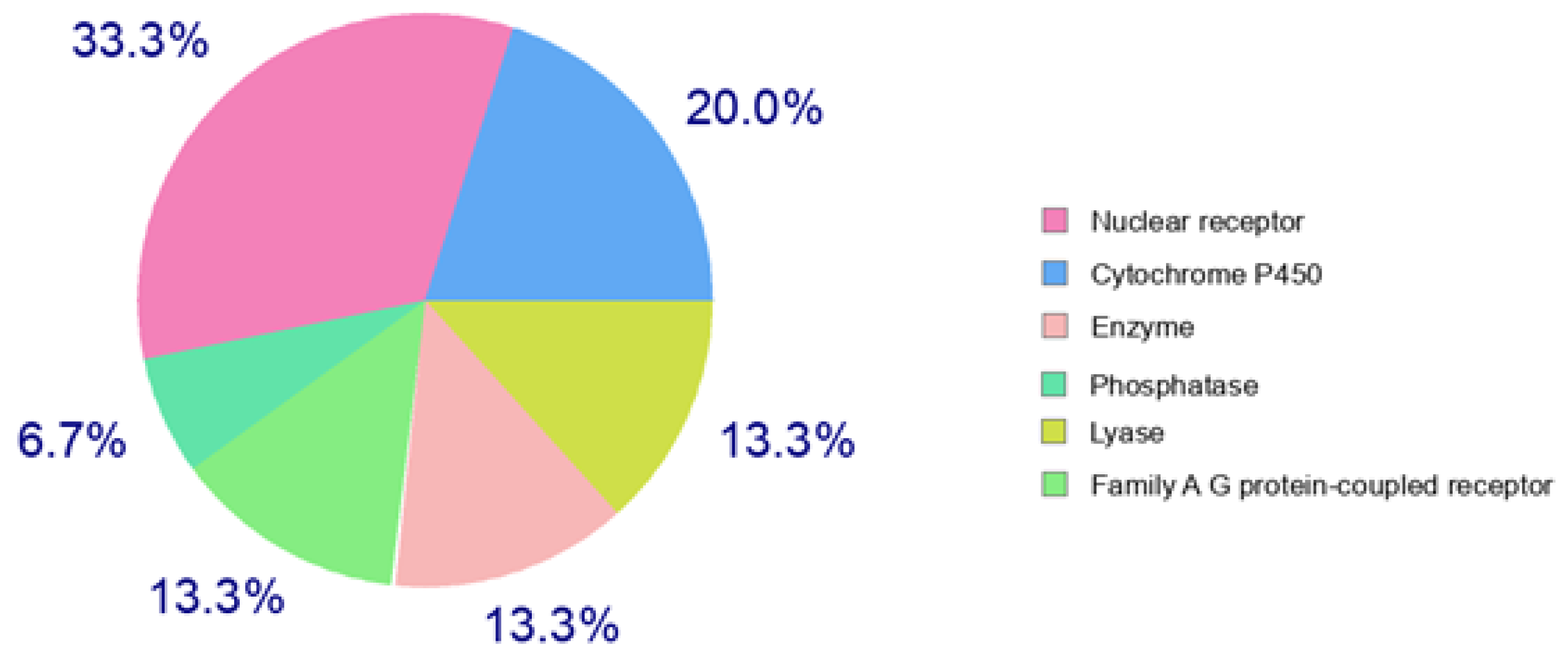
3.4. In Silico
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tilaoui, M.; Ait Mouse, H.; Zyad, A. Update and new insights on future cancer drug candidates from plant-based alkaloids. Front. Pharmacol. 2021, 12, 719694–719712. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Arora, S.; Singh, R. Isolation, characterization and biological activities of betulin from Acacia nilotica bark. Sci. Rep. 2022, 12, 9370–9379. [Google Scholar] [CrossRef] [PubMed]
- Król, S.K.; Kiełbus, M.; Rivero-Müller, A.; Stepulak, A. Comprehensive review on betulin as a potent anticancer agent. Biomed Res. Int. 2015, 2015, 584189. [Google Scholar] [CrossRef] [Green Version]
- Bębenek, E.; Chrobak, E.; Wietrzyk, J.; Kadela, M.; Chrobak, A.; Kusz, J.; Książek, M.; Jastrzębska, M.; Boryczka, S. Synthesis, structure and cytotoxic activity of acetylenic derivatives of betulonic and betulinic acids. J. Mol. Struct. 2016, 1106, 210–219. [Google Scholar] [CrossRef]
- Chrobak, E.; Jastrzębska, M.; Bębenek, E.; Kadela-Tomanek, M.; Marciniec, K.; Latocha, M.; Wrzalik, R.; Kusz, J.; Boryczka, S. Molecular structure, in vitro anticancer study and molecular docking of new phosphate derivatives of betulin. Molecules 2021, 26, 737. [Google Scholar] [CrossRef]
- Pęcak, P.; Orzechowska, B.; Chrobak, E.; Boryczka, S. Novel betulin dicarboxylic acid ester derivatives as potent antiviral agents: Design, synthesis, biological evaluation, structure-activity relationship and in-silico study. Eur. J. Med. Chem. 2021, 225, 113738–113750. [Google Scholar] [CrossRef]
- Wang, J.; Wu, J.; Han, Y.; Zhang, J.; Lin, Y.; Wang, H.; Wang, J.; Liu, J.; Bu, M. Design and synthesis of novel betulin derivatives containing thio-/semicarbazone moieties as apoptotic inducers through mitochindria-related pathways. Molecules 2021, 26, 6356. [Google Scholar] [CrossRef]
- Kirstgen, M.; Lowjaga, K.A.A.T.; Müller, S.F.; Goldmann, N.; Lehmann, F.; Alakurtti, S.; Yli-Kauhaluoma, J.; Glebe, D.; Geyer, J. Selective hepatitis B and D virus entry inhibitors from the group of pentacyclic lupane-type betulin-derived triterpenoids. Sci. Rep. 2020, 10, 21772–21787. [Google Scholar] [CrossRef]
- Heller, L.; Perl, V.; Wiemann, J.; Al-Harrasi, A.; Csuk, R. Amino(oxo)acetate moiety: A new functional group to improve the cytotoxicity of betulin derived carbamates. Bioorg. Med. Chem. Lett. 2016, 26, 2852–2854. [Google Scholar] [CrossRef]
- Grymel, M.; Lalik, A.; Kazek-Kęsik, A.; Szewczyk, M.; Grabiec, P.; Erfurt, K. Design, synthesis, and preliminary evaluation of the cytotoxicity and antibacterial activity of novel triphenylphosphonium derivatives of betulin. Molecules 2022, 27, 5156. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691–111708. [Google Scholar] [CrossRef] [PubMed]
- Sravanthi, T.V.; Manju, S.L. Indoles—A promising scaffold for drug development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [Google Scholar] [CrossRef]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wen, X.; Gong, Y.; Wang, X. Current scenario of indole derivatives with potential anti-drug resistant cancer activity. Eur. J. Med. Chem. 2020, 200, 112359. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Davletshin, E.V.; Tukhbatullin, A.A.; D’yakonov, V.A.; Yunusbaeva, M.M.; Dzhemileva, L.U.; Dzhemilev, U.M. Pentacyclic triterpene acid conjugated with mitochondria-targeting cation F16: Synthesis and evaluation of cytotoxic activities. Med. Chem. Res. 2021, 30, 940–951. [Google Scholar] [CrossRef]
- Li, A.L.; Hao, Y.; Wang, W.Y.; Liu, Q.S.; Sun, Y.; Gu, W. Design, synthesis, and anticancer evaluation of novel indole derivatives of ursolic acid as potential topoisomerase II inhibitors. Int. J. Mol. Sci. 2020, 21, 2876. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Xu, X.; Liu, Z.; Liu, J.; Hu, L. Synthesis and structure—Activity relationships of boswellic acid derivatives as potent VEGFR-2 inhibitors. Bioorg. Med. Chem. 2015, 23, 1982–1993. [Google Scholar] [CrossRef]
- Wang, W.Y.; Wu, W.Y.; Li, A.L.; Liu, Q.S.; Sun, Y.; Gu, W. Synthesis, anticancer evaluation and mechanism studies of novel indolequinone derivatives of ursolic acid. Bioorg. Chem. 2021, 109, 104705–104717. [Google Scholar] [CrossRef]
- Kumar, V.; Rani, N.; Aggarwal, P.; Sanna, V.K.; Singh, A.T.; Jaggi, M.; Joshi, N.; Sharma, P.K.; Irchhaiya, R.; Burman, A.C. Synthesis and cytotoxic activity of heterocyclic ring-substituted betulinic acid derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 5058–5062. [Google Scholar] [CrossRef]
- Bhandari, P.; Patel, N.K.; Gangwal, R.P.; Sangamwar, A.T.; Bhutani, K.K. Oleanolic acid analogs as NO, TNF-a and IL-1β inhibitors: Synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. Lett. 2014, 24, 4114–4119. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, P.; Patel, N.K.; Bhutani, K.K. Synthesis of new heterocyclic lupeol derivatives as nitric oxide and pro-inflammatory cytokine inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3596–3599. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, E.F.; Petrova, A.V.; Thu, H.N.T.; Tu, A.L.T.; Thanh, T.N.; Thi, C.B.; Babkov, D.A.; Kazakova, O.B. Structural modifications of 2,3-indolobetulinic acid: Design and synthesis of highly potent α-glucosidase inhibitors. Bioorg. Chem. 2019, 88, 102957–102968. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; He, H.; Ma, H.; Tu, B.; Li, J.; Guo, S.; Chen, S.; Cao, N.; Zheng, W.; Tang, X.; et al. Oleanolic acid indole derivatives as novel α-glucosidase inhibitors: Synthesis, biological evaluation, and mechanistic analysis. Bioorg. Chem. 2021, 107, 104580–104590. [Google Scholar] [CrossRef] [PubMed]
- Haavikko, R.; Nasereddin, A.; Sacerdoti-Sierra, N.; Kopelyanskiy, D.; Alakurtti, S.; Tikka, M.; Jaffe, C.L.; Yli-Kauhaluoma, J. Heterocycle-fused lupane triterpenoids inhibit Leishmania donovani amastigotes. MedChemcomm 2014, 5, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, Y.; Hu, H.; Persoons, L.; Daelemans, D.; Jonghe, S.D.; Luyten, W.; Krasniqi, B.; Dehaen, W. Antibacterial and antitumoral properties of 1,2,3-triazolo fused triterpenes and their mechanism of inhibiting the proliferation of HL-60 cells. Eur. J. Med. Chem. 2021, 224, 113727–113741. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, C.H.; Morris-Natschke, S.L.; Lee, K.H. Design, synthesis, and structure activity relationship analysis of new betulinic acid derivatives as potent HIV inhibitors. Eur. J. Med. Chem. 2021, 215, 113287–113301. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70. [Google Scholar] [CrossRef]
- Peng, Y.B.; Zhao, Z.L.; Liu, T.; Xie, G.J.; Jin, C.; Deng, T.G.; Sun, Y.; Li, X.; Hu, X.X.; Zhang, X.B.; et al. A multi-mitochondrial anticancer agent that selectively kills cancer cells and overcomes drug resistance. Chem. Med. Chem. 2017, 12, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; Leder, P. F16, a mitochondriotoxic compound, triggers apoptosis or necrosis depending on the genetic background of the target carcinoma cel. Cancer Res. 2004, 64, 329–336. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Penkov, N.V.; Nedopekina, D.A.; Sharapov, V.A.; Khoroshavina, E.I.; Davletshin, E.V.; Belosludtseva, N.V.; Spivak, A.Y.; et al. Mitochondria-targeted prooxidant effects of betulinic acid conjugated with delocalized lipophilic cation F16. Free Radic. Biol. Med. 2021, 168, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Semenova, A.A.; Nedopekina, D.A.; Davletshin, E.V.; Spivak, A.Y.; Belosludtsev, K.N. Effect of F16-betulin conjugate on mitochondrial membranes and its role in cell death initiation. Membranes 2021, 11, 352. [Google Scholar] [CrossRef] [PubMed]
- Pohjala, L.; Alakurtti, S.; Ahola, T.; Yli-Kauhaluoma, J.; Tammela, P. Betulin-derived compounds as inhibitors of alphavirus replication. J. Nat. Prod. 2009, 72, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Snyder, J.K. Preparation of a 24-nor-1,4-dien-3-one triterpene derivative from betulin: New route to 24-nortriterpene analogues. J. Org. Chem. 2002, 67, 2864–2873. [Google Scholar] [CrossRef]
- Rok, J.; Rzepka, Z.; Beberok, A.; Pawlik, J.; Wrześniok, D. Cellular and molecular aspects of anti-melanoma effect of minocycline-a study of cytotoxicity and apoptosis on human melanotic melanoma cells. Int. J. Mol. Sci. 2020, 21, 6917. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717–42729. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Ramos-Tomillero, I.; El-Faham, A.; Nicolas, E.; Rodriguez, H.; de la Torre, B.G.; Albericio, F. Understanding tetrahydropyranyl as a protecting group in peptide chemistry. ChemistryOpen 2017, 6, 168–177. [Google Scholar] [CrossRef]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel triazole hybrids of betulin: Synthesis and biological activity profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadela, M.; Jastrzębska, M.; Bębenek, E.; Chrobak, E.; Latocha, M.; Kusz, J.; Książek, M.; Boryczka, S. Synthesis, structure and cytotoxic activity of mono and dialkoxy derivatives of 5,8-quinolinedione. Molecules 2016, 21, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhadi, P.; Yarani, R.; Valipour, E.; Kiani, S.; Hoseinkhani, Z.; Mansouri, K. Cell line-directed breast cancer research based on glucose metabolism status. Biomed. Pharmacother. 2022, 146, 112526–112533. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reda, A.; Refaat, A.; Abd-Rabou, A.A.; Mahmoud, A.M.; Adel, M.; Sabet, S.; Ali, S.S. Role of mitochondria in rescuing glycolytically inhibited subpopulation of triple negative but not hormone-responsive breast cancer cells. Sci. Rep. 2019, 9, 13748–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. (Eds.) World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2003; Available online: http://publications.iarc.fr/586 (accessed on 12 October 2022).
- Cragg, G.M.; Newman, D.J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409–107447. [Google Scholar] [CrossRef]
- Kommera, H.; Kaluderović, G.N.; Kalbitz, J.; Paschke, R. Synthesis and anticancer activity of novel betulinic acid and betulin derivatives. Arch. Der Pharm. 2010, 343, 449–457. [Google Scholar] [CrossRef]
- Santos, R.C.; Salvador, J.A.; Marín, S.; Cascante, M.; Moreira, J.N.; Dinis, T.C. Synthesis and structure-activity relationship study of novel cytotoxic carbamate and N-acylheterocyclic bearing derivatives of betulin and betulinic acid. Bioorg. Med. Chem. 2010, 18, 4385–4396. [Google Scholar] [CrossRef]
- Güttler, A.; Eiselt, Y.; Funtan, A.; Thiel, A.; Petrenko, M.; Keßler, J.; Thondorf, I.; Paschke, R.; Vordermark, D.; Bache, M. Betulin sulfonamides as carbonic anhydrase inhibitors and anticancer agents in breast cancer cells. Int. J. Mol. Sci. 2021, 22, 8808. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Leong, N.; Zheng, D.; Ford, L.; Nguyen, T.H.; Williams, H.D.; Benameur, H.; Scammells, P.J.; Porter, C.J.H. Biocompatible cationic lipoamino acids as counterions for oral administration of API-ionic liquids. Pharm. Res. 2022, 39, 2405–2419. [Google Scholar] [CrossRef] [PubMed]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851–104865. [Google Scholar] [CrossRef] [PubMed]
- Yusof, I.; Segall, M.D. Considering the impact drug-like properties have on the chance of success. Drug Discov. Today 2013, 18, 659–666. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Jia, R.-B.; Li, Z.-R.; Wu, J.; Ou, Z.-R.; Zhu, Q.; Sun, B.; Lin, L.; Zhao, M. Physicochemical properties of polysaccharide fractions from Sargassum fusiforme and their hypoglycemic and hypolipidemic activities in type 2 diabetic rats. Int. J. Biol. Macromol. 2020, 147, 428–438. [Google Scholar] [CrossRef]
- Tinworth, C.P.; Young, R.J. Facts, patterns, and principles in drug discovery: Appraising the rule of 5 with measured physicochemical data. J. Med. Chem. 2020, 63, 10091–10108. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Smith, D.; Artursson, P.; Avdeef, A.; Di, L.; Ecker, G.F.; Faller, B.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Kramer, S.D.; et al. Passive lipoidal diffusion and carrier-mediated cell uptake are both important mechanisms of membrane permeation in drug disposition. Mol. Pharm. 2014, 11, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Oliver, S.G. How drugs get into cells: Tested and testable predictions to help discriminate between transporter-mediated uptake and lipoidal bilayer diffusion. Front. Pharmacol. 2014, 5, 231–262. [Google Scholar] [CrossRef] [Green Version]
- Amin, L.M. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J. 2006, 8, E101–E111. [Google Scholar] [CrossRef] [Green Version]
- Stieger, B.; Hagenbuch, B. Organic anion transporting polypeptides. Curr. Top. Membr. 2014, 73, 205–232. [Google Scholar] [CrossRef] [Green Version]
- Richard, A.M.; Huang, R.; Waidyanatha, S.; Shinn, P.; Collins, B.J.; Thillainadarajah, I.; Grulke, C.M.; Williams, A.J.; Lougee, R.R.; Judson, R.S.; et al. The Tox21 10k compound library: Collaborative chemistry advancing toxicology. Chem. Res. Toxicol. 2021, 34, 189–216. [Google Scholar] [CrossRef]
- Jäger, S.; Laszczyk, M.N.; Scheffler, A. A preliminary pharmacokinetic study of betulin, the main pentacyclic triterpene from extract of outer bark of birch (Betulae alba cortex). Molecules 2008, 13, 3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozharitskaya, O.N.; Karlina, M.V.; Shikov, A.N.; Kosman, V.M.; Makarov, V.G.; Casals, E.; Rosenholm, J.M. Pharmacokinetics and tissue disposition of nanosystem-entrapped betulin after endotracheal administration to rats. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Coricovac, D.; Dehelean, C.A.; Pinzaru, I.; Mioc, A.; Aburel, O.M.; Macasoi, I.; Draghici, G.A.; Petean, C.; Soica, C.; Boruga, M.; et al. Assessment of betulinic acid cytotoxicity and mitochondrial metabolism impairment in a human melanoma cell line. Int. J. Mol. Sci. 2021, 22, 4870. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of EB367 | Relative Cell Number [Mean Value ± SD] | Percentage of Cells with Damaged Cell Membrane [Mean Value ± SD] | Percentage of Cells with Fragmented DNA [Mean Value ± SD] |
|---|---|---|---|
| 0 µg/mL (control) | 100 ± 7 | 3.9 ± 1.9 | 1.3 ± 0.6 |
| 25 µg/mL | 45 ± 9 ** | 5.0 ± 1.0 | 2.7 ± 1.2 |
| 50 µg/mL | 43 ± 6 ** | 7.8 ± 1.6 * | 3.1 ± 0.9 |
| Parameter | Value |
|---|---|
| Formula | C40H57NO3 |
| Molecular weight (MW) | 599.89 g/mol |
| Fraction Csp3 (Fsp3) | 0.72 |
| Number of rotatable bonds (RB) | 6 |
| Number of H-bond acceptors (H-BA) | 3 |
| Number of H-bond donors (H-BD) | 2 |
| Molar Refractivity (MR) | 182.38 |
| Topological polar surface (TPSA) | 62.32 Ų |
| Log Po/w (WLOGP) | 9.27 |
| Log Po/w (XLOGP3) | 10.58 |
| Log Po/w (MLOGP) | 6.61 |
| Classification | Target | Prediction | Probability |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Inactive | 0.65 |
| Toxicity end points | Carcinogenicity | Inactive | 0.62 |
| Immunotoxicity | Active | 0.89 | |
| Mutagenicity | Inactive | 0.72 | |
| Cytotoxicity | Inactive | 0.78 | |
| Tox21-Nuclear receptor signaling pathways | Aryl hydrocarbon Receptor (AhR) | Inactive | 0.84 |
| Androgen Receptor (AR) | Inactive | 0.84 | |
| Androgen Receptor Ligand Binding Domain (AR-LBD) | Inactive | 0.78 | |
| Aromatase | Inactive | 0.82 | |
| Estrogen Receptor Alpha (ER) | Inactive | 0.73 | |
| Estrogen Receptor Ligand Binding Domain (ER-LBD) | Inactive | 0.88 | |
| Peroxisome Proliferator Activated Receptor Gamma (PPAR-Gamma) | Inactive | 0.92 | |
| Tox21-Stress response pathways | Nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (nrf2/ARE) | Inactive | 0.88 |
| Heat shock factor response element (HSE) | Inactive | 0.88 | |
| Mitochondrial Membrane Potential (MMP) | Inactive | 0.68 | |
| Phosphoprotein (Tumor Supressor) p53 | Inactive | 0.79 | |
| ATPase family AAA domain-containing protein 5 (ATAD5) | Inactive | 0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bębenek, E.; Chrobak, E.; Rzepka, Z.; Wrześniok, D. New Betulin Derivatives with Nitrogen Heterocyclic Moiety—Synthesis and Anticancer Activity In Vitro. Biomolecules 2022, 12, 1540. https://doi.org/10.3390/biom12101540
Bębenek E, Chrobak E, Rzepka Z, Wrześniok D. New Betulin Derivatives with Nitrogen Heterocyclic Moiety—Synthesis and Anticancer Activity In Vitro. Biomolecules. 2022; 12(10):1540. https://doi.org/10.3390/biom12101540
Chicago/Turabian StyleBębenek, Ewa, Elwira Chrobak, Zuzanna Rzepka, and Dorota Wrześniok. 2022. "New Betulin Derivatives with Nitrogen Heterocyclic Moiety—Synthesis and Anticancer Activity In Vitro" Biomolecules 12, no. 10: 1540. https://doi.org/10.3390/biom12101540