
TNP Analogues Inhibit the Virulence Promoting IP3-4 Kinase Arg1 in the Fungal Pathogen Cryptococcus neoformans
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. RNA Extraction and cDNA Synthesis
2.3. Cloning of IP3-4K into the pGEX-6P Expression Vector
2.4. Expression and Purification of IP3-4K Proteins
2.5. Determination of Km and Vmax for ATP
2.6. Enzyme Activity and Inhibition Assay to Screen for ATP-Competitive Inhibitors
2.7. Surface Plasmon Resonance (SPR) to Assess Binding Affinity of TNP Analogues
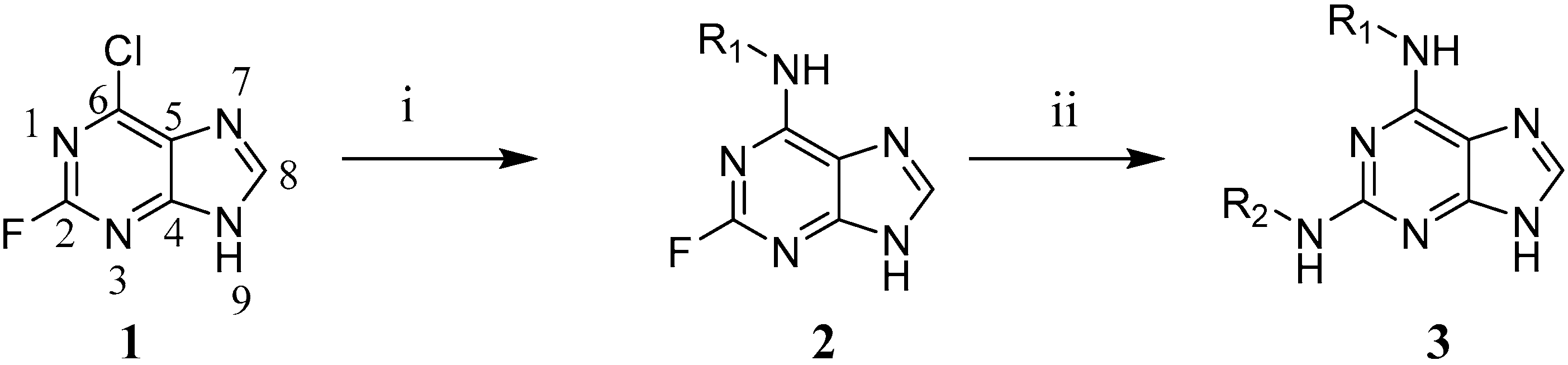
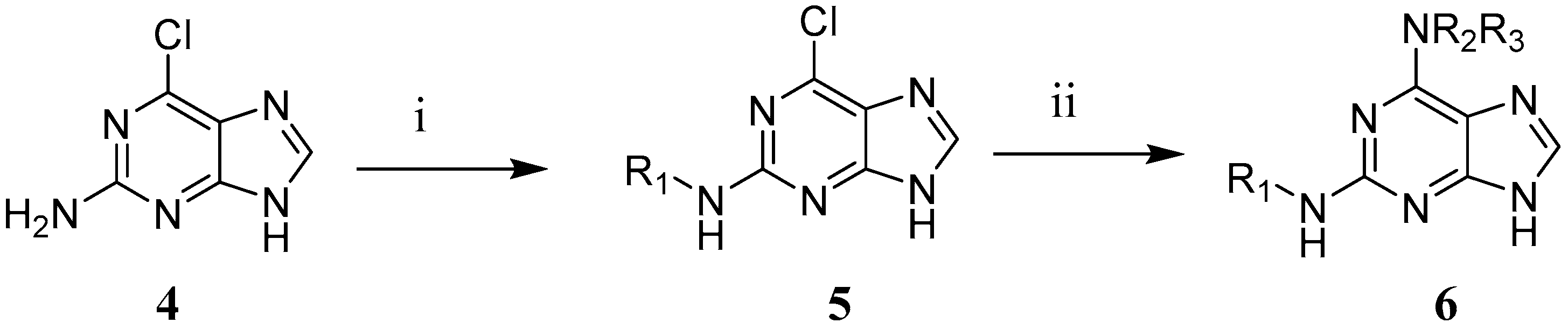
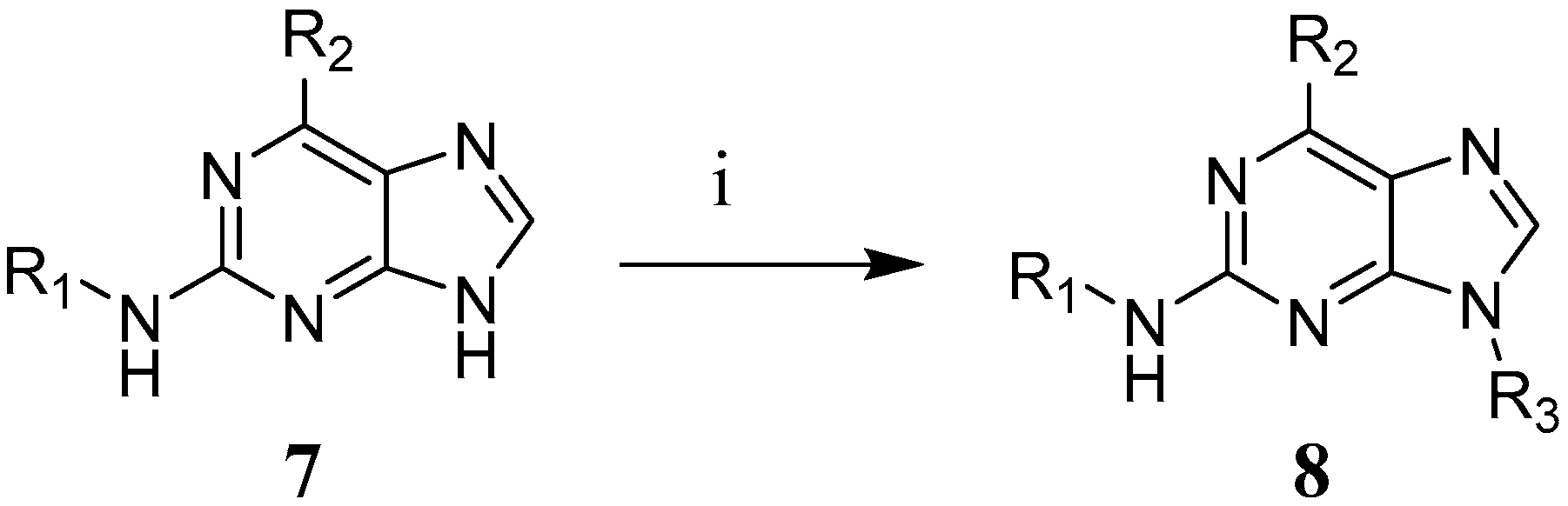
2.8. Synthesis of 2,6-Disubstituted Purine Analogues
2.8.1. General Experimental Procedure
2.8.2. General Procedure I for the First Nucleophilic Aromatic Substitution in the 6-Position (2a and 2b)
2.8.3. General Procedure II for the Second Nucleophilic Aromatic Substitution in the 2-Position (9 to 29 (Excluding 14 and 17), 32, 35, 38 and 39)
2.8.4. General Procedure III for the Reductive Amination in the 2-Position (5a to 5c)
2.8.5. General Procedure IV for the Nucleophilic Aromatic Substitution in the 6-Position (14, 30, 31, 36 and 41 to 44)
3. Results
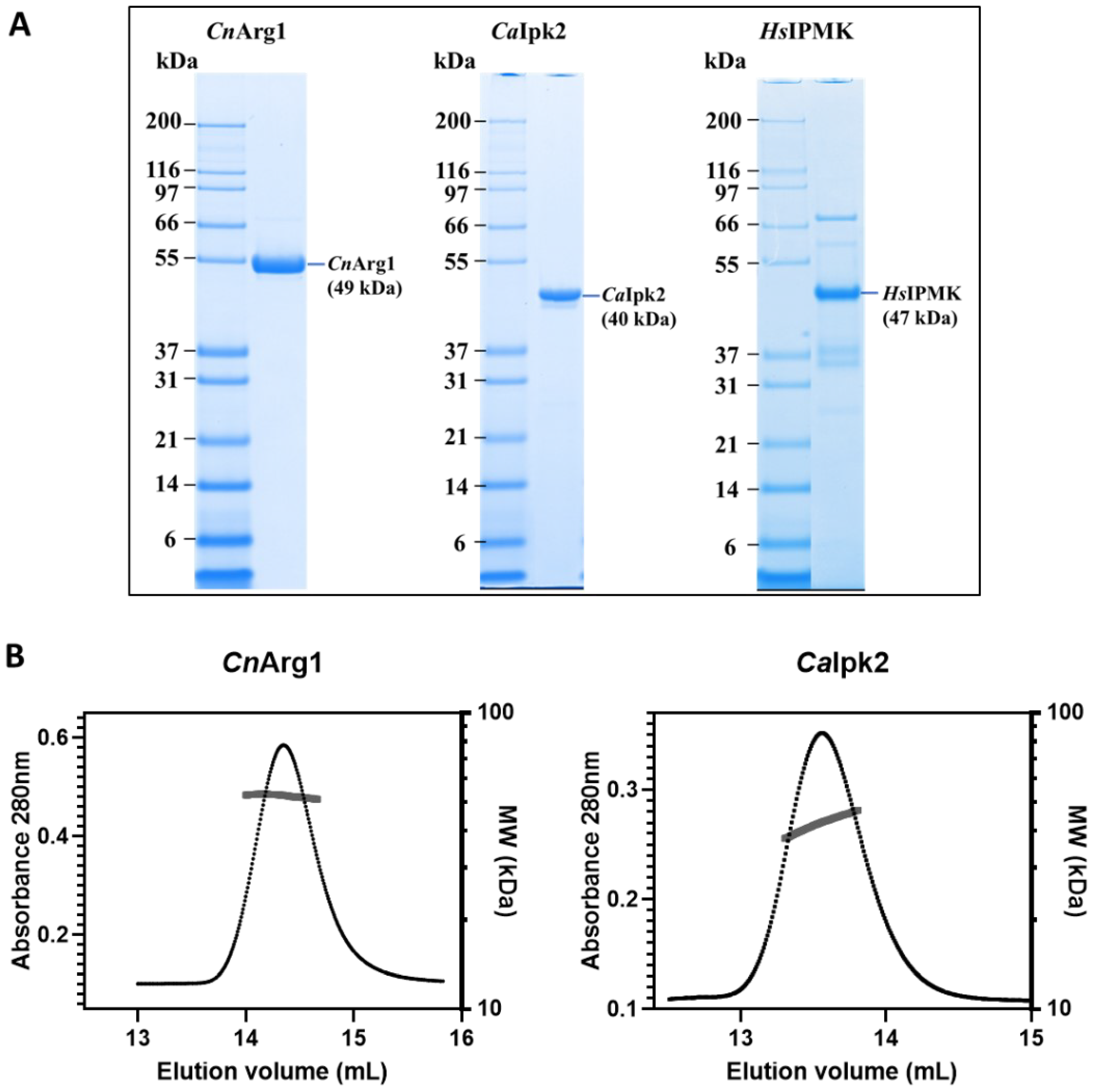
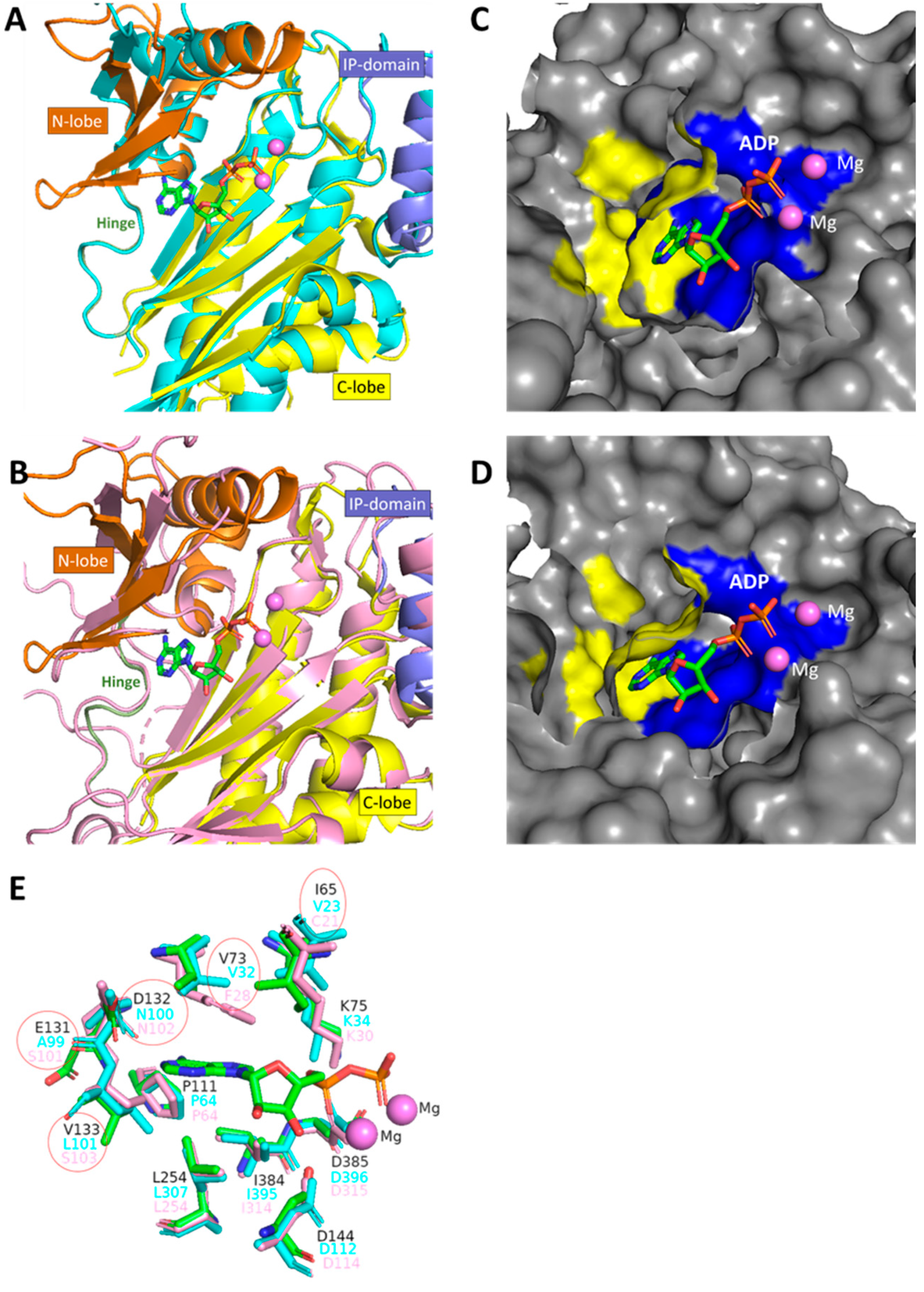
3.1. Purification of IP3-4K from C. neoformans, C. albicans and Human
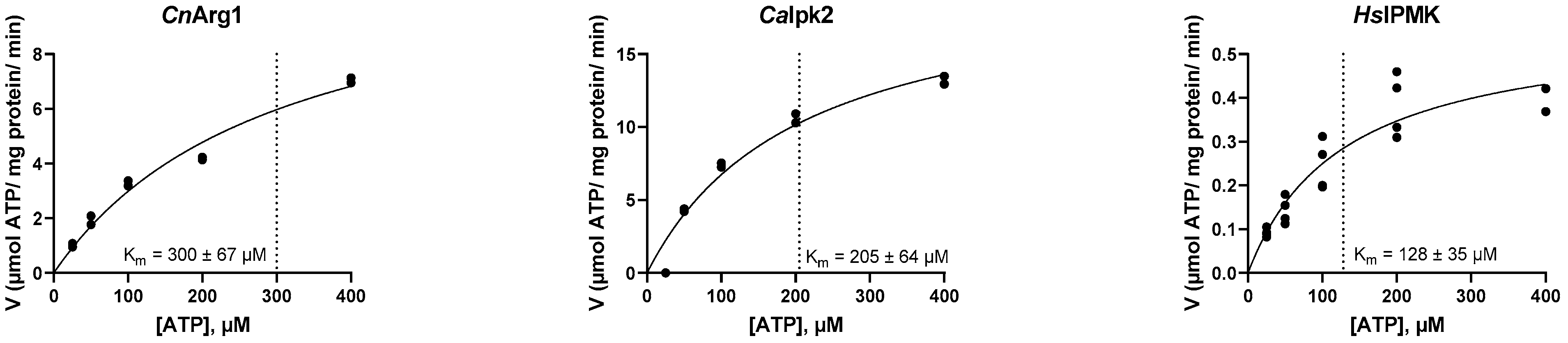
3.2. Establishing the Kinetic Properties of IP3-4K
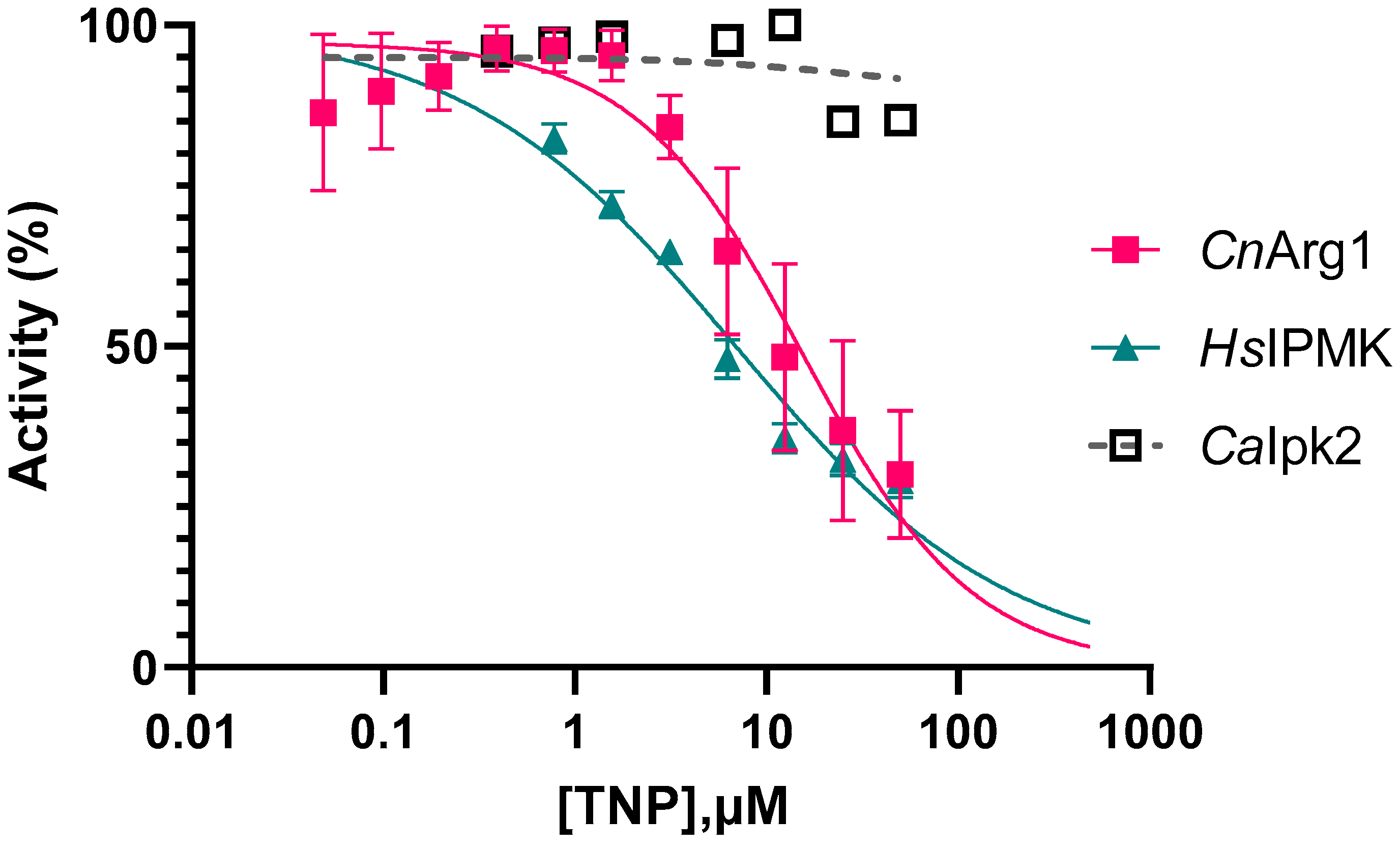
3.3. Optimizing the Enzyme Assay and Determining the IC50 of TNP
3.4. Synthesis of 2,6-Disubstituted Purine Analogues
3.5. Assessment of the Inhibitory Properties of 2,6-Disubstituted Purine Analogues
3.6. Assessing Binding Affinities of TNP and Its Analogues
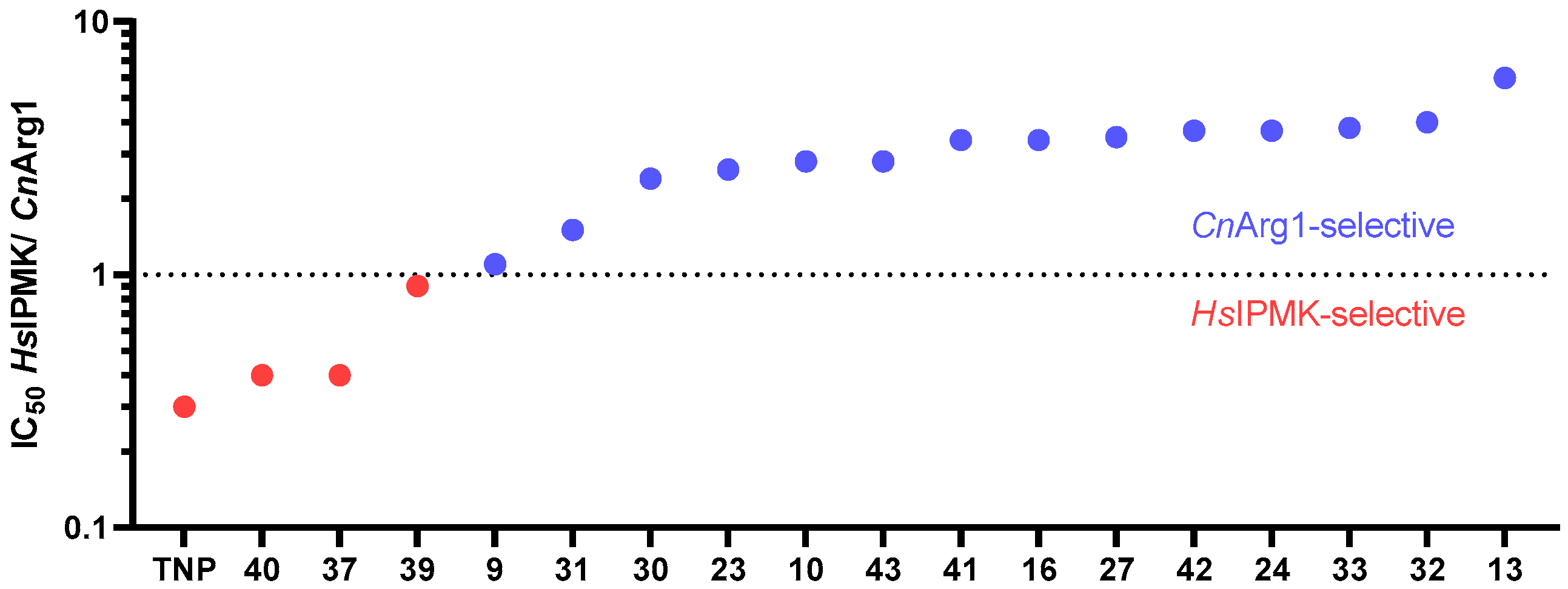
3.7. Comparing the Selectivity of TNP and Its Analogues for CnArg1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Gurr, S.J.; Cuomo, C.A.; Blehert, D.S.; Jin, H.; Stukenbrock, E.H.; Stajich, J.E.; Kahmann, R.; Boone, C.; Denning, D.W.; et al. Threats Posed by the Fungal Kingdom to Humans, Wildlife, and Agriculture. mBio 2020, 11, e00449-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Chang, C.-H.; Young-Xu, Y.; Chan, K.A. Systematic Review and Meta-Analysis of the Tolerability and Hepatotoxicity of Antifungals in Empirical and Definitive Therapy for Invasive Fungal Infection. Antimicrob. Agents Chemother. 2010, 54, 2409–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Cabañes, F.J. Aspergillosis, poultry farming and antifungal resistance. Rev. Iberoam. Micol. 2021, 38, 109–110. [Google Scholar] [CrossRef]
- Maligie, M.A.; Selitrennikoff, C.P. Cryptococcus neoformans Resistance to Echinocandins: (1,3)β-Glucan Synthase Activity Is Sensitive to Echinocandins. Antimicrob. Ag. Chemother. 2005, 49, 2851–2856. [Google Scholar] [CrossRef] [Green Version]
- Mroczyńska, M.; Brillowska-Dąbrowska, A. Review on Current Status of Echinocandins Use. Antibiotics 2020, 9, 227. [Google Scholar] [CrossRef]
- Chowdhary, A.; Prakash, A.; Sharma, C.; Kordalewska, M.; Kumar, A.; Sarma, S.; Tarai, B.; Singh, A.; Upadhyaya, G.; Upadhyay, S.; et al. A multicentre study of antifungal susceptibility patterns among 350 Candida auris isolates (2009–17) in India: Role of the ERG11 and FKS1 genes in azole and echinocandin resistance. J. Antimicrob. Chemother. 2018, 73, 891–899. [Google Scholar] [CrossRef]
- Li, C.; Lev, S.; Saiardi, A.; Desmarini, D.; Sorrell, T.C.; Djordjevic, J.T. Inositol Polyphosphate Kinases, Fungal Virulence and Drug Discovery. J. Fungi 2016, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Lev, S.; Bowring, B.; Desmarini, D.; Djordjevic, J.T. Inositol polyphosphate-protein interactions: Implications for microbial pathogenicity. Cell Microbiol. 2021, 23, e13325. [Google Scholar] [CrossRef]
- Saiardi, A.; Azevedo, C.; Desfougères, Y.; Portela-Torres, P.; Wilson, M.S.C. Microbial inositol polyphosphate metabolic pathway as drug development target. Adv. Biol. Regul. 2018, 67, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Lev, S.; Li, C.; Desmarini, D.; Sorrell, T.C.; Saiardi, A.; Djordjevic, J.T. Fungal Kinases with a Sweet Tooth: Pleiotropic Roles of Their Phosphorylated Inositol Sugar Products in the Pathogenicity of Cryptococcus neoformans Present Novel Drug Targeting Opportunities. Front Cell Infect. Microbiol. 2019, 9, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev, S.; Desmarini, D.; Li, C.; Chayakulkeeree, M.; Traven, A.; Sorrell, T.C.; Djordjevic, J.T. Phospholipase C of Cryptococcus neoformans regulates homeostasis and virulence by providing inositol trisphosphate as a substrate for Arg1 kinase. Infect. Immun. 2013, 81, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Lev, S.; Desmarini, D.; Kaufman-Francis, K.; Saiardi, A.; Silva, A.P.G.; Mackay, J.P.; Thompson, P.E.; Sorrell, T.C.; Djordjevic, J.T. IP3-4 kinase Arg1 regulates cell wall homeostasis and surface architecture to promote Cryptococcus neoformans infection in a mouse model. Virulence 2017, 8, 1833–1848. [Google Scholar] [CrossRef] [Green Version]
- Lev, S.; Li, C.; Desmarini, D.; Saiardi, A.; Fewings, N.L.; Schibeci, S.D.; Sharma, R.; Sorrell, T.C.; Djordjevic, J.T. Fungal Inositol Pyrophosphate IP7 Is Crucial for Metabolic Adaptation to the Host Environment and Pathogenicity. mBio 2015, 6, e00531-15. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Lev, S.; Saiardi, A.; Desmarini, D.; Sorrell, T.C.; Djordjevic, J.T. Identification of a major IP5 kinase in Cryptococcus neoformans confirms that PP-IP5/IP7, not IP6, is essential for virulence. Sci. Rep. 2016, 6, 23927. [Google Scholar] [CrossRef]
- Li, J.; Zhang, B.; Ma, T.; Wang, H.; Zhang, B.; Yu, Q.; Li, M. Role of the Inositol Polyphosphate Multikinase Ipk2 in Regulation of Hyphal Development, Calcium Signaling and Secretion in Candida albicans. Mycopathologia 2017, 182, 609–623. [Google Scholar] [CrossRef]
- Peng, X.; Yu, Q.; Liu, Y.; Ma, T.; Li, M. Study on the Function of the Inositol Polyphosphate Kinases Kcs1 and Vip1 of Candida albicans in Energy Metabolism. Front Microbiol. 2020, 11, 566069. [Google Scholar] [CrossRef]
- Ma, T.; Yu, Q.; Ma, C.; Mao, X.; Liu, Y.; Peng, X.; Li, M. Role of the inositol polyphosphate kinase Vip1 in autophagy and pathogenesis in Candida albicans. Future Microbiol. 2020, 15, 1363–1377. [Google Scholar] [CrossRef]
- Resnick, A.C.; Snowman, A.M.; Kang, B.N.; Hurt, K.J.; Snyder, S.H.; Saiardi, A. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc. Natl. Acad. Sci. USA 2005, 102, 12783–12788. [Google Scholar] [CrossRef]
- Wang, H.; Shears, S.B. Structural features of human inositol phosphate multikinase rationalize its inositol phosphate kinase and phosphoinositide 3-kinase activities. J. Biol. Chem. 2017, 292, 18192–18202. [Google Scholar] [CrossRef] [Green Version]
- Seacrist, C.D.; Blind, R.D. Crystallographic and kinetic analyses of human IPMK reveal disordered domains modulate ATP binding and kinase activity. Sci. Rep. 2018, 8, 16672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, S.F.; Maag, D.; Maxwell, M.J.; Resnick, A.C.; Juluri, K.R.; Chakraborty, A.; Koldobskiy, M.A.; Cha, S.H.; Barrow, R.; et al. Amino Acid Signaling to mTOR Mediated by Inositol Polyphosphate Multikinase. Cell Metab. 2011, 13, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Park, S.J.; Hong, S.; Kim, K.; Kim, S. Inositol polyphosphate multikinase signaling: Multifaceted functions in health and disease. Mol. Cells 2021, 44, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.H.; Haney, R.; Lamarre, E.; Drazba, J.; Prestwich, G.D.; Lindner, D.J. Gene deletion of inositol hexakisphosphate kinase 2 predisposes to aerodigestive tract carcinoma. Oncogene 2009, 28, 2383–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, F.; Cha, J.; Xu, J.; Xu, R.; Vandiver, M.S.; Tyagi, R.; Tokhunts, R.; Koldobskiy, M.A.; Fu, C.; Barrow, R.; et al. Inositol Pyrophosphates Mediate the DNA-PK/ATM-p53 Cell Death Pathway by Regulating CK2 Phosphorylation of Tti1/Tel2. Mol. Cell 2020, 79, 702. [Google Scholar] [CrossRef]
- Moritoh, Y.; Oka, M.; Yasuhara, Y.; Hozumi, H.; Iwachidow, K.; Fuse, H.; Tozawa, R. Inositol Hexakisphosphate Kinase 3 Regulates Metabolism and Lifespan in Mice. Sci. Rep. 2016, 6, 32072. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, S.; Zhu, Q.; Asteian, A.; Lin, H.; Xu, H.; Ernst, G.; Barrow, J.C.; Xu, B.; Cameron, M.D.; Kamenecka, T.M.; et al. TNP [N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol. Metab. 2016, 5, 903–917. [Google Scholar] [CrossRef]
- Kröber, T.; Bartsch, S.M.; Fiedler, D. Pharmacological tools to investigate inositol polyphosphate kinases—Enzymes of increasing therapeutic relevance. Adv. Biol. Regul. 2021, 83, 100836. [Google Scholar] [CrossRef]
- Moritoh, Y.; Abe, S.I.; Akiyama, H.; Kobayashi, A.; Koyama, R.; Hara, R.; Kasai, S.; Watanabe, M. The enzymatic activity of inositol hexakisphosphate kinase controls circulating phosphate in mammals. Nat. Commun. 2021, 12, 4847. [Google Scholar] [CrossRef]
- Liao, G.; Ye, W.; Heitmann, T.; Ernst, G.; DePasquale, M.; Xu, L.; Wormald, M.; Hu, X.; Ferrer, M.; Harmel, R.K.; et al. Identification of Small-Molecule Inhibitors of Human Inositol Hexakisphosphate Kinases by High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2021, 4, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Mukherjee, S.; Huang, D.; Chakraborty, M.; Gu, C.; Zong, G.; Stashko, M.A.; Pearce, K.H.; Shears, S.B.; Chakraborty, A.; et al. Development of Novel IP6K Inhibitors for the Treatment of Obesity and Obesity-Induced Metabolic Dysfunctions. J. Med. Chem. 2022, 65, 6869–6887. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, U.; Dollins, D.E.; Fridy, P.C.; York, J.D.; Downes, C.P. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: Use in defining biological roles and metabolic relationships of inositol pyrophosphates. J. Biol. Chem. 2009, 284, 10571–10582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.T.; Choi, G.; Bae, Y.S.; Burdett, M.; Moon, H.S.; Lee, J.W.; Gray, N.S.; Schultz, P.G.; Meijer, L.; Chung, S.K.; et al. Purine-based inhibitors of inositol-1,4,5-trisphosphate-3-kinase. Chembiochem 2002, 3, 897–901. [Google Scholar] [CrossRef]
- Lee, S.; Park, B.B.; Kwon, H.; Kim, V.; Jeon, J.S.; Lee, R.; Subedi, M.; Lim, T.; Ha, H.; An, D.; et al. TNP and its analogs: Modulation of IP6K and CYP3A4 inhibition. J. Enzym. Inhib. Med. Chem. 2022, 37, 269–279. [Google Scholar] [CrossRef]
- Kolozsvari, B.; Parisi, F.; Saiardi, A. Inositol phosphates induce DAPI fluorescence shift. Biochem. J. 2014, 460, 377–385. [Google Scholar] [CrossRef]
- Wormald, M.M.; Ernst, G.; Wei, H.; Barrow, J.C. Synthesis and characterization of novel isoform-selective IP6K1 inhibitors. Bioorganic Med. Chem. Lett. 2019, 29, 126628. [Google Scholar] [CrossRef]
- Dubois, E.; Scherens, B.; Vierendeels, F.; Ho, M.M.; Messenguy, F.; Shears, S.B. In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J. Biol. Chem. 2002, 277, 23755–23763. [Google Scholar] [CrossRef] [Green Version]
- Lev, S.; Kaufman-Francis, K.; Desmarini, D.; Juillard, P.G.; Li, C.; Stifter, S.A.; Feng, C.G.; Sorrell, T.C.; Grau, G.E.; Bahn, Y.S.; et al. Pho4 Is Essential for Dissemination of Cryptococcus neoformans to the Host Brain by Promoting Phosphate Uptake and Growth at Alkaline pH. mSphere 2017, 2, e00381-16. [Google Scholar] [CrossRef]
- Nalaskowski, M.M.; Deschermeier, C.; Fanick, W.; Mayr, G.W. The human homologue of yeast ArgRIII protein is an inositol phosphate multikinase with predominantly nuclear localization. Biochem. J. 2002, 366, 549–556. [Google Scholar] [CrossRef]
- Gu, C.; Stashko, M.A.; Puhl-Rubio, A.C.; Chakraborty, M.; Chakraborty, A.; Frye, S.V.; Pearce, K.H.; Wang, X.; Shears, S.B.; Wang, H. Inhibition of Inositol Polyphosphate Kinases by Quercetin and Related Flavonoids: A Structure–Activity Analysis. J. Med. Chem. 2019, 62, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Q.; Wu, K.; Guo, T.; Du, X.; Zhang, H.; Fang, L.; Zheng, N.; Zhang, Q.; Ye, F. Design, synthesis and activity of novel 2,6-disubstituted purine derivatives, potential small molecule inhibitors of signal transducer and activator of transcription 3. Eur. J. Med. Chem. 2019, 179, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Havlíček, L.; Hanuš, J.; Veselý, J.; Leclerc, S.; Meijer, L.; Shaw, G.; Strnad, M. Cytokinin-Derived Cyclin-Dependent Kinase Inhibitors: Synthesis and cdc2 Inhibitory Activity of Olomoucine and Related Compounds. J. Med. Chem. 1997, 40, 408–412. [Google Scholar] [CrossRef]
- Savelieva, E.M.; Oslovsky, V.E.; Karlov, D.S.; Kurochkin, N.N.; Getman, I.A.; Lomin, S.N.; Sidorov, G.V.; Mikhailov, S.N.; Osolodkin, D.I.; Romanov, G.A. Cytokinin activity of N6-benzyladenine derivatives assayed by interaction with the receptors in planta, in vitro, and in silico. Phytochemistry 2018, 149, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.S.; Wodicka, L.; Thunnissen, A.-M.W.H.; Norman, T.C.; Kwon, S.; Espinoza, F.H.; Morgan, D.O.; Barnes, G.; LeClerc, S.; Meijer, L.; et al. Exploiting Chemical Libraries, Structure, and Genomics in the Search for Kinase Inhibitors. Science 1998, 281, 533–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structure, Reactions, Synthesis, and Applications; Wiley-VCH: Wernheim, Germany, 2013. [Google Scholar]
- Huang, L.; Wang, Z.; Geng, L.; Chen, R.; Xing, W.; Wang, Y.; Huang, J. Selective and recyclable rhodium nanocatalysts for the reductive N-alkylation of nitrobenzenes and amines with aldehydes. RSC Adv. 2015, 5, 56936–56941. [Google Scholar] [CrossRef]
- Adamska, E.; Barciszewski, J.; Markiewicz, W.T. Convenient and Efficient Syntheses of N6- and N4- Substituted Adenines and Cytosines and their 2′-Deoxyribosides. Nucleosides Nucleotides Nucleic Acids 2012, 31, 861–871. [Google Scholar] [CrossRef]
- Desmarini, D.; Lev, S.; Furkert, D.; Crossett, B.; Saiardi, A.; Kaufman-Francis, K.; Li, C.; Sorrell, T.C.; Wilkinson-White, L.; Matthews, J.; et al. IP7-SPX Domain Interaction Controls Fungal Virulence by Stabilizing Phosphate Signaling Machinery. mBio 2020, 11, e01920-20. [Google Scholar] [CrossRef]
- Sparkes, M. DeepMind’s Protein-Folding AI Cracks Biology’s Biggest Problem. 2022. Available online: https://www.newscientist.com/article/2330866-deepminds-protein-folding-ai-cracks-biologys-biggest-problem/ (accessed on 29 July 2022).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- González, B.; Schell, M.J.; Letcher, A.J.; Veprintsev, D.B.; Irvine, R.F.; Williams, R.L. Structure of a Human Inositol 1,4,5-Trisphosphate 3-Kinase: Substrate Binding Reveals Why It Is Not a Phosphoinositide 3-Kinase. Mol. Cell 2004, 15, 689–701. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′ to 3′) | Description |
|---|---|---|
| ARG1-BglII-s | TACGagatctGACCTGCCCCTCACCCTCG | Sense primer to PCR-amplify CnArg1 cDNA with BglII recognition sequence and adapter sequence |
| ARG-EcoRI-a | CAAGgaattcTCAAACACAACCCCGTTCAACC | Antisense primer to PCR-amplify CnArg1 cDNA with EcoRI recognition sequence and adapter sequence |
| CaIPK2-BamHI-s | ACTCggatccATTCCCACTTTAAATTCACTCACTCCT | Sense primer to PCR-amplify CaIpk2 cDNA with BamHI recognition sequence and adapter sequence |
| CaIPK2-XhoI-a | TCTActcgagGTACAACCATTGCCATCGG | Antisense primer to PCR-amplify CaIpk2 cDNA with XhoI recognition sequence and adapter sequence |
| pGEX-6T-5’ | TTTTGCGCCGACATCATAACG | Sequencing primer 1 to confirm successful cloning of IP3-4K sequences |
| pGEX-Seq-s | GTGGCGACCATCCTCCAAA | Sequencing primer 2 to confirm successful cloning of IP3-4K sequences |
| pGEX-Seq-a | CAAGCTGTGACCGTCTCCG | Sequencing primer 3 to confirm successful cloning of IP3-4K sequences |
| Component | CnArg1 | CaIpk2 | HsIPMK |
|---|---|---|---|
| ATP (µM) | 10 | 10 | 10 |
| Enzyme (ng/µL) | 2.5 | 2.5 | 6 |
| IP3 (µM) | 50 | 25 | 25 |
 | ||||
|---|---|---|---|---|
| Compound | R1 | CnArg1 IC50 (µM) | HsIPMK IC50 (µM) | HsIPMK/CnArg1 |
| TNP | See above for structure | 21 ± 6 | 7 ± 0.5 | 0.3 |
| 9 |  | 28 ± 7 | 32 ± 3 | 1.1 |
| 10 |  | 20 ± 1 | 55 ± 2 | 2.8 |
| 11 |  | Low solubility—ND | ND | ND |
| 12 |  | 77 ± 14 | ND | ND |
| 13 |  | 25 ± 1 | 150 ± 30 | 6.0 |
| 14 |  | 44 ± 2 | ND | ND |
| 15 |  | 46 ± 22 | ND | ND |
| 16 |  | 14 ± 1 | 48 ± 2 | 3.4 |
| 17 |  | 45 ± 3 | ND | ND |
| 18 |  | Low solubility—ND | ND | ND |
| 19 |  | Low solubility—ND | ND | ND |
| 20 |  | 71 ± 12 | ND | ND |
| 21 (20 HCl salt) |  | 57 ± 6 | ND | ND |
| 22 |  | Not inhibitory | ND | ND |
| 23 |  | 21 ± 1 | 55 ± 4 | 2.6 |
| 24 |  | 24 ± 1 | 89 ± 9 | 3.7 |
| 25 |  | Not inhibitory | ND | ND |
| 26 |  | Not inhibitory | ND | ND |
| 27 |  | 35 ± 2 | 123 ± 80 | 3.5 |
| 28 |  | 127 ± 32 | ND | ND |
| 29 |  | 75 ± 9 | ND | ND |
 | ||||
|---|---|---|---|---|
| Compound | R1 | CnArg1 IC50 (µM) | HsIPMK IC50 (µM) | HsIPMK/CnArg1 |
| TNP | 21 ± 6 | 7 ± 0.5 | 0.3 | |
| 30 |  | 28 ± 1 | 67 ± 6 | 2.4 |
| 31 |  | 29 ± 3 | 43 ± 4 | 1.5 |
| 32 |  | 12 ± 1 | 48 ± 3 | 4.0 |
| 33 |  | 38 ± 5 | 144 ± 55 | 3.8 |
| 34 |  | Not inhibitory | ND | ND |
| 35 |  | 87 ± 9 | ND | ND |
| 36 |  | 55 ± 6 | ND | ND |
| 37 |  | 17 ± 2 | 7 ± 1 | 0.4 |
| 38 |  | 44 ± 6 | ND | ND |
| 39 |  | 22 ± 3 | 19 ± 1 | 0.9 |
| 40 |  | 14 ± 1 | 5 ± 1 | 0.4 |
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | CnArg1 IC50 (µM) | HsIPMK IC50 (µM) | HsIPMK/CnArg1 |
| TNP | 21 ± 6 | 7 ± 0.5 | 0.3 | ||
| 41 |  |  | 24 ± 1 | 82 ± 9 | 3.4 |
| 42 |  |  | 10 ± 0.5 | 37 ± 2 | 3.7 |
| 43 |  |  | 27 ± 2 | 75 ± 8 | 2.8 |
| 44 |  |  | 82 ± 13 | ND | ND |
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | CnArg1 IC50 (µM) | HsIPMK IC50 (µM) | HsIPMK/CnArg1 |
| TNP | 21 ± 6 | 7 ± 0.5 | 0.3 | |||
| 45 |  |  |  | Not inhibitory | ND | ND |
| 46 |  |  | CH3 | Not inhibitory | ND | ND |
| Compound | IC50 (µM) | KD (µM) |
|---|---|---|
| TNP | 21 ± 6 | Poor solubility—ND |
| 9 | 28 ± 7 | Poor solubility—ND |
| 10 | 20 ± 1 | 17 ± 5 |
| 13 | 25 ± 1 | 13 ± 3 |
| 23 | 21 ± 1 | 22 ± 4 |
| 24 | 24 ± 1 | 16 ± 9 |
| 28 | 127 ± 32 | 321 ± 10 |
| 29 | 75 ± 9 | 155 ± 27 |
| 30 | 28 ± 1 | 70 ± 13 |
| 31 | 29 ± 3 | 35 ± 9 |
| 32 | 12 ± 0.5 | 38 ± 11 |
| 36 | 55 ± 6 | 74 ± 10 |
| 37 | 17 ± 2 | No binding |
| 39 | 22 ± 3 | 188 ± 9 |
| 40 | 13 ± 0.5 | 316 ± 31 |
| 41 | 24 ± 1 | 96 ± 10 |
| 42 | 10 ± 0.5 | 48 ± 7 |
| 43 | 27 ± 2 | 21 ± 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desmarini, D.; Truong, D.; Wilkinson-White, L.; Desphande, C.; Torrado, M.; Mackay, J.P.; Matthews, J.M.; Sorrell, T.C.; Lev, S.; Thompson, P.E.; et al. TNP Analogues Inhibit the Virulence Promoting IP3-4 Kinase Arg1 in the Fungal Pathogen Cryptococcus neoformans. Biomolecules 2022, 12, 1526. https://doi.org/10.3390/biom12101526
Desmarini D, Truong D, Wilkinson-White L, Desphande C, Torrado M, Mackay JP, Matthews JM, Sorrell TC, Lev S, Thompson PE, et al. TNP Analogues Inhibit the Virulence Promoting IP3-4 Kinase Arg1 in the Fungal Pathogen Cryptococcus neoformans. Biomolecules. 2022; 12(10):1526. https://doi.org/10.3390/biom12101526
Chicago/Turabian StyleDesmarini, Desmarini, Daniel Truong, Lorna Wilkinson-White, Chandrika Desphande, Mario Torrado, Joel P. Mackay, Jacqueline M. Matthews, Tania C. Sorrell, Sophie Lev, Philip E. Thompson, and et al. 2022. "TNP Analogues Inhibit the Virulence Promoting IP3-4 Kinase Arg1 in the Fungal Pathogen Cryptococcus neoformans" Biomolecules 12, no. 10: 1526. https://doi.org/10.3390/biom12101526