
Molecular Regulation of Heme Oxygenase-1 Expression by E2F Transcription Factor 2 in Lung Fibroblast Cells: Relevance to Idiopathic Pulmonary Fibrosis
 and
and 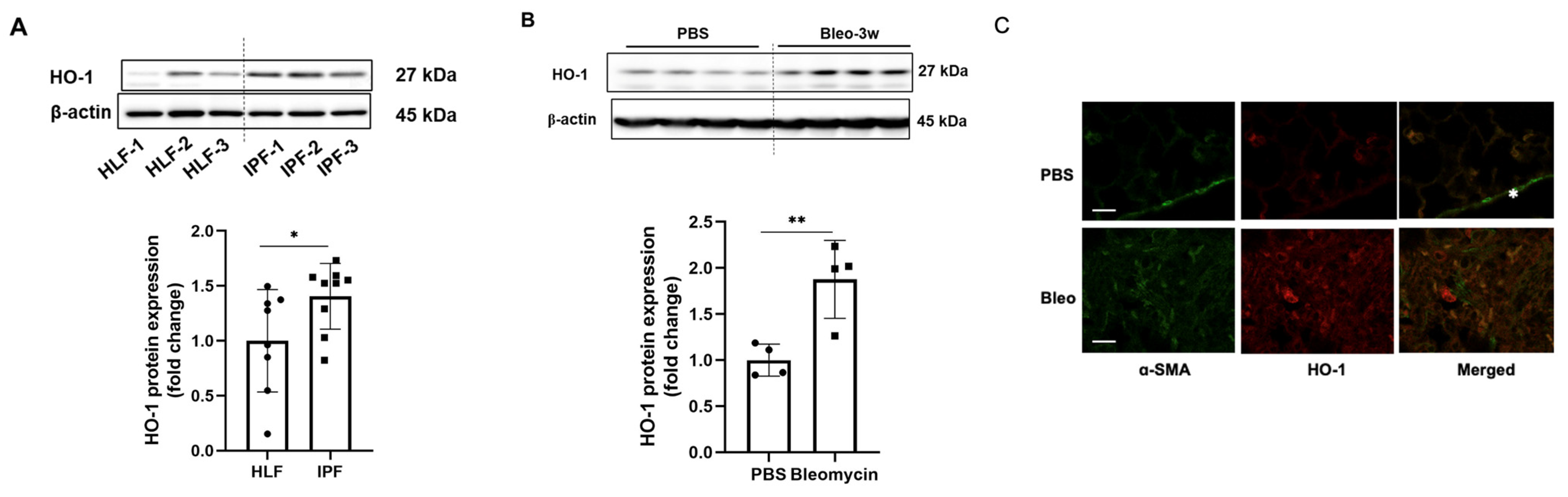{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Bleomycin-induced murine model of lung fibrosis
2.3. Western Blots
2.4. Plasmid and siRNA/esiRNA Transfection
2.5. RNA Extraction, RNA Sequencing, and Quantitative Real-Time PCR
2.6. Quantification and Statistical Analysis
3. Results
3.1. HO-1 Protein Levels Are Significantly Increased in Fibrotic Lungs
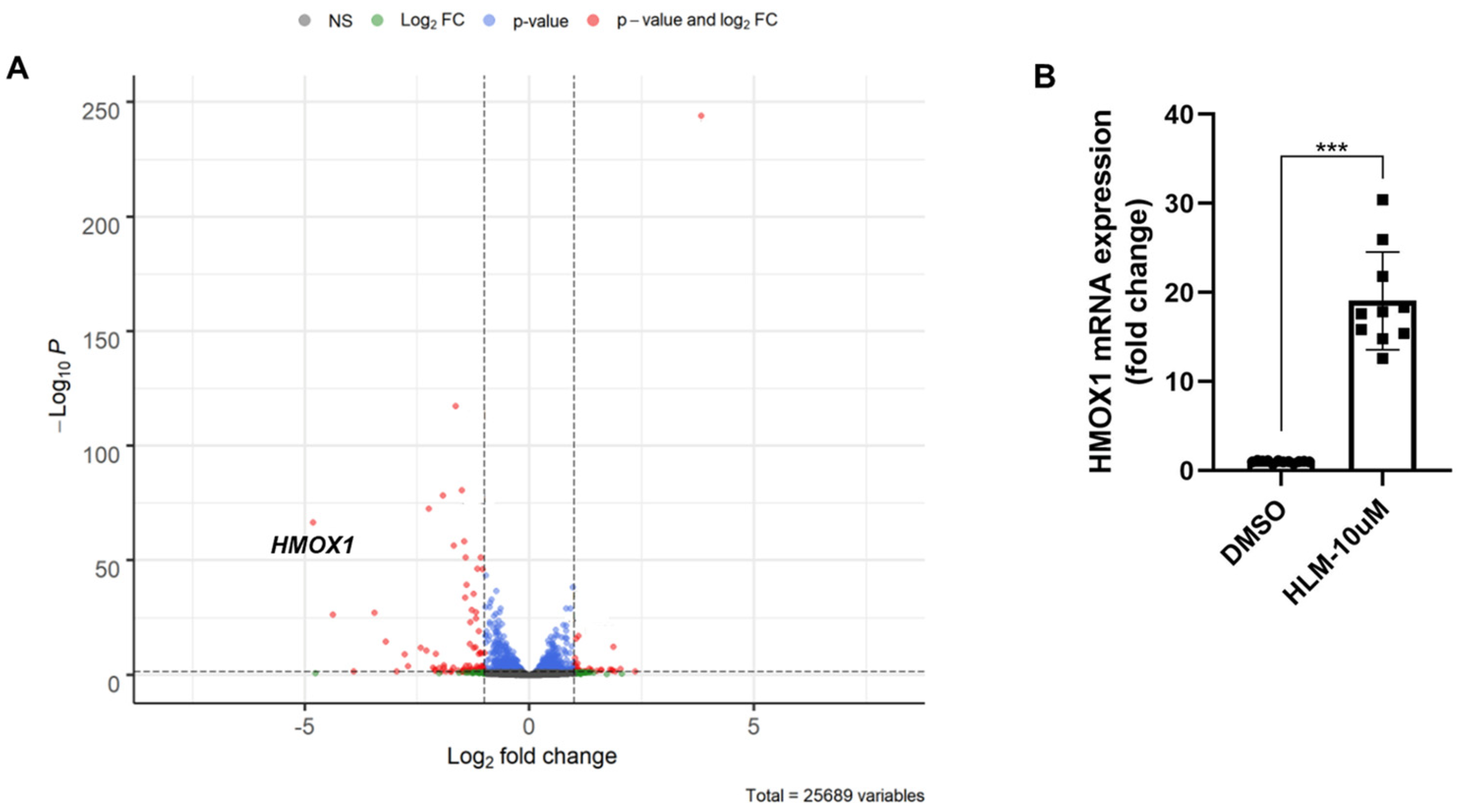
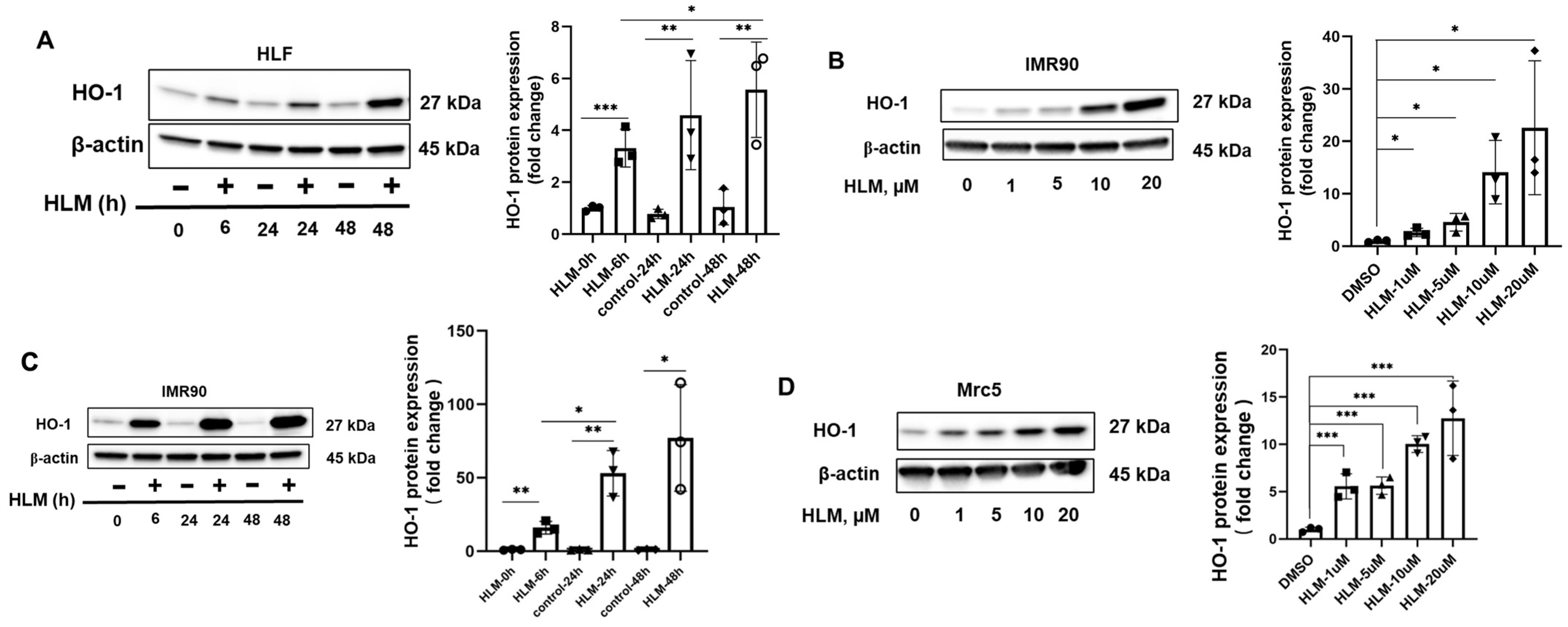
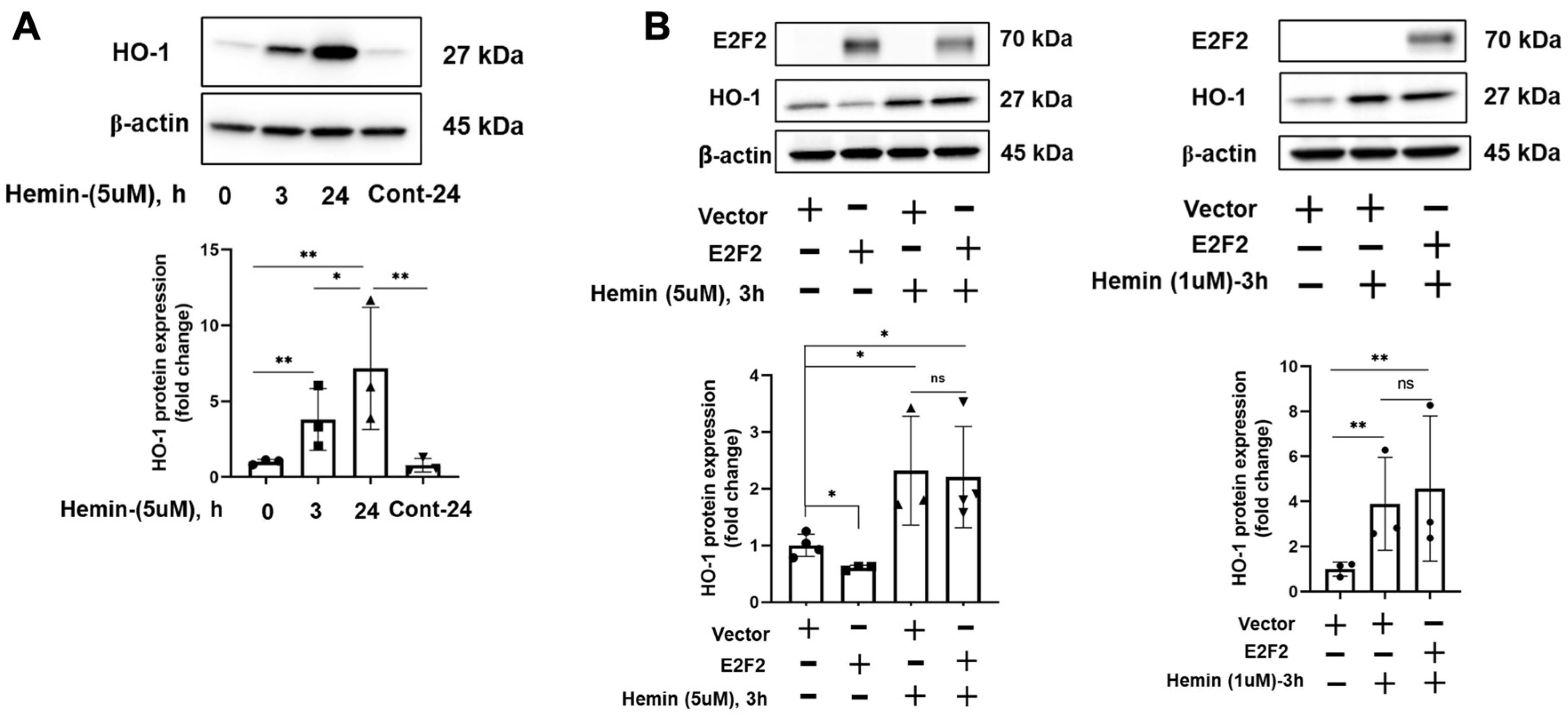
3.2. E2F Inhibitor Upregulates HO-1 mRNA and Protein Levels
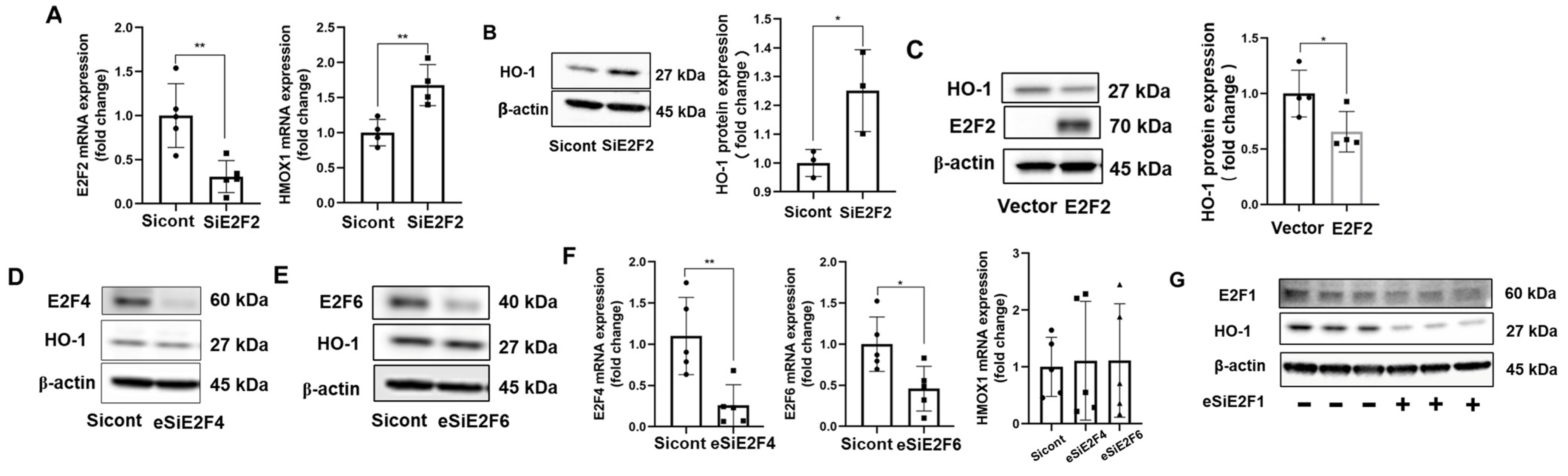
3.3. E2F2 Suppresses the Baseline Expression of HO-1
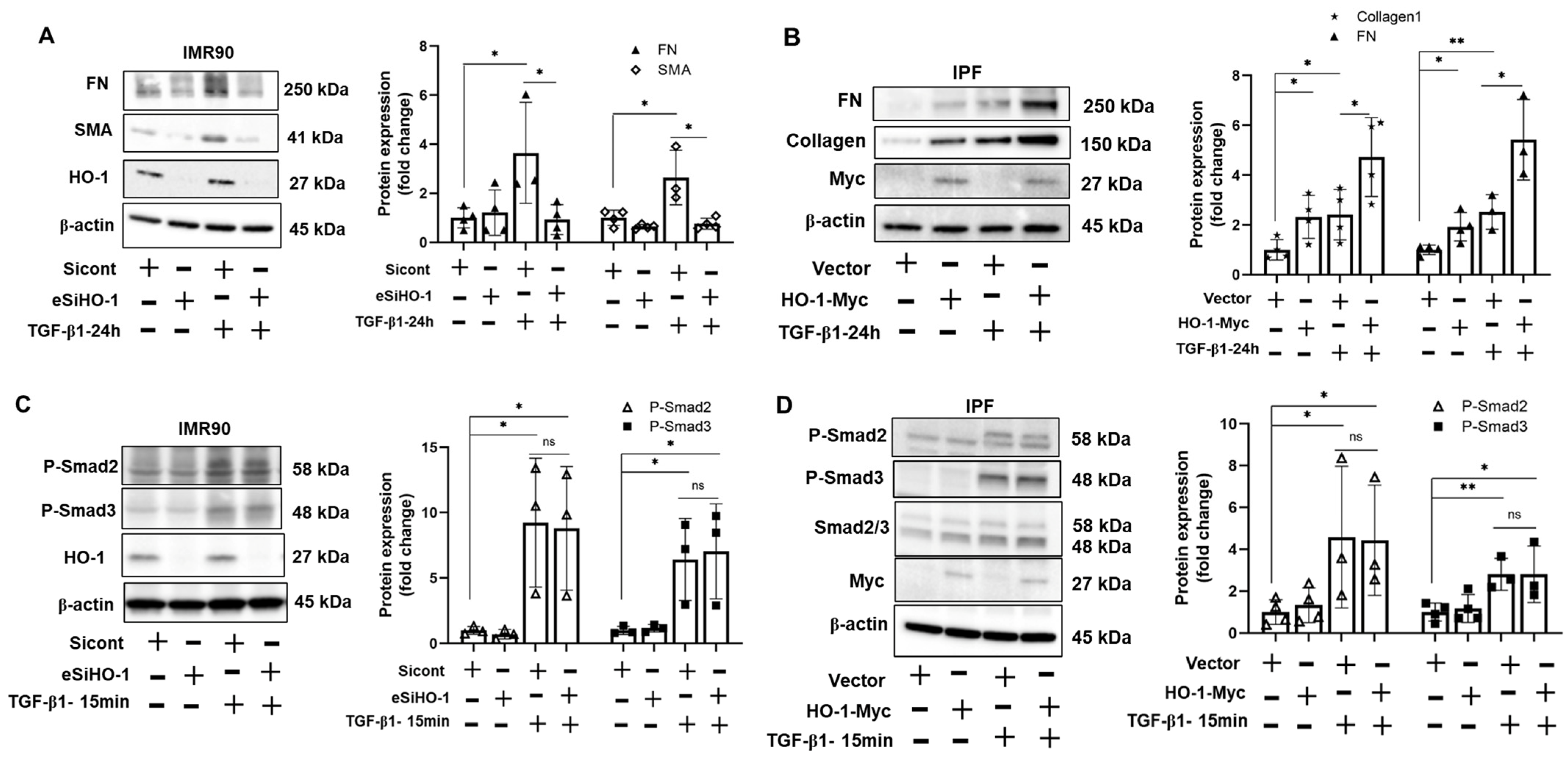
3.4. HO-1 Promotes TGF-β1-Induced Myofibroblast Differentiation without Altering Phosphorylation of Smad2/3
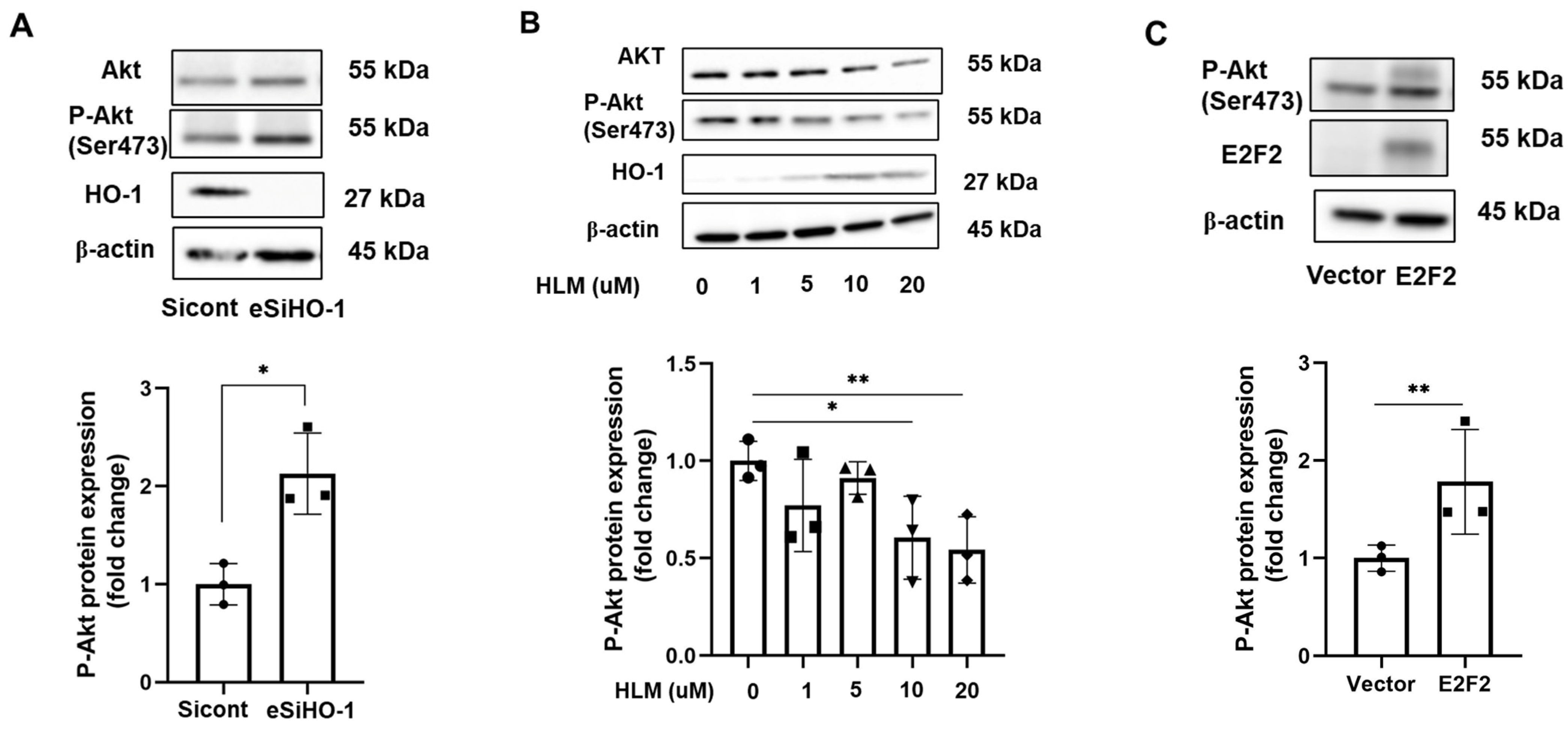
3.5. HO-1 Regulates Phosphorylation of AKT
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luppi, F.; Kalluri, M.; Faverio, P.; Kreuter, M.; Ferrara, G. Idiopathic pulmonary fibrosis beyond the lung: Understanding disease mechanisms to improve diagnosis and management. Respir. Res. 2021, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.L.; Perez, M.A.; Carrasco, M.T.L.; Gil, P.U. Lung transplantation in idiopathic pulmonary fibrosis. Med. Sci. 2018, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Horie, M.; Nagase, T. TGF-β signaling in lung health and disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [Green Version]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J. Adv. Res. 2021, 34, 43–63. [Google Scholar] [CrossRef]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef]
- Kliment, C.R.; Oury, T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef]
- Fois, A.G.; Paliogiannis, P.; Sotgia, S.; Mangoni, A.A.; Zinellu, E.; Pirina, P.; Carru, C.; Zinellu, A. Evaluation of oxidative stress biomarkers in idiopathic pulmonary fibrosis and therapeutic applications: A systematic review. Respir. Res. 2018, 19, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredenburgh, L.E.; Perrella, M.A.; Mitsialis, S.A. The role of heme oxygenase-1 in pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2007, 36, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.M.; Du, J.L.; Duan, Z.J.; Guo, S.B.; Sun, X.Y.; Liu, Z. Inhibiting heme oxygenase-1 attenuates rat liver fibrosis by removing iron accumulation. World J. Gastroenterol. 2013, 19, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Dalavanga, Y.; Poulakis, N.; Sixt, S.U.; Guzman, J.; Costabel, U. Decreased expression of haem oxygenase-1 by alveolar macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2008, 31, 1030–1036. [Google Scholar] [CrossRef] [Green Version]
- Gaubatz, S.; Lindeman, G.J.; Ishida, S.; Jakoi, L.; Nevins, J.R.; Livingston, D.M.; Rempel, R.E. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol. Cell. 2000, 6, 729–735. [Google Scholar] [CrossRef]
- Wang, F.; Li, P.; Li, F.S. Integrated analysis of a gene correlation network identifies critical regulation of fibrosis by lncRNAs and TFs in idiopathic pulmonary fibrosis. Biomed. Res. Int. 2020, 2020, 6537462. [Google Scholar] [CrossRef]
- Yoshihara, T.; Nanri, Y.; Nunomura, S.; Yamaguchi, Y.; Feghali-Bostwick, C.; Ajito, K.; Murakami, S.; Mawatari, M.; Izuhara, K. Periostin plays a critical role in the cell cycle in lung fibroblasts. Respir. Res. 2020, 21, 38. [Google Scholar] [CrossRef] [Green Version]
- Qian, Q.; Ma, Q.; Wang, B.; Qian, Q.; Zhao, C.; Feng, F.; Dong, X. MicroRNA-205-5p targets E2F1 to promote autophagy and inhibit pulmonary fibrosis in silicosis through impairing SKP2-mediated Beclin1 ubiquitination. J. Cell. Mol. Med. 2021, 25, 9214–9227. [Google Scholar] [CrossRef]
- Sozzani, R.; Maggio, C.; Varotto, S.; Canova, S.; Bergounioux, C.; Albani, D.; Cella, R. Interplay between Arabidopsis activating factors E2Fb and E2Fa in cell cycle progression and development. Plant Physiol. 2006, 140, 1355–1366. [Google Scholar] [CrossRef]
- Ma, Y.; Kurtyka, C.A.; Boyapalle, S.; Sung, S.-S.; Lawrence, H.; Guida, W.; Cress, W.D. A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. 2008, 68, 6292–6299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degryse, A.L.; Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Blackwell, T.S.; Lawson, W.E. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am. J. Physiol.-Lung Cell Mol. Physiol. 2010, 299, L442–L452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, I.; Goyal, A.; Singh, N.K.; Yadav, H.N.; Sharma, P.L. Hemin, a heme oxygenase-1 inducer, restores the attenuated cardioprotective effect of ischemic preconditioning in isolated diabetic rat heart. Hum. Exp. Toxicol. 2017, 36, 867–875. [Google Scholar] [CrossRef]
- Hsu, H.S.; Liu, C.C.; Lin, J.H.; Hsu, T.-W.; Hsu, J.-W.; Su, K.; Hung, S.-C. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.L.; Xing, R.G.; Chen, L.; Liu, C.R.; Miao, Z.G. PI3K/Akt signaling is involved in the pathogenesis of bleomycin-induced pulmonary fibrosis via regulation of epithelial-mesenchymal transition. Mol. Med. Rep. 2016, 14, 5699–5706. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hu, K.; Cai, X.; Yang, B.; He, Q.; Wang, J.; Weng, Q. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm. Sin. B 2022, 12, 18–32. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Atzori, L.; Chua, F.; Dunsmore, S.E.; Willis, D.; Barbarisi, M.; McAnulty, R.J.; Laurent, G.J. Attenuation of bleomycin induced pulmonary fibrosis in mice using the heme oxygenase inhibitor Zn-deuteroporphyrin IX-2,4-bisethylene glycol. Thorax 2004, 59, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Tsuburai, T.; Suzuki, M.; Nagashima, Y.; Suzuki, S.; Inoue, S.; Hashiba, T.; Ueda, A.; Ikehara, K.; Matsuse, T.; Ishigatsubo, Y. Adenovirus-mediated transfer and overexpression of heme oxygenase 1 cDNA in lung prevents bleomycin-induced pulmonary fibrosis via a Fas-Fas ligand-independent pathway. Hum. Gene Ther. 2002, 13, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Kim, K.C.; Kang, K.A.; Zhang, R.; Piao, M.J.; Kim, G.Y.; Kang, M.Y.; Lee, S.J.; Lee, N.H.; Surh, Y.-J.; Hyun, J.W. Up-regulation of Nrf2-mediated heme oxygenase-1 expression by eckol, a phlorotannin compound, through activation of Erk and PI3K/Akt. Int. J. Biochem. Cell Biol. 2010, 42, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Attwooll, C.; Denchi, E.L.; Helin, K. The E2F family: Specific functions and overlapping interests. EMBO J. 2004, 23, 4709–4716. [Google Scholar] [CrossRef] [PubMed]
- Ning, W.; Song, R.; Li, C.; Park, E.; Mohsenin, A.; Choi, A.M.K.; Choi, M.E. TGF-β1 stimulates HO-1 via the p38 mitogen-activated protein kinase in A549 pulmonary epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2002, 283, L1094–L1102. [Google Scholar] [CrossRef]
- Shi, J.; Yu, J.; Zhang, Y.; Wu, L.; Dong, S.; Wu, L.; Wu, L.; Du, S.; Zhang, Y.; Ma, D. PI3K/Akt pathway-mediated HO-1 induction regulates mitochondrial quality control and attenuates endotoxin-induced acute lung injury. Lab. Investig. 2019, 99, 1795–1809. [Google Scholar] [CrossRef]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; de Galarreta, C.M.R.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [Green Version]
- Potteti, H.R.; Tamatam, C.R.; Marreddy, R.; Reddy, N.M.; Noel, S.; Rabb, H.; Reddy, S.P. Nrf2-AKT interactions regulate heme oxygenase 1 expression in kidney epithelia during hypoxia and hypoxia-reoxygenation. Am. J. Physiol.-Renal Physiol. 2016, 311, F1025–F1034. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Joe, Y.; Kong, J.S.; Jeong, S.-O.; Cho, G.J.; Ryter, S.W.; Chung, H.T. Carbon monoxide protects against hepatic ischemia/reperfusion injury via ROS-dependent Akt signaling and inhibition of glycogen synthase kinase 3β. Oxidative Med. Cell. Longev. 2013, 2013, 306421. [Google Scholar] [CrossRef] [Green Version]
- Al-Tamari, H.M.; Debral, S.; Schmall, A.; Sarvari, P.; Ruppert, C.; Paik, J.; DePinho, R.; Grimminger, F.; Eickelberg, O.; Guenther, A.; et al. FoxO3 an important player in fibrogenesis and therapeutic target for idiopathic pulmonary fibrosis. EMBO Mol. Med. 2018, 10, 276–293. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Taleb, S.J.; Wang, H.; Parinandi, N.L.; Kass, D.J.; Rojas, M.; Wang, C.; Ma, Q.; Zhao, J.; Zhao, Y. Molecular Regulation of Heme Oxygenase-1 Expression by E2F Transcription Factor 2 in Lung Fibroblast Cells: Relevance to Idiopathic Pulmonary Fibrosis. Biomolecules 2022, 12, 1531. https://doi.org/10.3390/biom12101531
Ye Q, Taleb SJ, Wang H, Parinandi NL, Kass DJ, Rojas M, Wang C, Ma Q, Zhao J, Zhao Y. Molecular Regulation of Heme Oxygenase-1 Expression by E2F Transcription Factor 2 in Lung Fibroblast Cells: Relevance to Idiopathic Pulmonary Fibrosis. Biomolecules. 2022; 12(10):1531. https://doi.org/10.3390/biom12101531
Chicago/Turabian StyleYe, Qinmao, Sarah J. Taleb, Heather Wang, Narasimham L. Parinandi, Daniel J. Kass, Mauricio Rojas, Cankun Wang, Qin Ma, Jing Zhao, and Yutong Zhao. 2022. "Molecular Regulation of Heme Oxygenase-1 Expression by E2F Transcription Factor 2 in Lung Fibroblast Cells: Relevance to Idiopathic Pulmonary Fibrosis" Biomolecules 12, no. 10: 1531. https://doi.org/10.3390/biom12101531