
The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program
Abstract
:1. Introduction
2. Methods
2.1. Membrane Proteins Study
2.2. Structural and Functional Study of the Luminal Domain of h-SV2C
2.3. Soluble Proteins Study
2.4. Docking of BoNT/B with its Membrane Receptors
2.5. TM-Score and Root-Mean-Square Deviation
3. Results
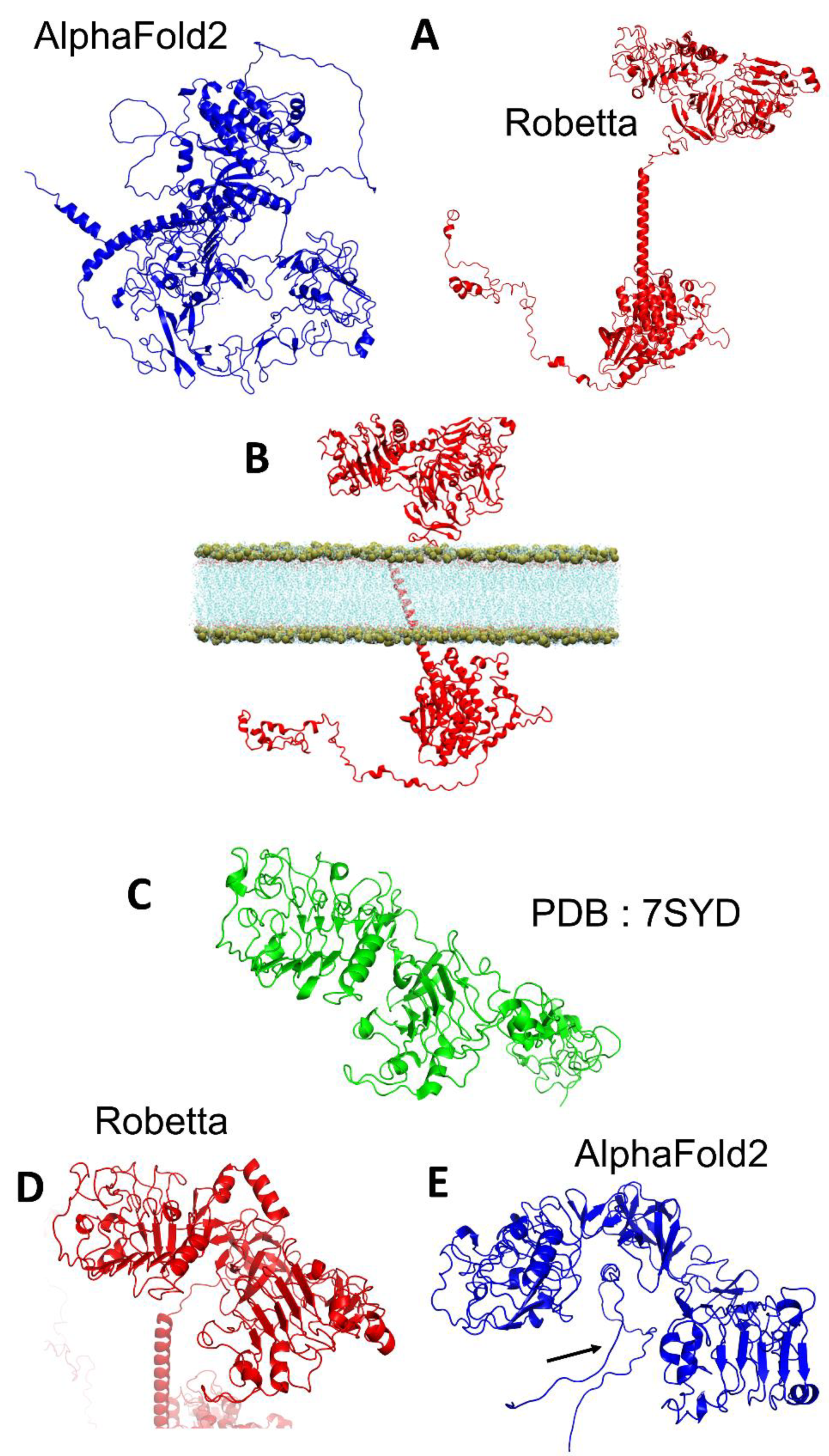
3.1. EGFR
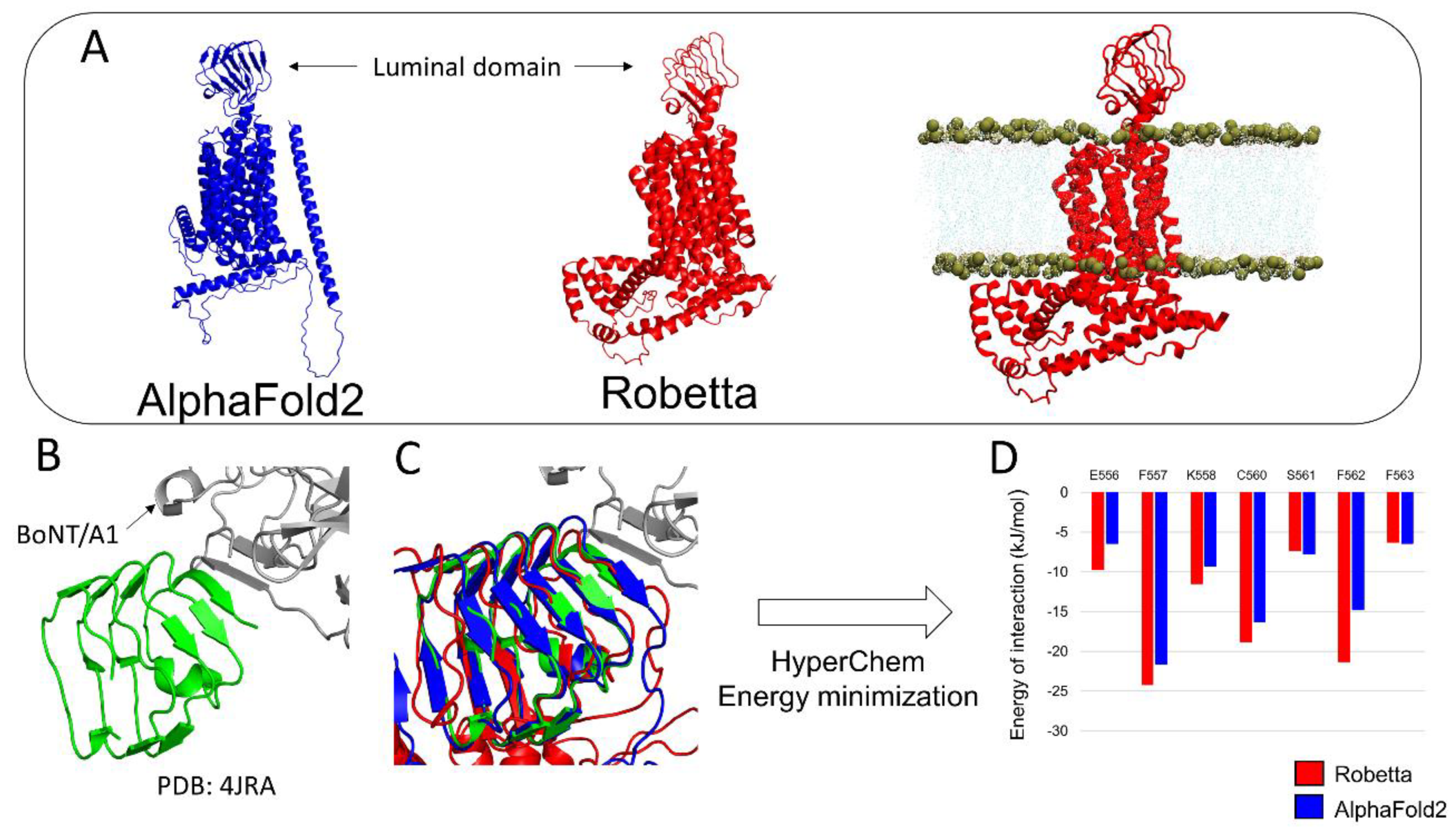
3.2. h-SV2C
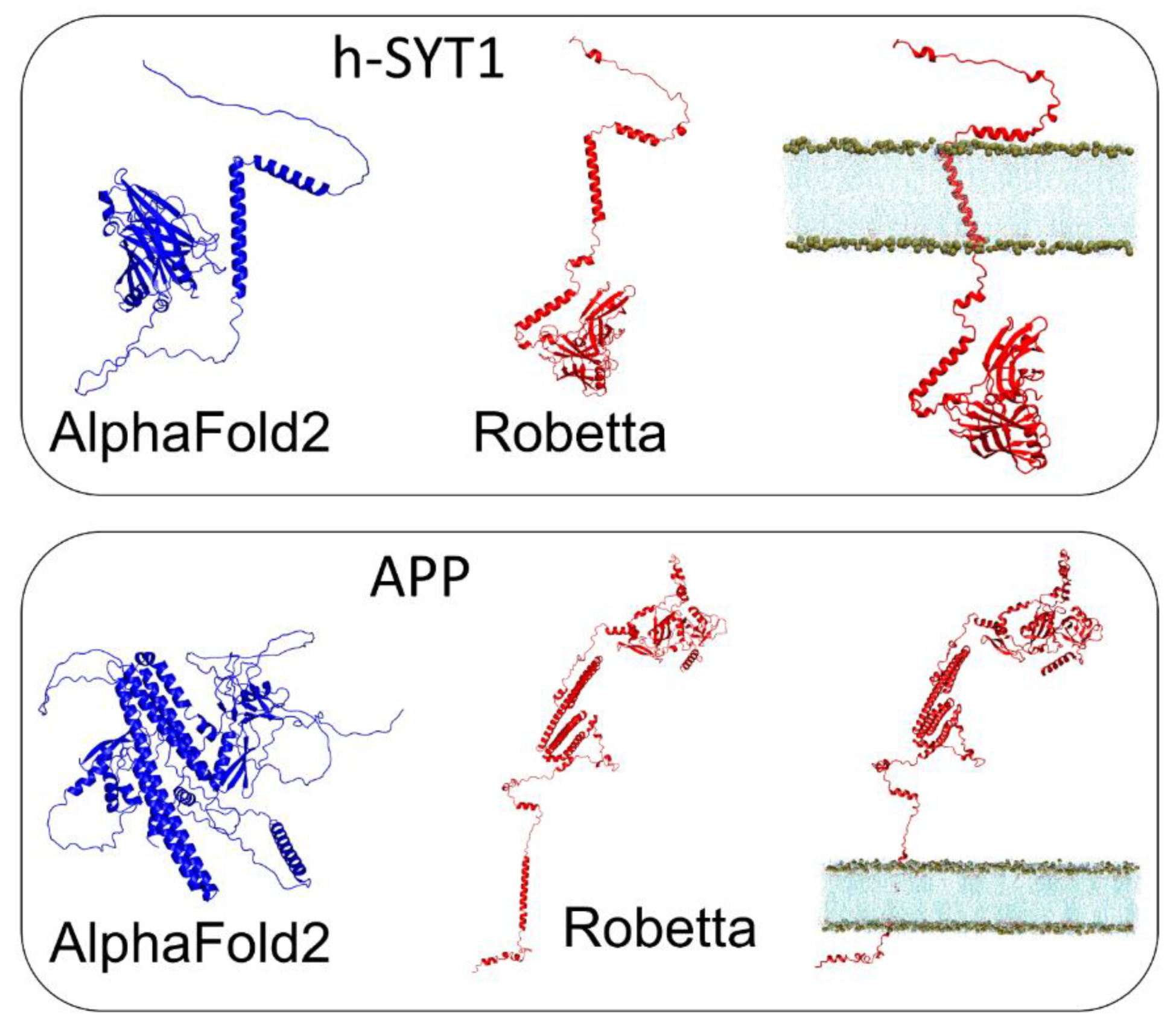
3.3. h-SYT1 and APP
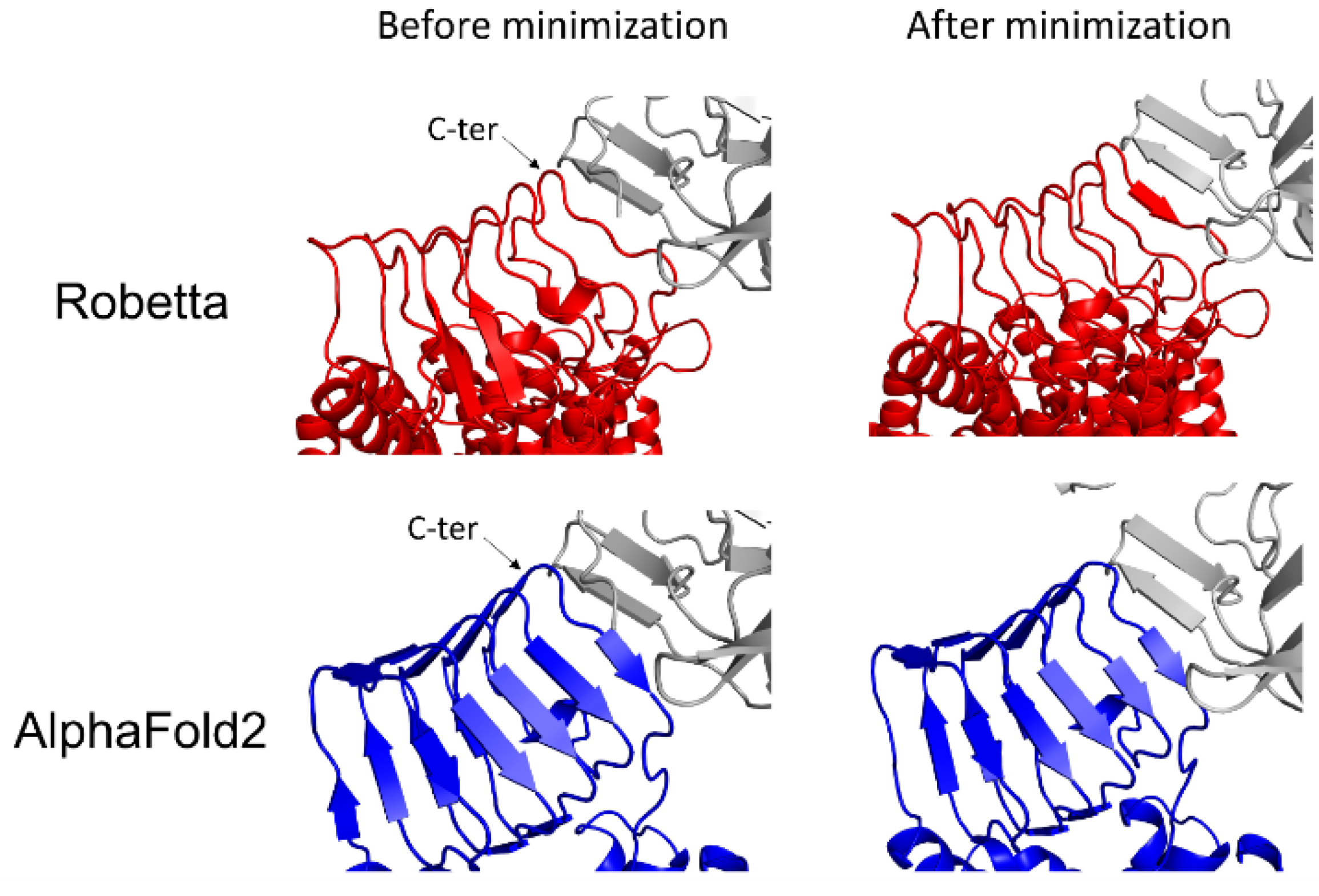
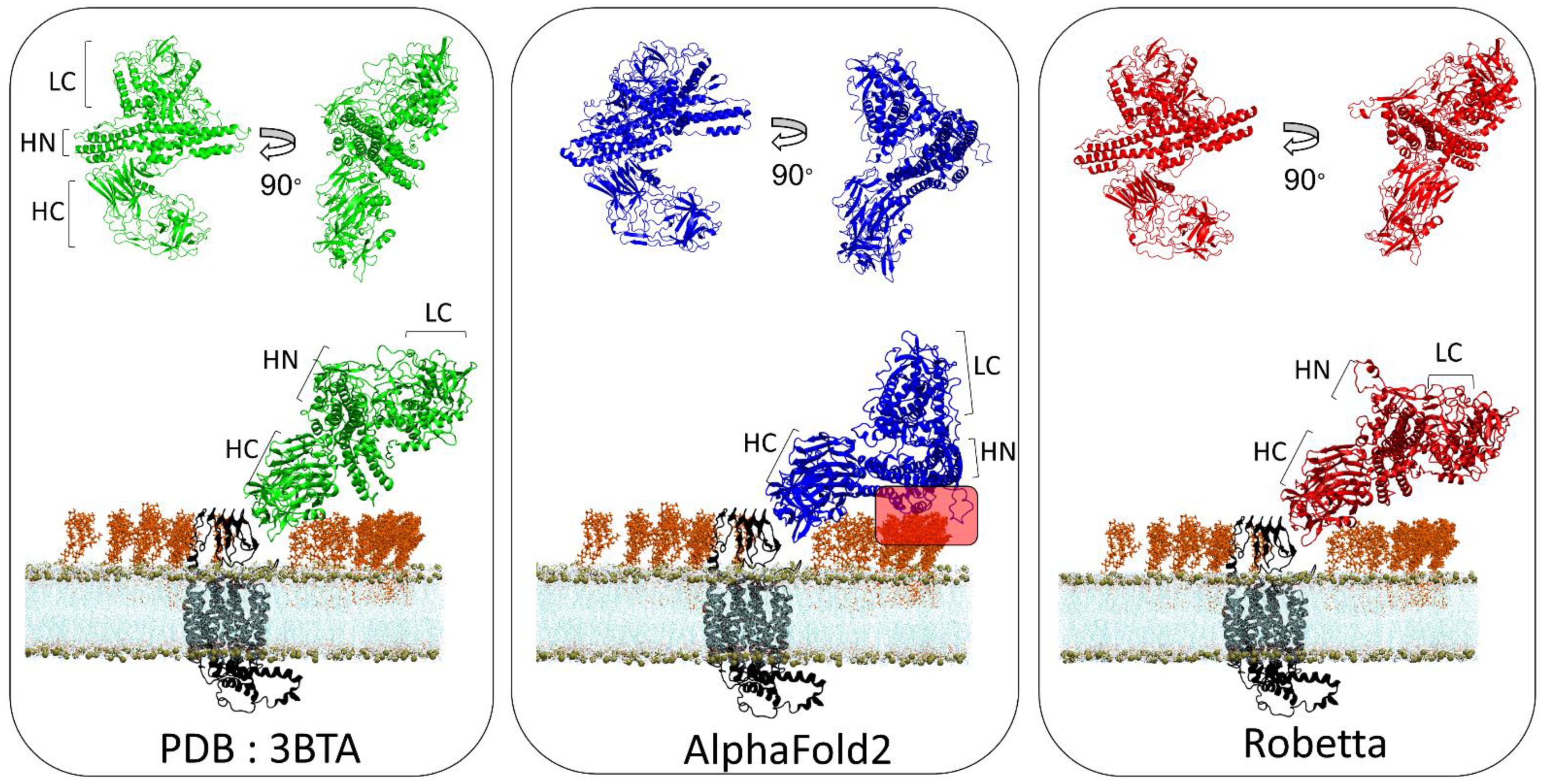
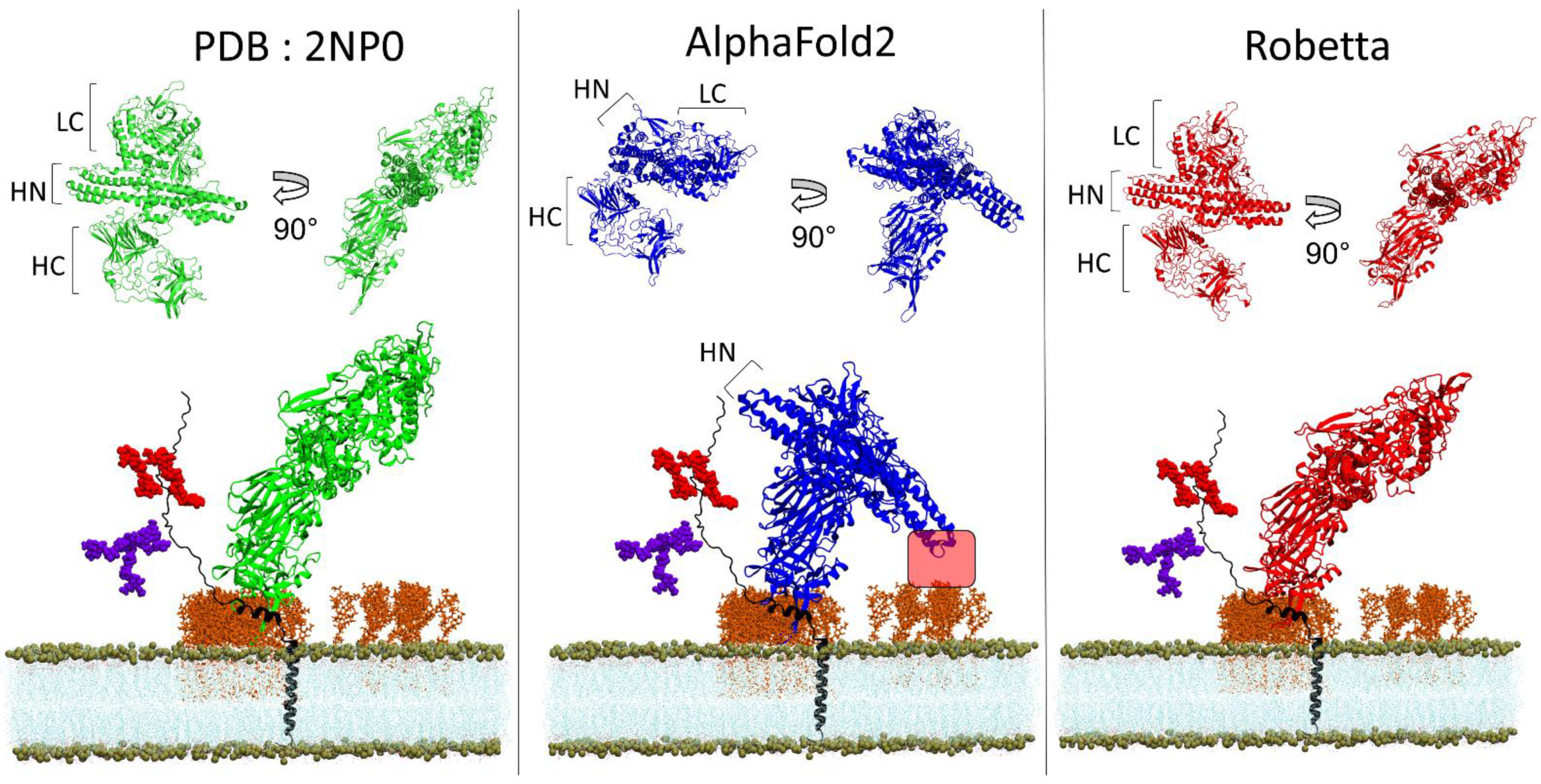
3.4. BoNT/A1 and BoNT/B1
3.5. TM-Score and Root-Mean-Square Deviation of AlphaFold2 and Robetta Models
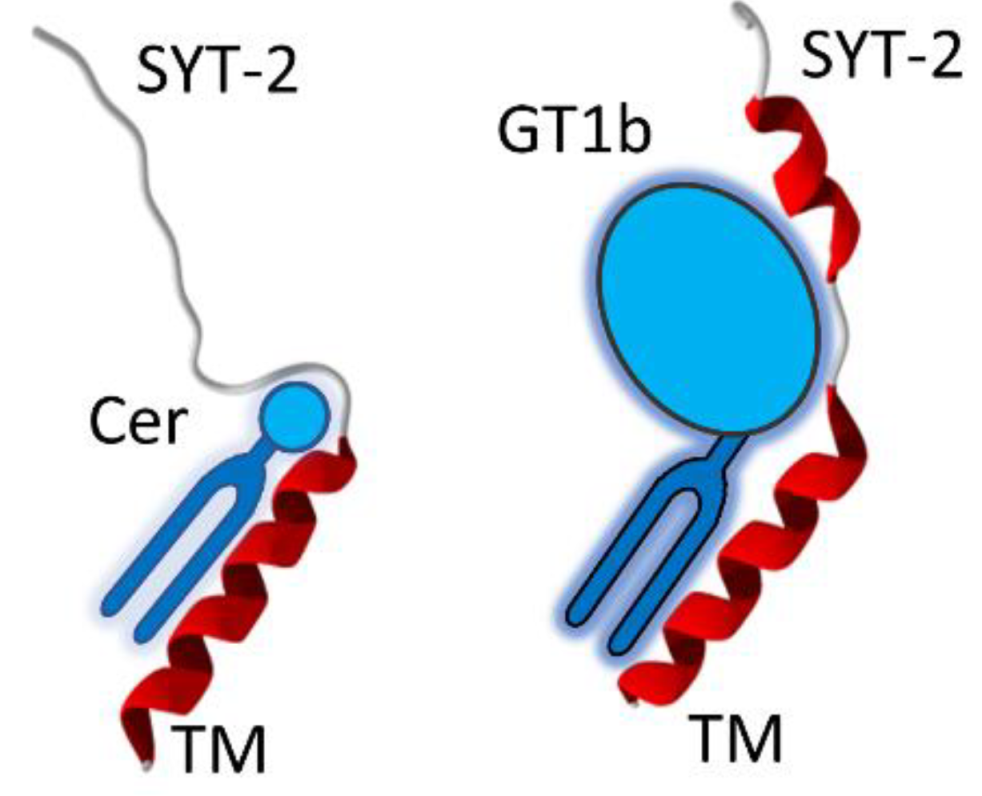
3.6. A Chaperone Activity in Lipid Rafts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pearce, R.; Zhang, Y. Toward the solution of the protein structure prediction problem. J. Biol. Chem. 2021, 297, 100870. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Azzaz, F.; Fantini, J. The epigenetic dimension of protein structure. Biomol. Concepts 2022, 13, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Anfinsen, C.B. The formation and stabilization of protein structure. Biochem. J. 1972, 128, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Norn, C.; Wicky, B.I.M.; Juergens, D.; Liu, S.; Kim, D.; Tischer, D.; Koepnick, B.; Anishchenko, I.; Baker, D.; Ovchinnikov, S. Protein sequence design by conformational landscape optimization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017228118. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Dong, M.; Liu, H.; Tepp, W.H.; Johnson, E.A.; Janz, R.; Chapman, E.R. Glycosylated SV2A and SV2B Mediate the Entry of Botulinum Neurotoxin E into Neurons. Mol. Biol. Cell 2008, 19, 5226–5237. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.-J.; Kern, N.R.; Kim, S.H.; Lee, J.; Cheng, X.; Valvano, M.A.; Holst, O.; et al. CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J. Chem. Theory Comput. 2018, 15, 775–786. [Google Scholar] [CrossRef]
- Flores, A.; Ramirez-Franco, J.; Desplantes, R.; Debreux, K.; Ferracci, G.; Wernert, F.; Blanchard, M.-P.; Maulet, Y.; Youssouf, F.; Sangiardi, M.; et al. Gangliosides interact with synaptotagmin to form the high-affinity receptor complex for botulinum neurotoxin B. Proc. Natl. Acad. Sci. USA 2019, 116, 18098–18108. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.; Favre-Guilmard, C.; Liu, S.M.; Maignel, J.; Masuyer, G.; Beard, M.; Boone, C.; Carré, D.; Kalinichev, M.; Lezmi, S.; et al. Engineered botulinum neurotoxin B with improved binding to human receptors has enhanced efficacy in preclinical models. Sci. Adv. 2019, 5, eaau7196. [Google Scholar] [CrossRef] [Green Version]
- Berntsson, R.P.-A.; Peng, L.; Svensson, L.M.; Dong, M.; Stenmark, P. Crystal Structures of Botulinum Neurotoxin DC in Complex with Its Protein Receptors Synaptotagmin I and II. Structure 2013, 21, 1602–1611. [Google Scholar] [CrossRef] [Green Version]
- Stern, D.; Weisemann, J.; Le Blanc, A.; Von Berg, L.; Mahrhold, S.; Piesker, J.; Laue, M.; Luppa, P.B.; Dorner, M.B.; Dorner, B.G.; et al. A lipid-binding loop of botulinum neurotoxin serotypes B, DC and G is an essential feature to confer their exquisite potency. PLOS Pathog. 2018, 14, e1007048. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2017, 12, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Dunn, A.R.; Stout, K.A.; Ozawa, M.; Lohr, K.M.; Hoffman, C.A.; Bernstein, A.I.; Li, Y.; Wang, M.; Sgobio, C.; Sastry, N.; et al. Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc. Natl. Acad. Sci. USA 2017, 114, E2253–E2262. [Google Scholar] [CrossRef] [Green Version]
- Benoit, R.; Frey, D.; Hilbert, M.; Kevenaar, J.T.; Wieser, M.M.; Stirnimann, C.; McMillan, D.; Ceska, T.; Lebon, F.; Jaussi, R.; et al. Structural basis for recognition of synaptic vesicle protein 2C by botulinum neurotoxin A. Nature 2014, 505, 108–111. [Google Scholar] [CrossRef]
- Arnon, S.S.; Schechter, R.; Inglesby, T.V.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Fine, A.D.; Hauer, J.; Layton, M.; et al. Botulinum toxin as a biological weapon: Medical and public health management. Jama 2001, 285, 1059–1070. [Google Scholar] [CrossRef]
- Poulain, B.; Popoff, M.R. Why are botulinum neurotoxin-producing bacteria so diverse and botulinum neurotoxins so toxic? Toxins 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Yahi, N. Brain Lipids in Synaptic Function and Neurological Disease: Clues to Innovative Therapeutic Strategies for Brain Disorders; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Aslam, M.; Perkins, S.J. Folded-back solution structure of monomeric factor H of human complement by synchrotron X-ray and neutron scattering, analytical ultracentrifugation and constrained molecular modelling. J. Mol. Biol. 2001, 309, 1117–1138. [Google Scholar] [CrossRef]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Garmy, N.; Mahfoud, R.; Yahi, N. Lipid rafts: Structure, function and role in HIV. Alzheimer’s and prion diseases. Expert Rev. Mol. Med. 2002, 4, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.A.; Fu, Z.; Kim, J.-J.P.; Baldwin, M.R. Unique Ganglioside Recognition Strategies for Clostridial Neurotoxins. J. Biol. Chem. 2011, 286, 34015–34022. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Zhang, S.; Mahrhold, S.; Lam, K.H.; Stern, D.; Bagramyan, K.; Perry, K.; Kalkum, M.; Rummel, S.M.A.; Dong, S.Z.M.; et al. N-linked glycosylation of SV2 is required for binding and uptake of botulinum neurotoxin A. Nat. Struct. Mol. Biol. 2016, 23, 656–662. [Google Scholar] [CrossRef]
- Strotmeier, J.; Willjes, G.; Binz, T.; Rummel, A. Human synaptotagmin-II is not a high affinity receptor for botulinum neurotoxin B and G: Increased therapeutic dosage and immunogenicity. FEBS Lett. 2012, 586, 310–313. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J. How sphingolipids bind and shape proteins: Molecular basis of lipid-protein interactions in lipid shells, rafts and related biomembrane domains. Cell. Mol. Life Sci. CMLS 2003, 60, 1027–1032. [Google Scholar] [CrossRef]
- Gil, C.; Soler-Jover, A.; Blasi, J.; Aguilera, J. Synaptic proteins and SNARE complexes are localized in lipid rafts from rat brain synaptosomes. Biochem. Biophys. Res. Commun. 2005, 329, 117–124. [Google Scholar] [CrossRef]
- Lv, J.-H.; He, L.; Sui, S.-F. Lipid rafts association of synaptotagmin I on synaptic vesicles. Biochem. Biokhimiia 2008, 73, 283–288. [Google Scholar] [CrossRef]
- Jia, J.Y.; Lamer, S.; Schümann, M.; Schmidt, M.R.; Krause, E.; Haucke, V. Quantitative proteomics analysis of detergent-resistant membranes from chemical synapses: Evidence for cholesterol as spatial organizer of synaptic vesicle cycling. Mol. Cell. Proteom. MCP 2006, 5, 2060–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J. Biol. Chem. 2016, 291, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. Intrinsically disordered proteins play diverse roles in cell signaling. Cell Commun. Signal. 2022, 20, 20. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their environment: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein, J. 2009, 28, 305–325. [Google Scholar] [CrossRef]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Kallberg, Y.; Gustafsson, M.; Persson, B.; Thyberg, J.; Johansson, J. Prediction of Amyloid Fibril-forming Proteins. J. Biol. Chem. 2001, 276, 12945–12950. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, F.; Santos, J.; Ventura, S. AlphaFold and the amyloid landscape. J. Mol. Biol. 2021, 433, 167059. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 2010, 12, e27. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, M.F.; Lolicato, F.; Tempra, C.; Scollo, F.; Sahoo, B.R.; Watson, M.D.; García-Viñuales, S.; Milardi, D.; Raudino, A.; Lee, J.C.; et al. Lipid-Chaperone Hypothesis: A Common Molecular Mechanism of Membrane Disruption by Intrinsically Disordered Proteins. ACS Chem. Neurosci. 2020, 11, 4336–4350. [Google Scholar] [CrossRef]
- Fantini, J. Interaction of Proteins with Lipid Rafts Through Glycolipid-Binding Domains:Biochemical Background and Potential Therapeutic Applications. Curr. Med. Chem. 2007, 14, 2911–2917. [Google Scholar] [CrossRef]
- El-Battari, A.; Rodriguez, L.; Chahinian, H.; Delézay, O.; Fantini, J.; Yahi, N.; Di Scala, C. Gene Therapy Strategy for Alzheimer’s and Parkinson’s Diseases Aimed at Preventing the Formation of Neurotoxic Oligomers in SH-SY5Y Cells. Int. J. Mol. Sci. 2021, 22, 11550. [Google Scholar] [CrossRef]
- Popelka, H.; Uversky, V.N. Theater in the Self-Cleaning Cell: Intrinsically Disordered Proteins or Protein Regions Acting with Membranes in Autophagy. Membranes 2022, 12, 457. [Google Scholar] [CrossRef]
- Opekarová, M.; Tanner, W. Specific lipid requirements of membrane proteins—A putative bottleneck in heterologous expression. Biochim. Biophys. Acta (BBA)-Biomembr. 2003, 1610, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zheng, J. Cholesterol Promotes the Interaction of Alzheimer β-Amyloid Monomer with Lipid Bilayer. J. Mol. Biol. 2012, 421, 561–571. [Google Scholar] [CrossRef]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [Green Version]
- Di Scala, C.; Yahi, N.; Flores, A.; Boutemeur, S.; Kourdougli, N.; Chahinian, H.; Fantini, J. Broad neutralization of calcium-permeable amyloid pore channels with a chimeric Alzheimer/Parkinson peptide targeting brain gangliosides. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 213–222. [Google Scholar] [CrossRef]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry 2004, 53, 4489–4502. [Google Scholar] [CrossRef]
- Burley, S.K.; Arap, W.; Pasqualini, R. Predicting Proteome-Scale Protein Structure with Artificial Intelligence. N. Engl. J. Med. 2021, 385, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Su, B.-H.; Tseng, Y.J. Comparative studies of AlphaFold, RoseTTAFold and Modeller: A case study involving the use of G-protein-coupled receptors. Briefings Bioinform. 2022, 23, bbac308. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Eliezer, D. Membrane interactions of intrinsically disordered proteins: The example of alpha-synuclein. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Elenis, D.; Papasilekas, T.; Stranjalis, G.; Gerozissis, K.; Ioannou, P.C.; Vekrellis, K. Assessment of α-synuclein secretion in mouse and human brain parenchyma. PLoS ONE 2011, 6, e22225. [Google Scholar] [CrossRef]
- Marques, O.; Outeiro, T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012, 3, e350. [Google Scholar] [CrossRef] [Green Version]
- Yahi, N.; Di Scala, C.; Chahinian, H.; Fantini, J. Innovative treatment targeting gangliosides aimed at blocking the formation of neurotoxic α-synuclein oligomers in Parkinson’s disease. Glycoconj. J. 2021, 39, 1–11. [Google Scholar] [CrossRef]
- Fantini, J.; Chahinian, H.; Yahi, N. Progress toward Alzheimer’s disease treatment: Leveraging the Achilles’ heel of Aβ oligomers? Protein Sci. A Publ. Protein 2020, 29, 1748–1759. [Google Scholar] [CrossRef]
- Yahi, N.; Fantini, J. Deciphering the Glycolipid Code of Alzheimer’s and Parkinson’s Amyloid Proteins Allowed the Creation of a Universal Ganglioside-Binding Peptide. PLoS ONE 2014, 9, e104751. [Google Scholar] [CrossRef] [Green Version]
- Di Scala, C.; Fantini, J. Hybrid In Silico/In Vitro Approaches for the Identification of Functional Cholesterol-Binding Domains in Membrane Proteins. Methods Mol. Biol. 2017, 1583, 7–19. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular Basis for the Glycosphingolipid-Binding Specificity of α-Synuclein: Key Role of Tyrosine 39 in Membrane Insertion. J. Mol. Biol. 2011, 408, 654–669. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. The Driving Force of Alpha-Synuclein Insertion and Amyloid Channel Formation in the Plasma Membrane of Neural Cells: Key Role of Ganglioside- and Cholesterol-Binding Domains. Adv. Exp. Med. Biol. 2013, 991, 15–26. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry 1974, 13, 211–222. [Google Scholar] [CrossRef]
- Matsubara, T.; Iida, M.; Tsumuraya, T.; Fujii, I.; Sato, T. Selection of a carbohydrate-binding domain with a helix-loop-helix structure. Biochemistry 2008, 47, 6745–6751. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Carson, G.S.; Seo, H.C.; Hiraiwa, M.; Weiler, S.; Tomich, J.M.; Barranger, J.A.; Kahn, M.; Azuma, N.; Kishimoto, Y. Identification of the neurotrophic factor sequence of prosaposin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 681–685. [Google Scholar] [CrossRef]
- Ikeda, K.; Yamaguchi, T.; Fukunaga, S.; Hoshino, M.; Matsuzaki, K. Mechanism of Amyloid β-Protein Aggregation Mediated by GM1 Ganglioside Clusters. Biochemistry 2011, 50, 6433–6440. [Google Scholar] [CrossRef]
- Choo-Smith, L.P.; Garzon-Rodriguez, W.; Glabe, C.G.; Surewicz, W.K. Acceleration of amyloid fibril formation by specific binding of Abeta-(1-40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 1997, 272, 22987–22990. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Yoda, M.; Takaku, N.; Hirose, T.; Takeuchi, H. Clustered negative charges on the lipid membrane surface induce beta-sheet formation of prion protein fragment 106-126. Biochemistry 2007, 46, 11589–11597. [Google Scholar] [CrossRef]
- Luo, X.; Sharma, D.; Inouye, H.; Lee, D.; Avila, R.L.; Salmona, M.; Kirschner, D.A. Cytoplasmic domain of human myelin protein zero likely folded as beta-structure in compact myelin. Biophys. J. 2007, 92, 1585–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordes, F.S.; Bright, J.N.; Sansom, M.S. Proline-induced Distortions of Transmembrane Helices. J. Mol. Biol. 2002, 323, 951–960. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein |
Template PDB |
RMSD Robetta |
RMSD AlphaFold2 |
TM-Score Robetta |
Tm-Score AlphaFold2 |
|---|---|---|---|---|---|
| EGFR (25–638) | 7SYD | 21.1 | 27.085 | 0.29 | 0.23 |
| EGFR (25–309) | 7SYD | 2.64 | 2.36 | 0.86 | 0.91 |
| EGFR (366–492) | 7SYD | 0.88 | 0.44 | 0.96 | 0.99 |
| h-SYT1 (141–419) | 2R83 | 12.19 | 17.482 | 0.44 | 0.41 |
| h-SYT1 (143–265) | 2R83 | 1.31 | 1.27 | 0.93 | 0.95 |
| h-SYT1 (274–419) | 2R83 | 0.88 | 0.47 | 0.95 | 0.97 |
| APP (30–123) | 1MWP | 1.08 | 0.6 | 0.90 | 0.92 |
| APP (290–342) | 1APP | 1.2 | 0.57 | 0.85 | 0.95 |
| BoNT/A (whole protein) | 3BTA | 4.327 | 25.47 | 0.77 | 0.61 |
| BoNT/A LC (0–441) | 3BTA | 2 | 0.7 | 0.92 | 0.97 |
| BoNT/A HN (442–850) | 3BTA | 2.46 | 1.58 | 0.9 | 0.95 |
| BoNT/A HC (851–end) | 3BTA | 2.02 | 1.55 | 0.92 | 0.96 |
| BoNT/B (whole protein) | 2NP0 | 5.298 | 22.558 | 0.78 | 0.684 |
| BoNT/B LC (0–441) | 2NP0 | 1.75 | 1.41 | 0.96 | 0.97 |
| BoNT/B HN (442–850) | 2NP0 | 2.56 | 1.98 | 0.91 | 0.95 |
| BoNT/B HC (851–end) | 2NP0 | 2.37 | 1.65 | 0.91 | 0.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzaz, F.; Yahi, N.; Chahinian, H.; Fantini, J. The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program. Biomolecules 2022, 12, 1527. https://doi.org/10.3390/biom12101527
Azzaz F, Yahi N, Chahinian H, Fantini J. The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program. Biomolecules. 2022; 12(10):1527. https://doi.org/10.3390/biom12101527
Chicago/Turabian StyleAzzaz, Fodil, Nouara Yahi, Henri Chahinian, and Jacques Fantini. 2022. "The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program" Biomolecules 12, no. 10: 1527. https://doi.org/10.3390/biom12101527