
EP-Pred: A Machine Learning Tool for Bioprospecting Promiscuous Ester Hydrolases
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Esterase Dataset
2.2. Lipase Engineering and Uniref 50 Databases
2.3. Feature Extraction
2.4. Feature Selection
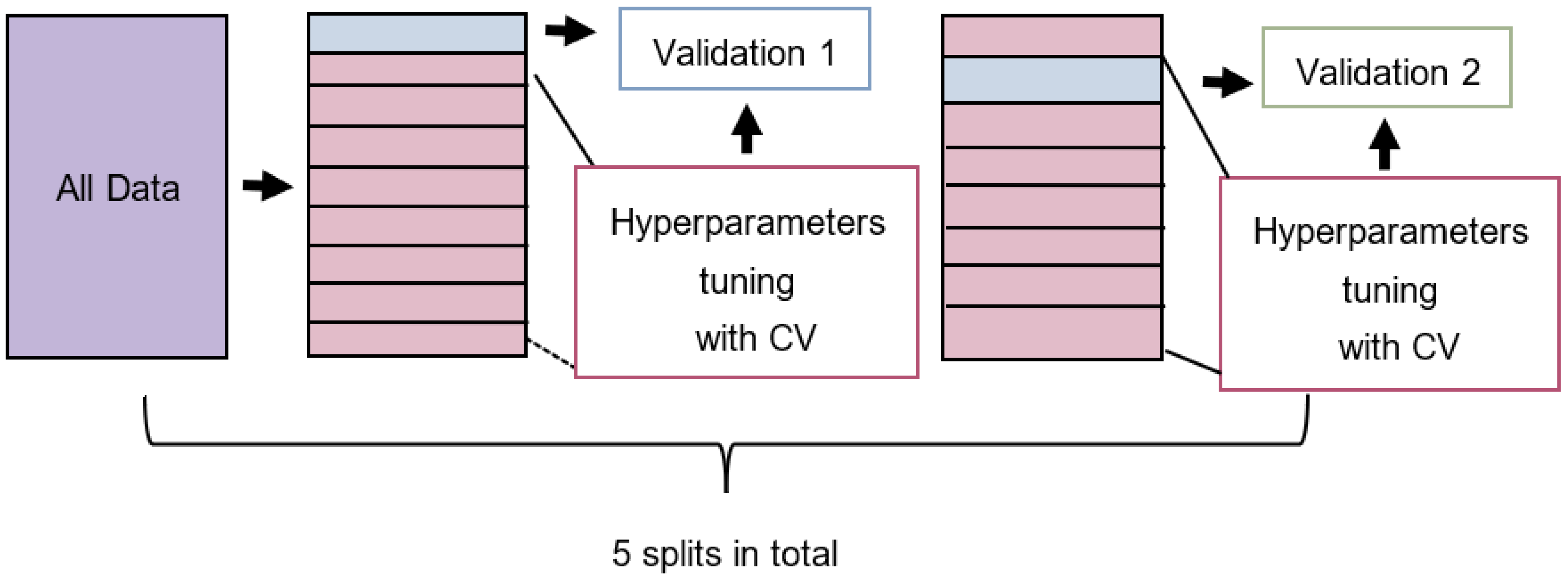
2.5. Model Training
2.6. Performance Metrics
2.7. Applicability Domain
2.8. Hidden Markov Model (HMM) Profiles
2.9. Homology Modelling (HM) and Active Site Analysis
2.10. Enzyme Source, Production, and Purification
2.11. Activity Tests
3. Results and Discussion
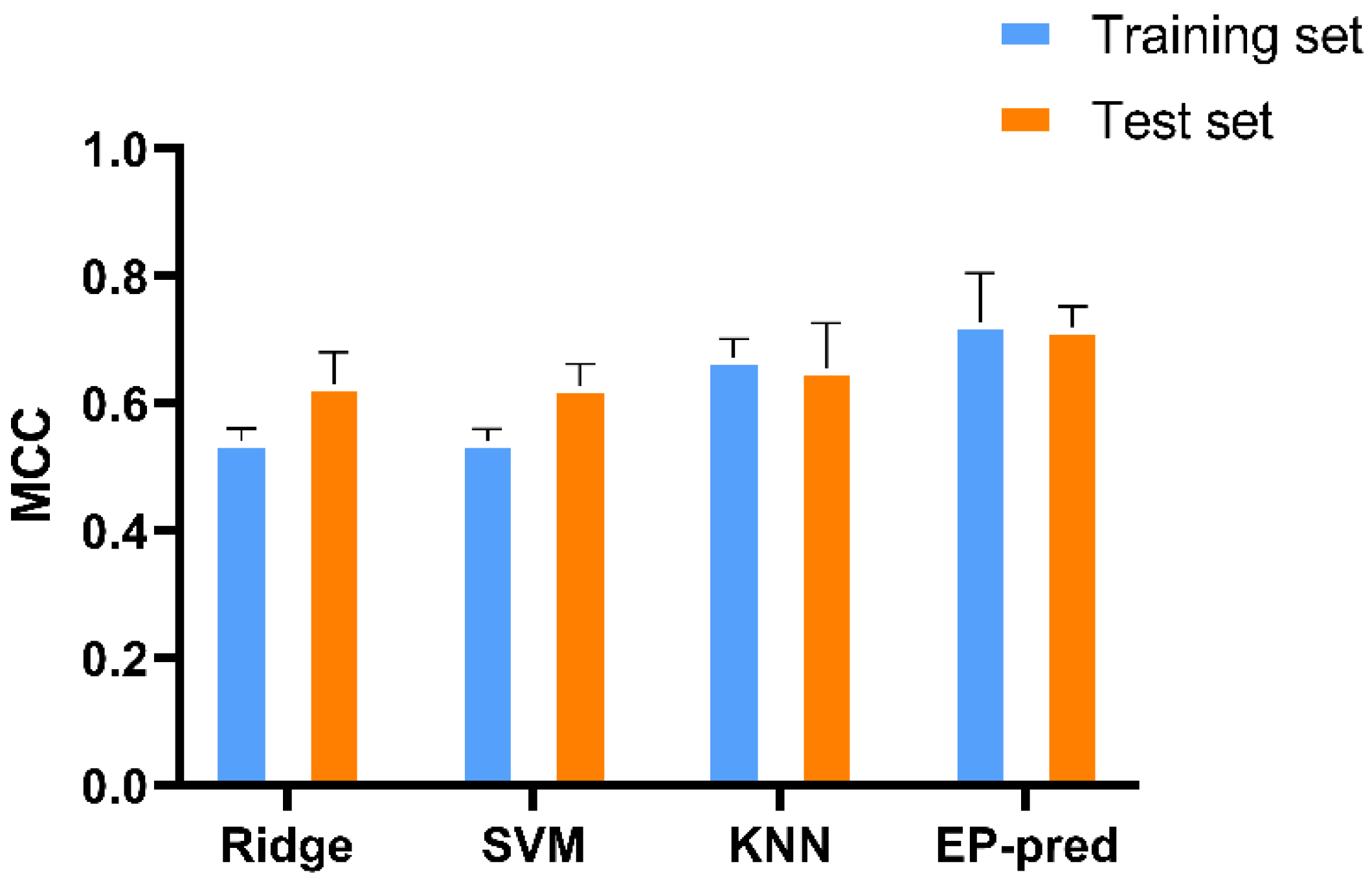
3.1. Model Buildup
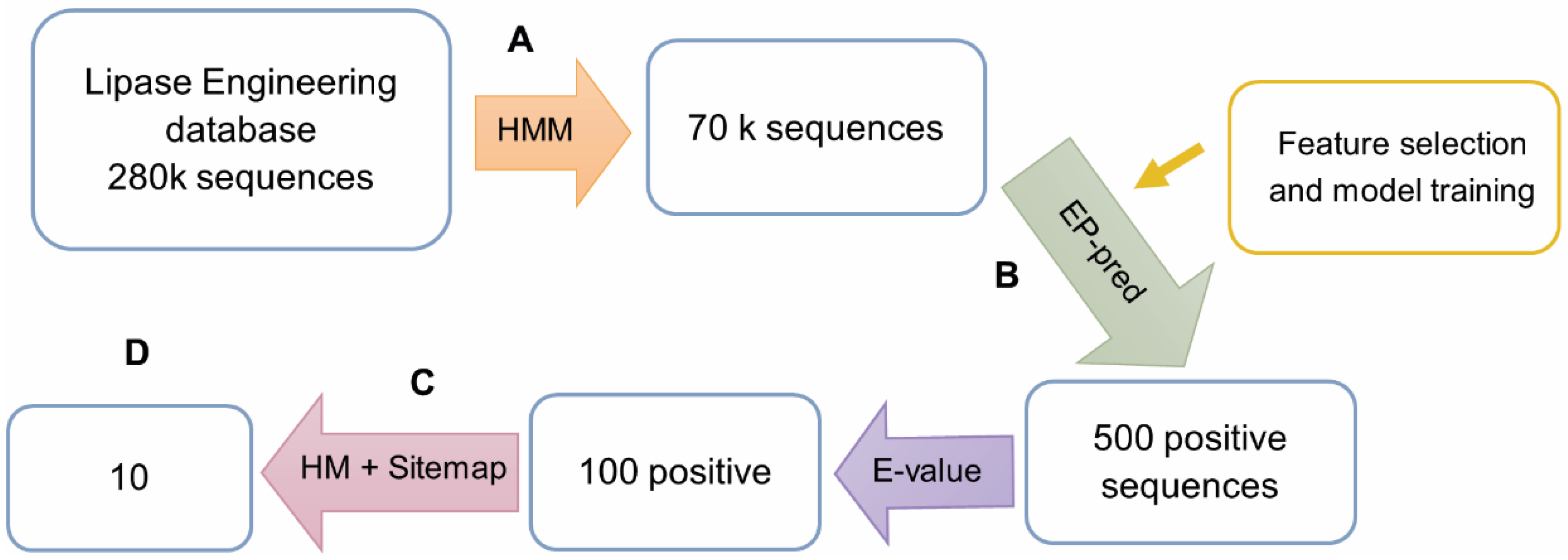
3.2. The Workflow for In Silico Bioprospecting
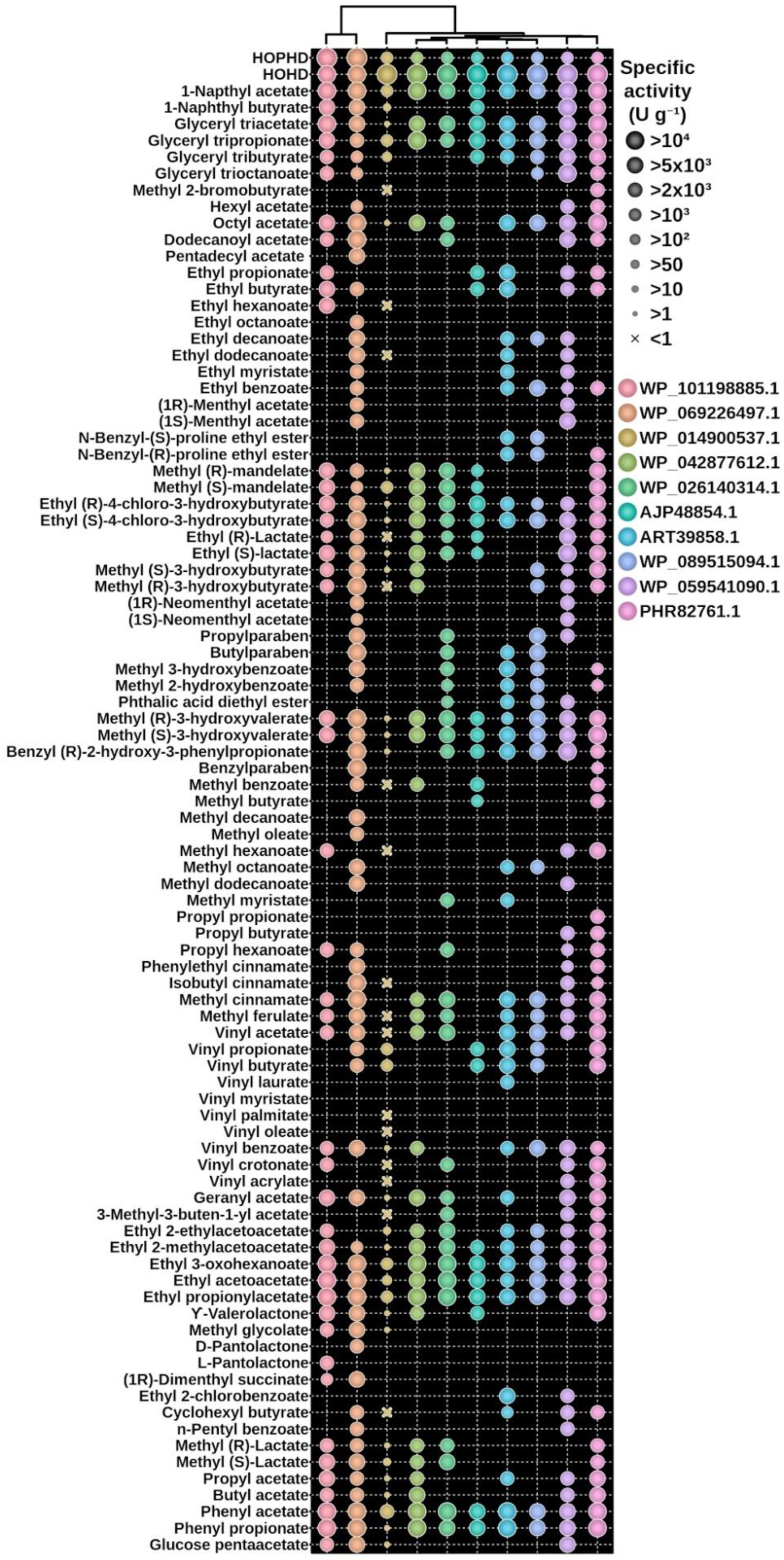
3.3. The Experimental Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alcántara, A.; Hernaiz, M.-J.; Sinisterra, J.-V. 3.28 Biocatalyzed Production of Fine Chemicals. Compr. Biotechnol. 2017, 3, 334–373. [Google Scholar] [CrossRef]
- Panda, T.; Gowrishankar, B.S. Production and applications of esterases. Appl. Microbiol. Biotechnol. 2005, 67, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kamble, A.; Srinivasan, S.; Singh, H. In-Silico Bioprospecting: Finding Better Enzymes. Mol. Biotechnol. 2019, 61, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martínez, M.; Coscolín, C.; Santiago, G.; Chow, J.; Stogios, P.J.; Bargiela, R.; Gertler, C.; Navarro-Fernández, J.; Bollinger, A.; Thies, S.; et al. Determinants and Prediction of Esterase Substrate Promiscuity Patterns. ACS Chem. Biol. 2018, 13, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Schmid, A.; Dordick, J.S.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt, B. Industrial biocatalysis today and tomorrow. Nature 2001, 409, 258–268. [Google Scholar] [CrossRef]
- Braakman, R.; Smith, E. Metabolic evolution of a deep-branching hyperthermophilic chemoautotrophic bacterium. PLoS ONE 2014, 9, e87950. [Google Scholar] [CrossRef] [Green Version]
- Giunta, C.I.; Cea-Rama, I.; Alonso, S.; Briand, M.L.; Bargiela, R.; Coscolín, C.; Corvini, P.F.-X.; Ferrer, M.; Sanz-Aparicio, J.; Shahgaldian, P. Tuning the Properties of Natural Promiscuous Enzymes by Engineering Their Nano-environment. ACS Nano 2020, 14, 17652–17664. [Google Scholar] [CrossRef]
- Roda, S.; Fernandez-Lopez, L.; Cañadas, R.; Santiago, G.; Ferrer, M.; Guallar, V. Computationally Driven Rational Design of Substrate Promiscuity on Serine Ester Hydrolases. ACS Catal. 2021, 11, 3590–3601. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. The Lipase Engineering Database: A navigation and analysis tool for protein families. Nucleic Acids Res. 2003, 31, 319–321. [Google Scholar] [CrossRef]
- Zhang, Y.; Vass, M.; Shi, D.; Abualrous, E.; Chambers, J.; Chopra, N.; Higgs, C.; Kasavajhala, K.; Li, H.; Nandekar, P.; et al. Benchmarking Refined and Unrefined AlphaFold2 Structures for Hit Discovery. ChemRxiv 2022. This content is a preprint and has not been peer-reviewed. [Google Scholar] [CrossRef]
- Li, F.; Yuan, L.; Lu, H.; Li, G.; Chen, Y.; Engqvist, M.K.M.; Kerkhoven, E.J.; Nielsen, J. Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction. Nat. Catal. 2022, 5, 662–672. [Google Scholar] [CrossRef]
- Freund, G.S.; Yao, A.; Mak, W.S.; Tagkopoulos, I.; Facciotti, M.T.; Tantillo, D.J.; Siegel, J.B. Elucidating Substrate Promiscuity within the FabI Enzyme Family. ACS Chem. Biol. 2017, 12, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, P.; Faulon, J.-L. Molecular signatures-based prediction of enzyme promiscuity. Bioinformatics 2010, 26, 2012–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertusi, D.A.; Moura, M.E.; Jeffryes, J.G.; Prabhu, S.; Walters Biggs, B.; Tyo, K.E.J. Predicting novel substrates for enzymes with minimal experimental effort with active learning. Metab. Eng. 2017, 44, 171–181. [Google Scholar] [CrossRef]
- Goldman, S.; Das, R.; Yang, K.K.; Coley, C.W. Machine learning modeling of family wide enzyme-substrate specificity screens. PLoS Comput. Biol. 2022, 18, e1009853. [Google Scholar] [CrossRef]
- Uniref. Available online: https://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref50/README (accessed on 3 August 2022).
- Wang, J.; Yang, B.; Revote, J.; Leier, A.; Marquez-Lago, T.T.; Webb, G.; Song, J.; Chou, K.-C.; Lithgow, T. POSSUM: A bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017, 33, 2756–2758. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.-C.; et al. iFeature: A Python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef] [Green Version]
- Vabalas, A.; Gowen, E.; Poliakoff, E.; Casson, A.J. Machine learning algorithm validation with a limited sample size. PLoS ONE 2019, 14, e0224365. [Google Scholar] [CrossRef]
- Chou, K.-C.; Shen, H.-B. MemType-2L: A Web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem. Biophys. Res. Commun. 2007, 360, 339–345. [Google Scholar] [CrossRef]
- Zahiri, J.; Yaghoubi, O.; Mohammad-Noori, M.; Ebrahimpour, R.; Masoudi-Nejad, A. PPIevo: Protein–protein interaction prediction from PSSM based evolutionary information. Genomics 2013, 102, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Elisseeff, A. An Introduction to Feature Extraction. In Feature Extraction; Guyon, I., Nikravesh, M., Gunn, S., Zadeh, L.A., Eds.; Studies in Fuzziness and Soft Computing; Springer: Berlin/Heidelberg, Germany, 2006; Volume 207, pp. 1–25. [Google Scholar] [CrossRef] [Green Version]
- Pilnenskiy, N.; Smetannikov, I. Feature Selection Algorithms as One of the Python Data Analytical Tools. Future Internet 2020, 12, 54. [Google Scholar] [CrossRef] [Green Version]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cheng, K.; Wang, S.; Morstatter, F.; Trevino, R.P.; Tang, J.; Liu, H. Feature Selection: A Data Perspective. ACM Comput. Surv. 2018, 50, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef] [Green Version]
- Kubat, M. Performance Evaluation. In An Introduction to Machine Learning; Springer International Publishing: Cham, Switzerland, 2017; pp. 211–229. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [Green Version]
- Sahigara, F.; Ballabio, D.; Todeschini, R.; Consonni, V. Defining a novel k-nearest neighbours approach to assess the applicability domain of a QSAR model for reliable predictions. J. Cheminformatics 2013, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-García, P.; Danso, D.; Zhang, H.; Chow, J.; Streit, W.R. Exploring the global metagenome for plastic-degrading enzymes. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 648, pp. 137–157. [Google Scholar] [CrossRef]
- ModWeb. Available online: https://modbase.compbio.ucsf.edu/modweb/ (accessed on 22 July 2022).
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Roda, S.; Fernandez-Lopez, L.; Benedens, M.; Bollinger, A.; Thies, S.; Schumacher, J.; Coscolín, C.; Kazemi, M.; Santiago, G.; Gertzen, C.G.W.; et al. A Plurizyme with Transaminase and Hydrolase Activity Catalyzes Cascade Reactions. Angew. Chem. 2022, 134, e202207344. [Google Scholar] [CrossRef]
- Vidal, P.; Martínez-Martínez, M.; Fernandez-Lopez, L.; Roda, S.; Méndez-García, C.; Golyshina, O.V.; Guallar, V.; Peláez, A.I.; Ferrer, M. Metagenomic Mining for Esterases in the Microbial Community of Los Rueldos Acid Mine Drainage Formation. Front. Microbiol. 2022, 13, 868839. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Tornés, J.; Stogios, P.J.; Xu, X.; Gertler, C.; Di Leo, R.; Bargiela, R.; Lafraya, Á.; Guazzaroni, M.-E.; López-Cortés, N.; et al. Single residues dictate the co-evolution of dual esterases: MCP hydrolases from the α/β hydrolase family. Biochem. J. 2013, 454, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutschel, C.; Coscolín, C.; David, B.; Mulnaes, D.; Ferrer, M.; Jaeger, K.-E.; Gohlke, H. Promiscuous Esterases Counterintuitively Are Less Flexible than Specific Ones. J. Chem. Inf. Model. 2021, 61, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Fushinobu, S.; Saku, T.; Hidaka, M.; Jun, S.-Y.; Nojiri, H.; Yamane, H.; Shoun, H.; Omori, T. Takayoshi Wakagi Crystal structures of a meta-cleavage product hydrolase from Pseudomo. Protein Sci. 2009, 11, 2184–2195. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
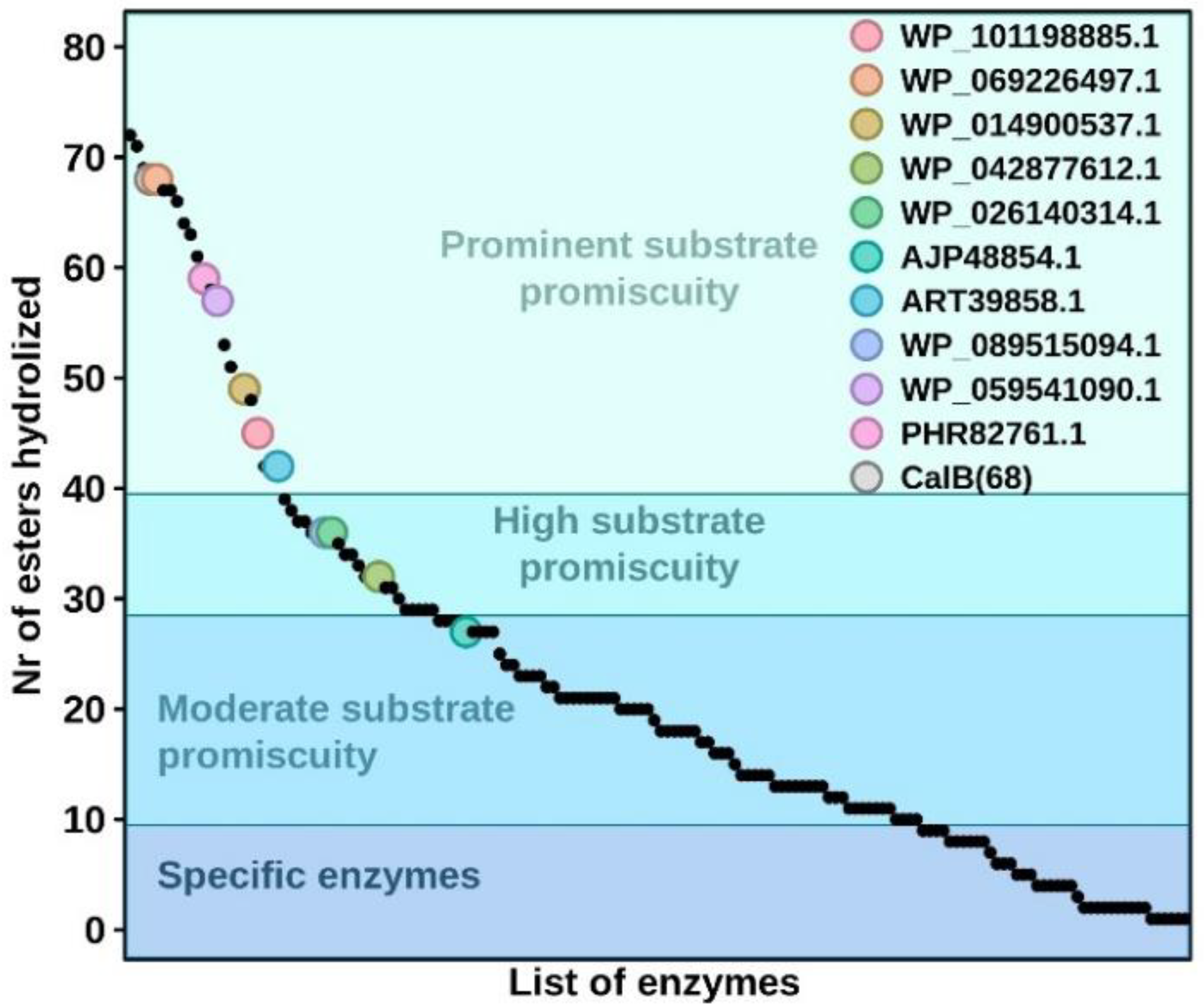{kind=link}
| Specific Activity (Units mg−1) | |||
|---|---|---|---|
| Enzyme | p-NP Acetate | p-NP Propionate | p-NP Butyrate |
| AJP48854.1 | 2.96 ± 0.36 | 3.99 ± 0.25 | 1.68 ± 0.10 |
| ART39858.1 | 2.53 ± 0.39 | 3.63 ± 0.26 | 1.48 ± 0.13 |
| PHR82761.1 | 4.39 ± 0.44 | 2.75 ± 0.19 | 2.41 ± 0.25 |
| WP_014900537.1 | 0.64 ± 0.02 | 2.19 ± 0.17 | 1.31 ± 0.18 |
| WP_026140314.1 | 1.47 ± 0.05 | 3.33 ± 0.14 | 1.22 ± 0.09 |
| WP_042877612.1 | 0.51 ± 0.01 | 2.57 ± 0.24 | 0.99 ± 0.06 |
| WP_059541090.1 | 0.97 ± 0.02 | 2.96 ± 0.23 | 1.06 ± 0.08 |
| WP_069226497.1 | 0.56 ± 0.01 | 2.26 ± 0.13 | 1.01 ± 0.04 |
| WP_089515094.1 | 3.75 ± 0.17 | 4.46 ± 0.19 | 2.20 ± 0.15 |
| WP_101198885.1 | 4.32 ± 0.14 | 5.85 ± 0.08 | 3.15 ± 0.25 |
| Enzyme | kcat (min−1) | Km (µM) |
|---|---|---|
| AJP48854.1 | n.d. | n.d. |
| ART39858.1 | 33.1 ± 0.1 | 33.3 ± 3.1 |
| PHR82761.1 | 2569.3 ± 6.3 | 33.7 ± 3.6 |
| WP_014900537.1 | 46.8 ± 0.0 | 16.9 ± 1.6 |
| WP_026140314.1 | 5.9 ± 0.0 | 14.5 ± 1.1 |
| WP_042877612.1 | 5043.0 ± 1268 | 65.8 ± 26.1 |
| WP_059541090.1 | 3246.8 ± 10.6 | 31 ± 3.3 |
| WP_069226497.1 | 3775.1 ± 37.2 | 46.5 ± 11.8 |
| WP_089515094.1 | 2060.8 ± 5.7 | 23.5 ± 3.5 |
| WP_101198885.1 | 3452 ± 15.9 | 48.7 ± 7.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, R.; Fernandez-Lopez, L.; Robles-Martín, A.; Ferrer, M.; Guallar, V. EP-Pred: A Machine Learning Tool for Bioprospecting Promiscuous Ester Hydrolases. Biomolecules 2022, 12, 1529. https://doi.org/10.3390/biom12101529
Xiang R, Fernandez-Lopez L, Robles-Martín A, Ferrer M, Guallar V. EP-Pred: A Machine Learning Tool for Bioprospecting Promiscuous Ester Hydrolases. Biomolecules. 2022; 12(10):1529. https://doi.org/10.3390/biom12101529
Chicago/Turabian StyleXiang, Ruite, Laura Fernandez-Lopez, Ana Robles-Martín, Manuel Ferrer, and Victor Guallar. 2022. "EP-Pred: A Machine Learning Tool for Bioprospecting Promiscuous Ester Hydrolases" Biomolecules 12, no. 10: 1529. https://doi.org/10.3390/biom12101529