
Gerstmann–Sträussler–Scheinker Disease with F198S Mutation Induces Independent Tau and Prion Protein Pathologies in Bank Voles
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introductions
2. Materials and Methods
2.1. Ethics Statement
2.2. Inocula
2.3. Bioassays, Clinical Examination and Sampling
2.4. Histology, Immunohistochemistry and Biochemistry
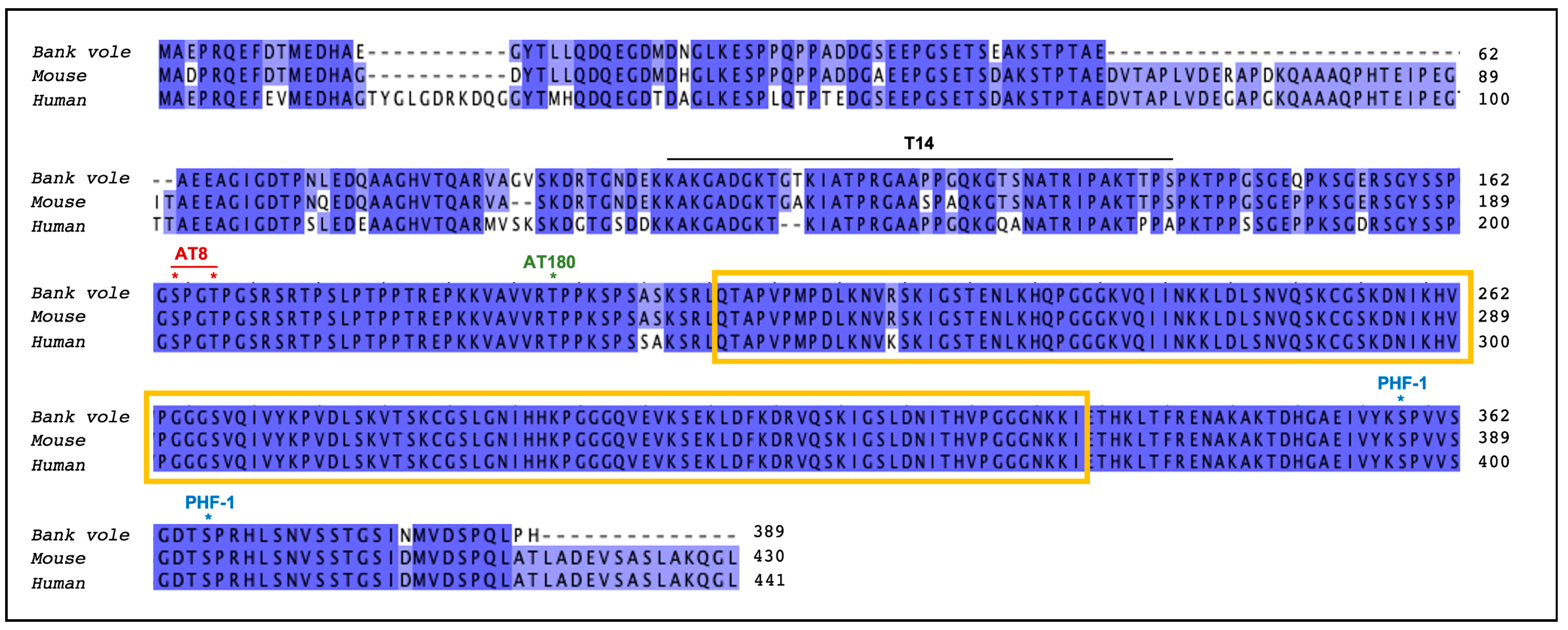
2.5. Sequence of Tau Gene
3. Results and Discussion
3.1. Resistance of BvM to GSS-F198S Prions
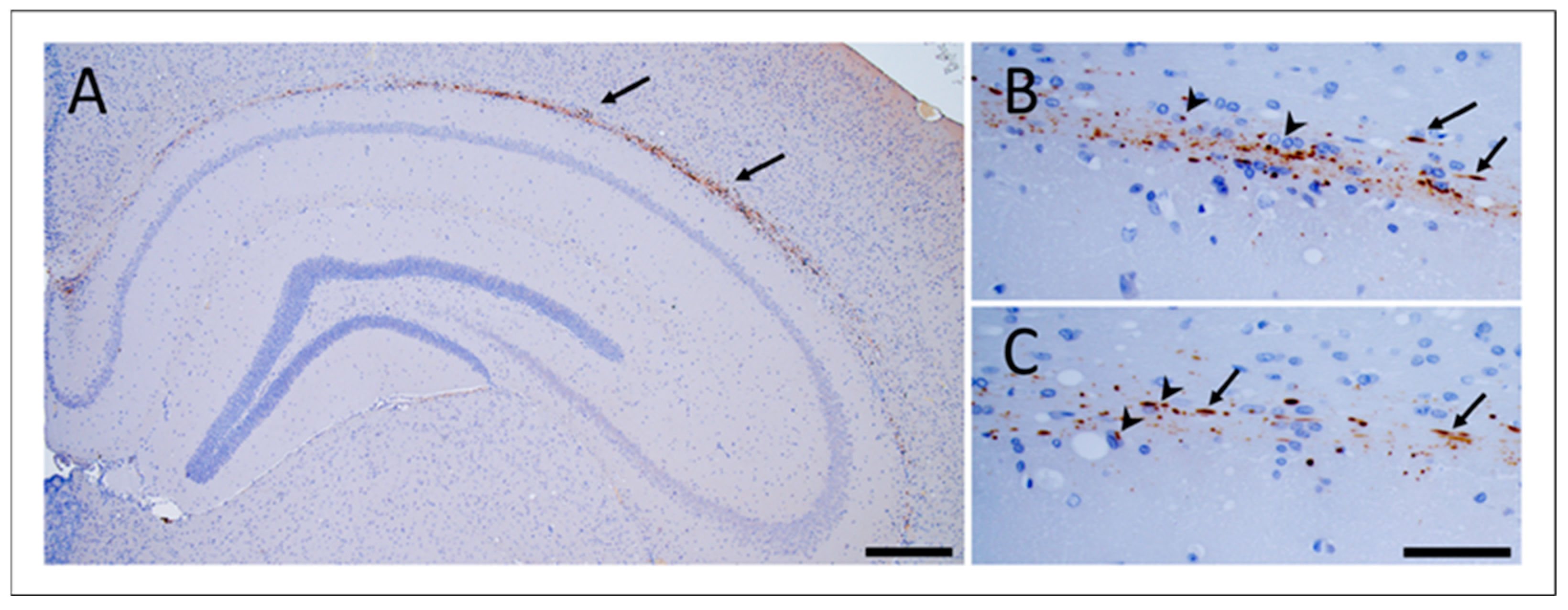
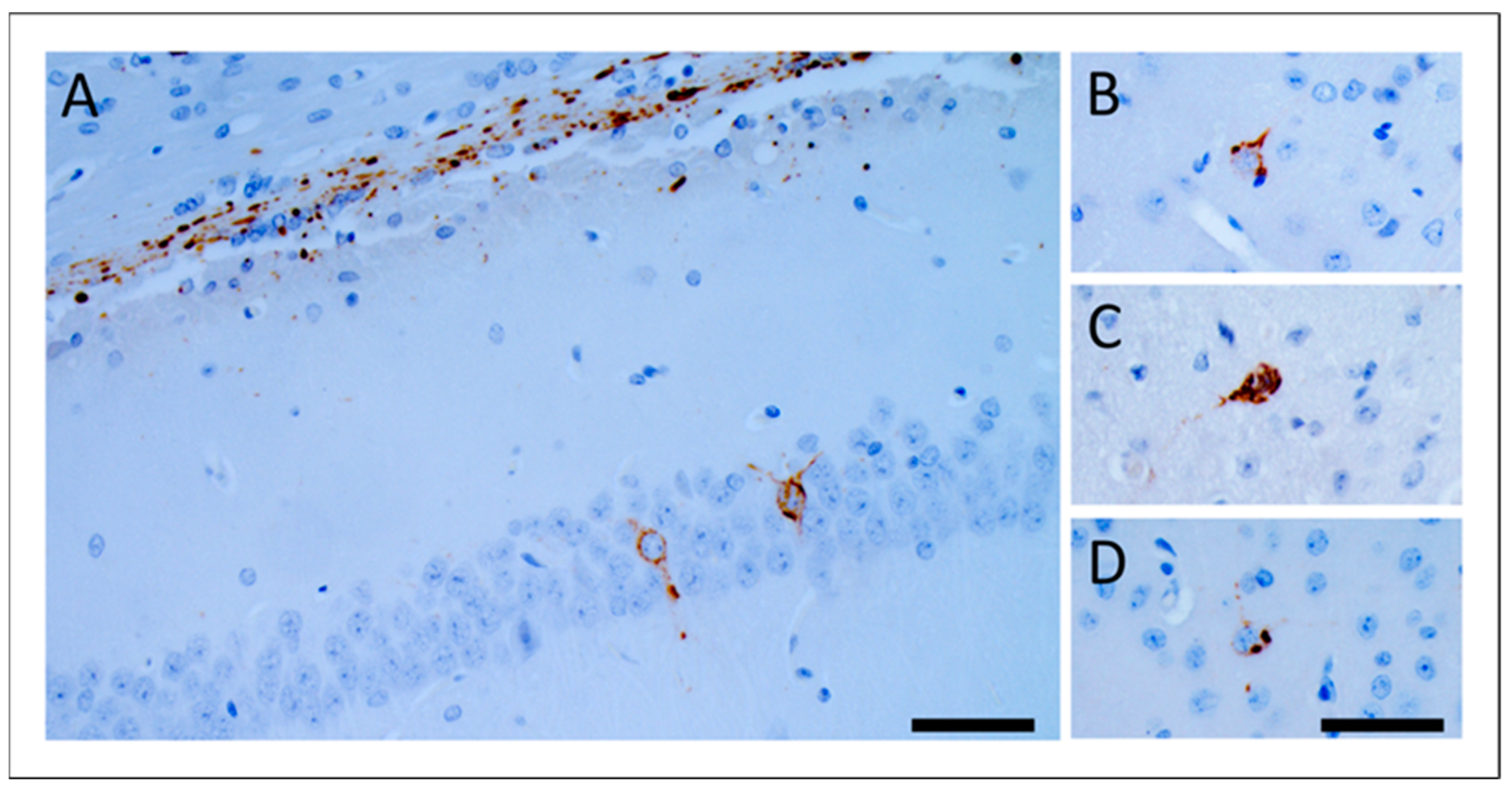
3.2. Propagation of GSS-F198S-Derived pTau in Bank Voles Is Independent of GSS-F198S Prions
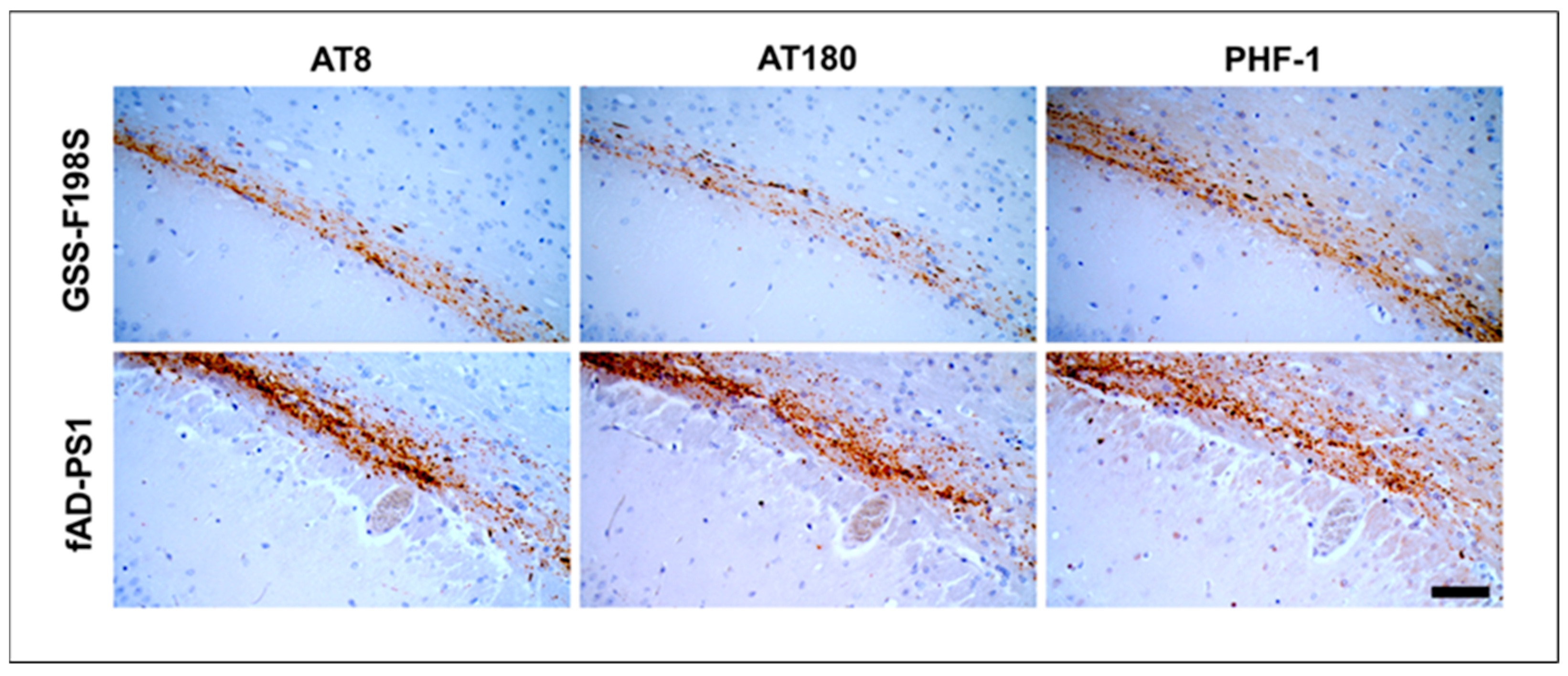
3.3. FAD-PS1 Induce pTau Pathology in Voles Similarly to GSS-F198S
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BvI | bank voles carrying isoleucine at codon 109 of cellular prion protein |
| BvM | bank voles carrying methionine at codon 109 of cellular prion protein |
| pTau | hyperphosphorylated Tau |
| bv-pTau | hyperphosphorylated Tau deposition of vole |
| GSS-F198S | Gerstmann–Sträussler–Scheinker disease with F198S mutation |
| fAD-PS1 | familial Alzheimer’s disease carrying Presenilin-1 mutation |
References
- Farlow, M.R. Gerstmann–Sträussler–Scheinker Syndrome. In Encyclopedia of the Neurological Sciences; Elsevier: Amsterdam, The Netherlands, 2014; pp. 421–425. [Google Scholar]
- Dlouhy, S.R.; Hsiao, K.; Farlow, M.R.; Foroud, T.; Conneally, M.; Johnson, P.; Prusiner, S.B.; Hodes, M.E.; Ghetti, B. Linkage of the Indiana kindred of Gerstmann-Sträussler-Scheinker disease to the prion protein gene. Nat. Genet. 1992, 1, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Tagliavini, F.; Masters, C.L.; Beyreuther, K.; Ciaccone, G.; Verga, L.; Farlow, M.; Conneally, P.; Dlouhy, S.; Azzarelli, B.; et al. Gerstmann-sträussler-scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology 1989, 39, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Tagliavini, F.; Giaccone, G.; Bugiani, O.; Frangione, B.; Farlow, M.R.; Dlouhy, S.R. Familial Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Mol. Neurobiol. 1994, 8, 41–48. [Google Scholar] [CrossRef]
- Bugiani, O.; Giaccone, G.; Piccardo, P.; Morbin, M.; Tagliavini, F.; Ghetti, B.; Words, K.E.Y. Neuropathology of Gerstmann-Straussler-Scheinker Disease. Microsc. Res. Tech. 2000, 15, 10–15. [Google Scholar] [CrossRef]
- Ishizawa, K.; Komori, T.; Shimazu, T.; Yamamoto, T.; Kitamoto, T.; Shimazu, K.; Hirose, T. Hyperphosphorylated tau deposition parallels prion protein burden in a case of Gerstmann-Sträussler-Scheinker syndrome P102L mutation complicated with dementia. Acta Neuropathol. 2002, 104, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-F.; Dong, C.-F.; Zhang, J.; Wan, Y.-Z.; Li, F.; Huang, Y.-X.; Han, L.; Shan, B.; Gao, C.; Han, J.; et al. Human tau protein forms complex with PrP and some GSS- and fCJD-related PrP mutants possess stronger binding activities with tau in vitro. Mol. Cell. Biochem. 2008, 310, 49–55. [Google Scholar] [CrossRef]
- Risacher, S.L.; Farlow, M.R.; Bateman, D.R.; Epperson, F.; Tallman, E.F.; Richardson, R.; Murrell, J.R.; Unverzagt, F.W.; Apostolova, L.G.; Bonnin, J.M.; et al. Detection of tau in Gerstmann-Sträussler-Scheinker disease (PRNP F198S) by [18F]Flortaucipir PET. Acta Neuropathol. Commun. 2018, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Tagliavini, F.; Giaccone, G.; Prelli, F.; Verga, L.; Porro, M.; Trojanowski, J.Q.; Farlow, M.R.; Frangione, B.; Ghetti, B.; Bugiani, O. A68 is a component of paired helical filaments of Gerstmann-Sträussler-Scheinker disease, Indiana kindred. Brain Res. 1993, 616, 325–329. [Google Scholar] [CrossRef]
- Hallinan, G.I.; Hoq, M.R.; Ghosh, M.; Vago, F.S.; Fernandez, A.; Garringer, H.J.; Vidal, R.; Jiang, W.; Ghetti, B. Structure of Tau filaments in Prion protein amyloidoses. Acta Neuropathol. 2021, 142, 227–241. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Walker, L.C.; Jucker, M. Neurodegenerative Diseases: Expanding the Prion Concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bari, M.A.; Chianini, F.; Vaccari, G.; Esposito, E.; Conte, M.; Eaton, S.L.; Hamilton, S.; Finlayson, J.; Steele, P.J.; Dagleish, M.P.; et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J. Gen. Virol. 2008, 89, 2975–2985. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.A.; Nonno, R.; Castilla, J.; D’Agostino, C.; Pirisinu, L.; Riccardi, G.; Conte, M.; Richt, J.; Kunkle, R.; Langeveld, J.; et al. Chronic Wasting Disease in Bank Voles: Characterisation of the Shortest Incubation Time Model for Prion Diseases. PLoS Pathog. 2013, 9, e1003219. [Google Scholar] [CrossRef]
- Galeno, R.; Di Bari, M.A.; Nonno, R.; Cardone, F.; Sbriccoli, M.; Graziano, S.; Ingrosso, L.; Fiorini, M.; Valanzano, A.; Pasini, G.; et al. Prion Strain Characterization of a Novel Subtype of Creutzfeldt-Jakob Disease. J. Virol. 2017, 91, e02390-16. [Google Scholar] [CrossRef] [Green Version]
- Nonno, R.; Di Bari, M.A.; Cardone, F.; Vaccari, G.; Fazzi, P.; Dell’Omo, G.; Cartoni, C.; Ingrosso, L.; Boyle, A.; Galeno, R.; et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006, 2, e12. [Google Scholar] [CrossRef]
- Nonno, R.; Notari, S.; Di Bari, M.A.; Cali, I.; Pirisinu, L.; D’Agostino, C.; Cracco, L.; Kofskey, D.; Vanni, I.; Lavrich, J.; et al. Variable Protease-Sensitive Prionopathy Transmission to Bank Voles. Emerg. Infect. Dis. 2019, 25, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Pirisinu, L.; Di Bari, M.A.; D’Agostino, C.; Marcon, S.; Riccardi, G.; Poleggi, A.; Cohen, M.L.; Appleby, B.S.; Gambetti, P.; Ghetti, B.; et al. Gerstmann-Sträussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci. Rep. 2016, 6, 20443. [Google Scholar] [CrossRef] [Green Version]
- Cartoni, C.; Schininà, M.E.; Maras, B.; Nonno, R.; Vaccari, G.; Di Bari, M.; Conte, M.; De Pascalis, A.; Principe, S.; Cardone, F.; et al. Quantitative profiling of the pathological prion protein allotypes in bank voles by liquid chromatography-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 849, 302–306. [Google Scholar] [CrossRef]
- Nonno, R.; Di Bari, M.A.; Pirisinu, L.; D’Agostino, C.; Vanni, I.; Chiappini, B.; Marcon, S.; Riccardi, G.; Tran, L.; Vikøren, T.; et al. Studies in bank voles reveal strain differences between chronic wasting disease prions from Norway and North America. Proc. Natl. Acad. Sci. USA 2020, 117, 31417–31426. [Google Scholar] [CrossRef] [PubMed]
- Nonno, R.; Marin-Moreno, A.; Carlos Espinosa, J.; Fast, C.; Van Keulen, L.; Spiropoulos, J.; Lantier, I.; Andreoletti, O.; Pirisinu, L.; Di Bari, M.A.; et al. Characterization of goat prions demonstrates geographical variation of scrapie strains in Europe and reveals the composite nature of prion strains. Sci. Rep. 2020, 10, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, J.C.; Nonno, R.; Di Bari, M.; Aguilar-Calvo, P.; Pirisinu, L.; Fernández-Borges, N.; Vanni, I.; Vaccari, G.; Marín-Moreno, A.; Frassanito, P.; et al. PrP C Governs Susceptibility to Prion Strains in Bank Vole, While Other Host Factors Modulate Strain Features. J. Virol. 2016, 90, 10660–10669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirisinu, L.; Di Bari, M.A.; D’Agostino, C.; Vanni, I.; Riccardi, G.; Marcon, S.; Vaccari, G.; Chiappini, B.; Benestad, S.L.; Agrimi, U.; et al. A single amino acid residue in bank vole prion protein drives permissiveness to Nor98/atypical scrapie and the emergence of multiple strain variants. PLOS Pathog. 2022, 18, e1010646. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Nonno, R.; Agrimi, U. The Mouse Model for Scrapie: Inoculation, Clinical Scoring, and Histopathologi-cal Techniques. Methods Mol. Biol. 2012, 849, 453–471. [Google Scholar] [CrossRef]
- Bautista, M.J.; Gutiérrez, J.; Salguero, F.J.; Fernández de Marco, M.M.; Romero-Trevejo, J.L.; Gómez-Villamandos, J.C. BSE infection in bovine PrP transgenic mice leads to hyperphosphorylation of tau-protein. Vet. Microbiol. 2006, 115, 293–301. [Google Scholar] [CrossRef]
- Piccardo, P.; King, D.; Brown, D.; Barron, R.M. Variable tau accumulation in murine models with abnormal prion protein deposits. J. Neurol. Sci. 2017, 383, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Asher, D.M.; Belay, E.; Bigio, E.; Brandner, S.; Brubaker, S.A.; Caughey, B.; Clark, B.; Damon, I.; Diamond, M.; Freund, M.; et al. Risk of transmissibility from neurodegenerative disease-associated proteins: Experimental knowns and unknowns. J. Neuropathol. Exp. Neurol. 2021, 79, 1141–1146. [Google Scholar] [CrossRef]
- Cali, I.; Cohen, M.L.; Haik, S.; Parchi, P.; Giaccone, G.; Collins, S.J.; Kofskey, D.; Wang, H.; McLean, C.A.; Brandel, J.P.; et al. Iatrogenic Creutzfeldt-Jakob disease with Amyloid-β pathology: An international study. Acta Neuropathol. Commun. 2018, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef]
- Ritchie, D.L.; Adlard, P.; Peden, A.H.; Lowrie, S.; Le Grice, M.; Burns, K.; Jackson, R.J.; Yull, H.; Keogh, M.J.; Wei, W.; et al. Amyloid-β accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol. 2017, 134, 221–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghetti, B.; Piccardo, P.; Spillantini, M.; Ichimiya, Y.; Porro, M.; Perini, F.; Kitamoto, T.; Tateishi, J.; Seiler, C.; Frangione, B.; et al. Vascular variant of prion protein cerebral amyloidosis with τ-positive neurofibrillary tangles: The phenotype of the stop codon 145 mutation in PRNP. Proc. Natl. Acad. Sci. USA 1996, 93, 744–748. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, R.; Pirisinu, L.; Riccardi, G.; D’Agostino, C.; De Cecco, E.; Legname, G.; Cardone, F.; Gambetti, P.; Nonno, R.; Agrimi, U.; et al. Gerstmann–Sträussler–Scheinker Disease with F198S Mutation Induces Independent Tau and Prion Protein Pathologies in Bank Voles. Biomolecules 2022, 12, 1537. https://doi.org/10.3390/biom12101537
Bruno R, Pirisinu L, Riccardi G, D’Agostino C, De Cecco E, Legname G, Cardone F, Gambetti P, Nonno R, Agrimi U, et al. Gerstmann–Sträussler–Scheinker Disease with F198S Mutation Induces Independent Tau and Prion Protein Pathologies in Bank Voles. Biomolecules. 2022; 12(10):1537. https://doi.org/10.3390/biom12101537
Chicago/Turabian StyleBruno, Rosalia, Laura Pirisinu, Geraldina Riccardi, Claudia D’Agostino, Elena De Cecco, Giuseppe Legname, Franco Cardone, Pierluigi Gambetti, Romolo Nonno, Umberto Agrimi, and et al. 2022. "Gerstmann–Sträussler–Scheinker Disease with F198S Mutation Induces Independent Tau and Prion Protein Pathologies in Bank Voles" Biomolecules 12, no. 10: 1537. https://doi.org/10.3390/biom12101537