
An Overview of Molecular Mechanisms in Fabry Disease

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Phenotypic Variability
3. Molecular Mechanisms of Fabry Disease
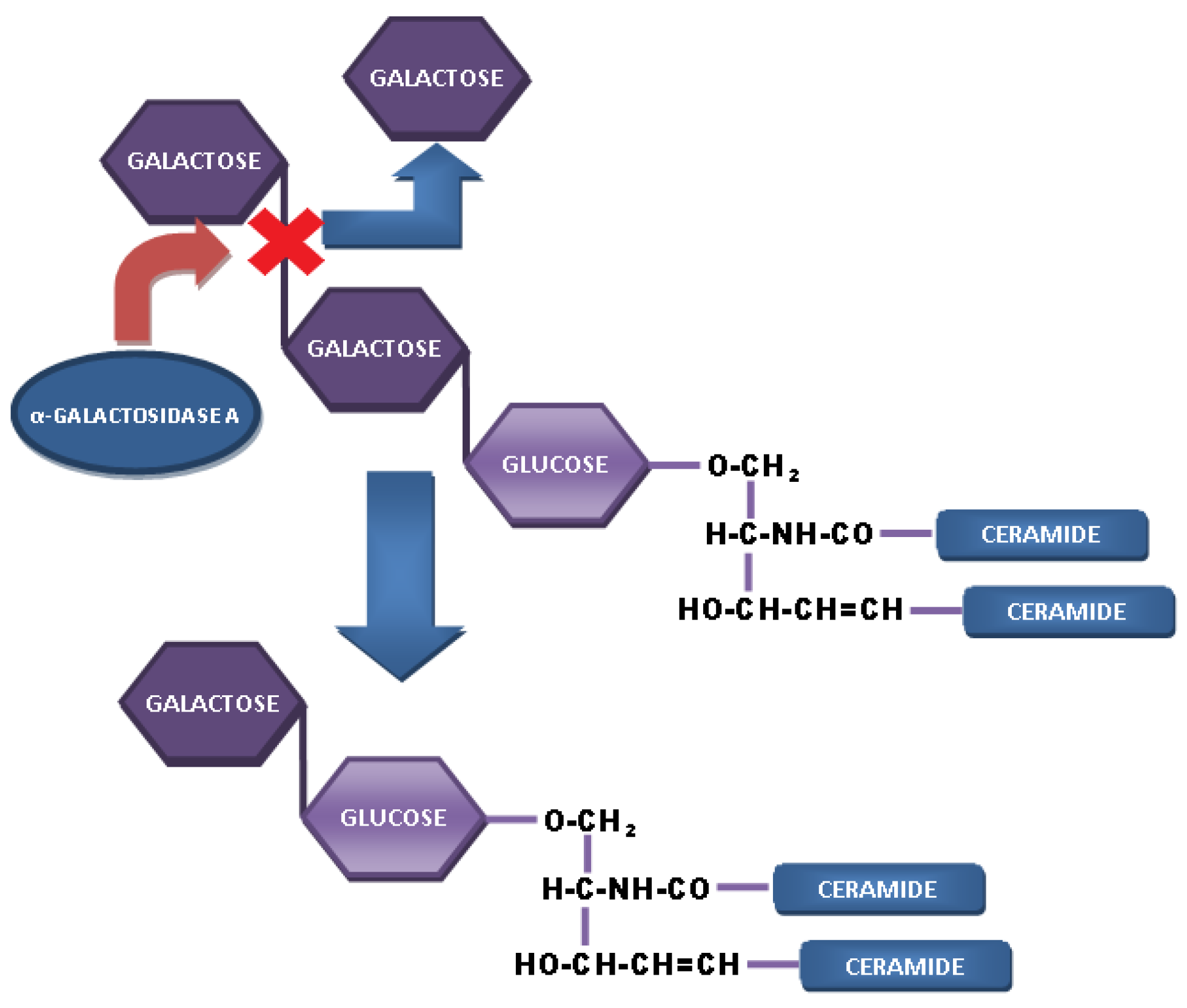
4. The Role of Gb3 in FD
5. Inheritance
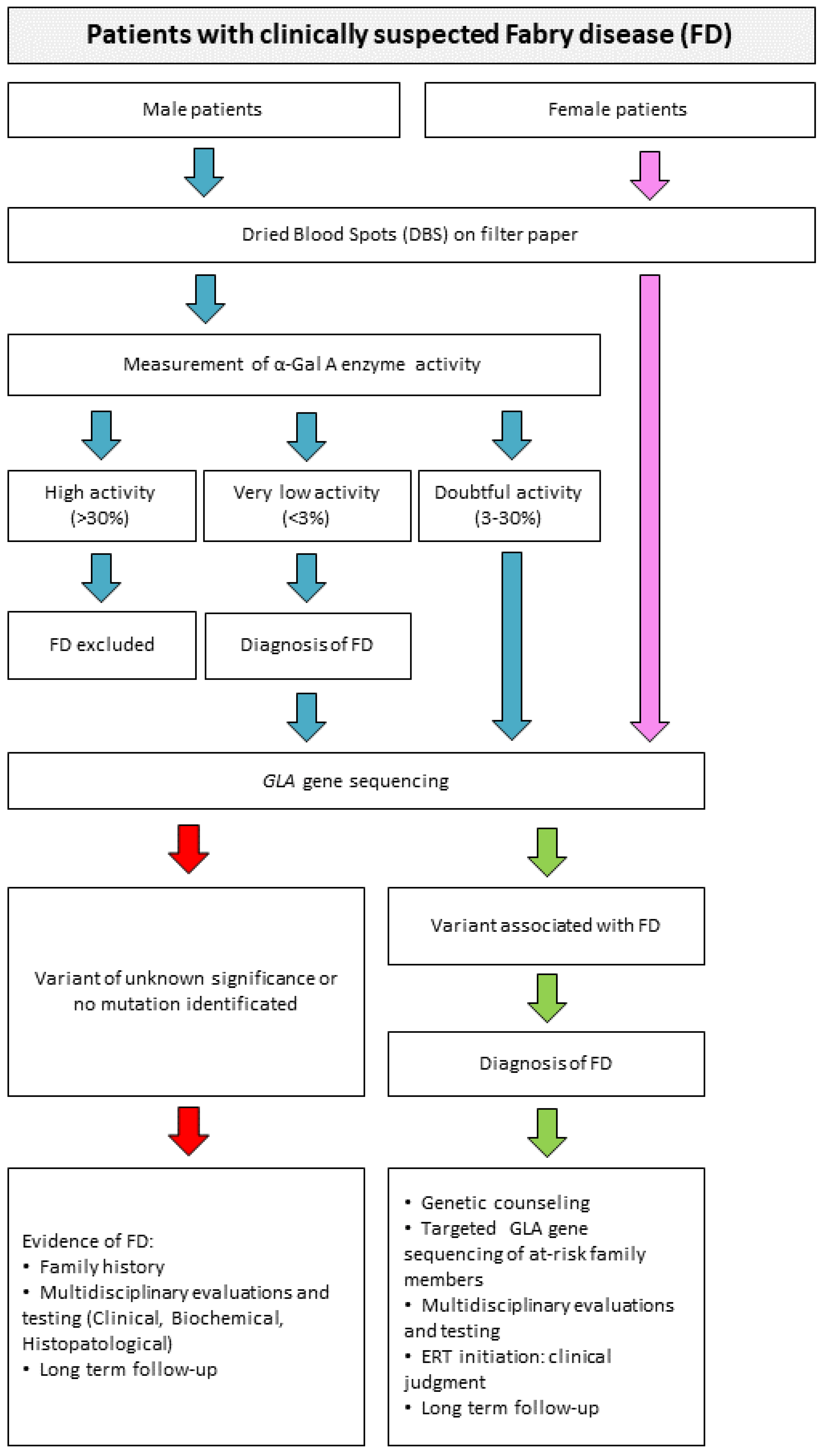
6. Diagnosis Algorithm
7. Biomarkers
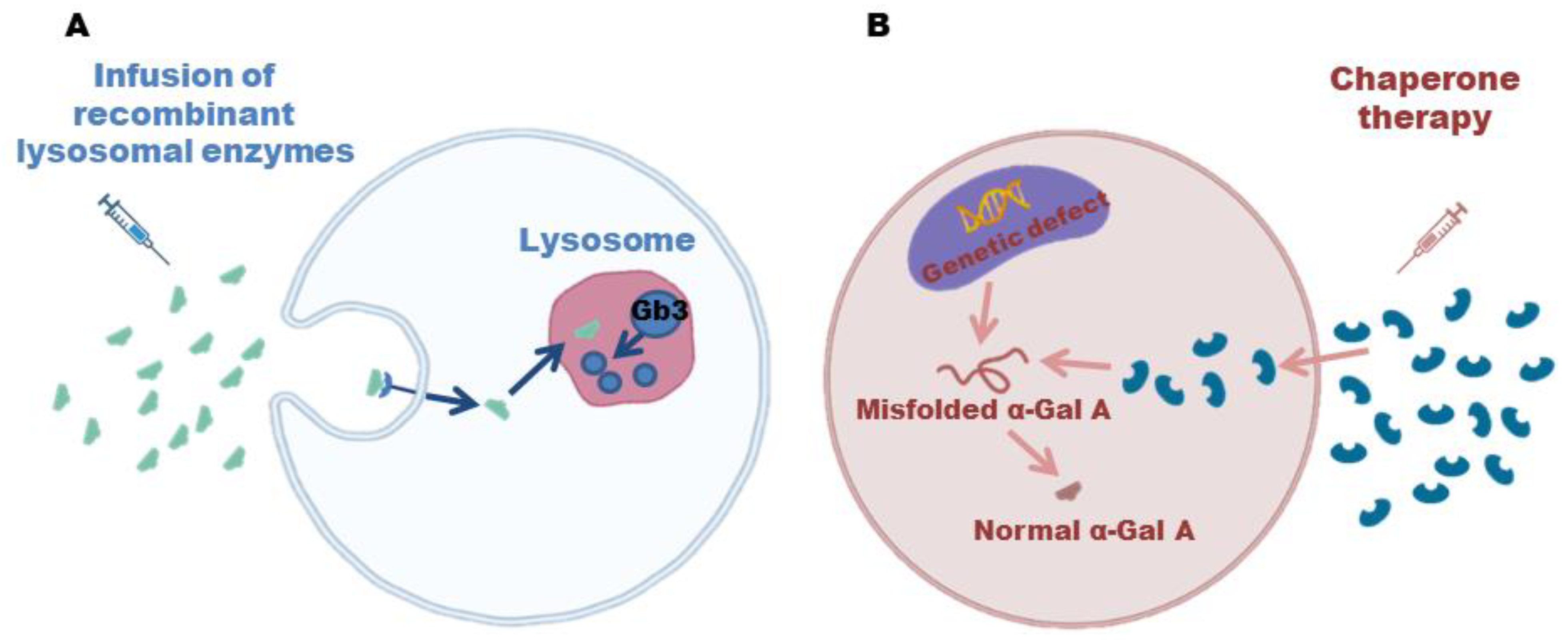
8. Therapies for FD
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Linthorst, G.E.; Hollak, C.E.M.; Korevaar, J.C.; Van Manen, J.G.; Aerts, J.; Boeschoten, E.W. alpha-Galactosidase A deficiency in Dutch patients on dialysis: A critical appraisal of screening for Fabry disease. Nephrol. Dial. Transplant. 2003, 18, 1581–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; De Lorenzo, A.G.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur. J. Clin. Investig. 2004, 34, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, M.P.; Iacobellis, F.; Acampora, E.; Caiazza, M.; Rubino, M.; Monda, E.; Magaldi, M.R.; Tarallo, A.; Sasso, M.; De Pasquale, V.; et al. Aortopathies in mouse models of Pompe, Fabry and Mucopolysaccharidosis IIIB lysosomal storage diseases. PLoS ONE 2020, 15, e0233050. [Google Scholar] [CrossRef]
- Ioannou, Y.A.; Zeidner, K.M.; Gordon, R.E.; Desnick, R.J. Fabry Disease: Preclinical Studies Demonstrate the Effectiveness of α-Galactosidase A Replacement in Enzyme-Deficient Mice. Am. J. Hum. Genet. 2001, 68, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef]
- Hagège, A.A.; Caudron, E.; Damy, T.; Roudaut, R.; Millaire, A.; Etchecopar-Chevreuil, C.; Tran, T.-C.; Jabbour, F.; Boucly, C.; Prognon, P.; et al. Screening patients with hypertrophic cardiomyopathy for Fabry disease using a filter-paper test: The FOCUS study. Heart 2010, 97, 131–136. [Google Scholar] [CrossRef] [Green Version]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med Genet. 2001, 38, 769–775. [Google Scholar] [CrossRef] [Green Version]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J. Med. Genet. 2001, 38, 750–760. [Google Scholar] [CrossRef]
- Reuser, A.J.; Verheijen, F.W.; Bali, D.; van Diggelen, O.P.; Germain, D.; Hwu, W.-L.; Lukacs, Z.; Mühl, A.; Olivova, P.; Piraud, M.; et al. The use of dried blood spot samples in the diagnosis of lysosomal storage disorders—Current status and perspectives. Mol. Genet. Metab. 2011, 104, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Linthorst, G.E.; Bouwman, M.G.; Wijburg, F.A.; Aerts, J.; Poorthuis, B.J.H.M.; Hollak, C.E.M. Screening for Fabry disease in high-risk populations: A systematic review. J. Med. Genet. 2010, 47, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Rubino, M.; Monda, E.; Lioncino, M.; Caiazza, M.; Palmiero, G.; Dongiglio, F.; Fusco, A.; Cirillo, A.; Cesaro, A.; Capodicasa, L.; et al. Diagnosis and Management of Cardiovascular Involvement in Fabry Disease. Heart Fail. Clin. 2022, 18, 39–49. [Google Scholar] [CrossRef]
- Monda, E.; Palmiero, G.; Lioncino, M.; Rubino, M.; Cirillo, A.; Fusco, A.; Caiazza, M.; Verrillo, F.; Diana, G.; Mauriello, A.; et al. Multimodality Imaging in Cardiomyopathies with Hypertrophic Phenotypes. J. Clin. Med. 2022, 11, 868. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef]
- Limongelli, G.; Adorisio, R.; Baggio, C.; Bauce, B.; Biagini, E.; Castelletti, S.; Favilli, S.; Imazio, M.; Lioncino, M.; Merlo, M.; et al. Diagnosis and Management of Rare Cardiomyopathies in Adult and Paediatric Patients. A Position Paper of the Italian Society of Cardiology (SIC) and Italian Society of Paediatric Cardiology (SICP). Int. J. Cardiol. 2022, 357, 55–71. [Google Scholar] [CrossRef]
- Waldek, S.; Patel, M.R.; Banikazemi, M.; Lemay, R.; Lee, P. Life expectancy and cause of death in males and females with Fabry disease: Findings from the Fabry Registry. Genet. Med. 2009, 11, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Bodamer, O.; Ratschmann, R.; Paschke, E.; Voigtländer, T.; Stockler-Ipsiroglu, S. Recurrent acroparaesthesia during febrile infections. Lancet 2004, 363, 1698. [Google Scholar] [CrossRef]
- Ries, M.; Ramaswami, U.; Parini, R.; Lindblad, B.; Whybra, C.; Willers, I.; Gal, A.; Beck, M. The early clinical phenotype of Fabry disease: A study on 35 European children and adolescents. Eur. J. Pediatr. 2003, 162, 767–772. [Google Scholar] [CrossRef]
- Desnick, R.J.; Brady, R.O. Fabry disease in childhood. J. Pediatr. 2004, 144, S20–S26. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Gupta, S.; Moore, D.F.; Sachdev, V.; Quirk, J.M.; Murray, G.J.; Rosing, D.R.; Robinson, C.; Schaefer, E.; Gal, A.; et al. Pediatric Fabry Disease. Pediatrics 2005, 115, e344–e355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswami, U.; Whybra, C.; Parini, R.; Pintos-Morell, G.; Mehta, A.; Sunder-Plassmann, G.; Widmer, U.; Beck, M. FOS European Investigators. Clinical manifestations of Fabry disease in children: Data from the Fabry Outcome Survey. Acta Paediatr. 2006, 95, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Kodama, C.; Takenaka, T.; Tanaka, A.; Yasumoto, Y.; Yoshida, A.; Kanzaki, T.; Enriquez, A.L.; Eng, C.M.; Tanaka, H.; et al. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003, 64, 801–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branton, M.H.; Schiffmann, R.; Sabnis, S.G.; Murray, G.J.; Quirk, J.M.; Altarescu, G.; Goldfarb, L.; Brady, R.O.; Balow, J.E.; Austin Iii, H.A.; et al. Natural history of Fabry renal disease: Influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine 2002, 81, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Cianciaruso, B.; Pisani, A.; Andreucci, M.V.; Parente, N.; Andria, G.; Federico, S.; Sabbatini, M.; Sessa, A. Malattia di Anderson-Fabry: Problematiche diagnostiche, attualità terapeutiche ed esperienza clinica nel trattamento della malattia con terapia enzimatica sostitutiva in pazienti nefropatici [Anderson-Fabry’s disease: Diagnostic problems, therapeutic relevance, and clinical experience in the treatment of the disease with enzyme replacement therapy in nephropathic patients]. G. Ital. Nefrol. 2003, 20, 113–119. [Google Scholar] [PubMed]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- Dobyns, W.B.; Filauro, A.; Tomson, B.N.; Chan, A.S.; Ho, A.; Ting, N.T.; Oosterwijk, J.C.; Ober, C. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am. J. Med. Genet. A 2004, 129A, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS—Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauhin, W.; Lidove, O.; Masat, E.; Mingozzi, F.; Mariampillai, K.; Ziza, J.-M.; Benveniste, O. Innate and Adaptive Immune Response in Fabry Disease. JIMD Rep. 2015, 22, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanos, J.; Nicholls, K.; Grigg, L.; Kiers, L.; Crawford, A.; Becker, G. Clinical features of Fabry’s disease in Australian patients. Intern. Med. J. 2002, 32, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef]
- Maier, E.M.; Osterrieder, S.; Whybra, C.; Ries, M.; Gal, A.; Beck, M.; Roscher, A.A.; Muntau, A.C. Disease manifestations and X inactivation in heterozygous females with Fabry disease. Acta Paediatr. Suppl. 2006, 95, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, E.; Baron, K.; Widmer, U.; Deegan, P.; Neumann, H.P.; Sunder-Plassmann, G.; Johansson, J.-O.; Whybra, C.; Ries, M.; Pastores, G.M.; et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum. Mutat. 2005, 25, 412. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K.; et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genomic. Med. 2018, 12, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.P.; Winchester, B.G.; Malcolm, S. Sequence variations in the first exon of alpha-galactosidase A. J. Med. Genet. 1993, 30, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Beck, M.; Höppner, W.; Germain, D.P. Clinical utility gene card for: Fabry disease—Update 2016. Eur. J. Hum. Genet. 2017, 25, e1–e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med. Genet. 2018, 55, 261–268. [Google Scholar] [CrossRef]
- Ishii, S.; Sakuraba, H.; Suzuki, Y. Point mutations in the upstream region of the α-galactosidase A gene exon 6 in an atypical variant of Fabry disease. Hum. Genet. 1992, 89, 29–32. [Google Scholar] [CrossRef]
- Altarescu, G.M.; Goldfarb, L.G.; Park, K.Y.; Kaneski, C.; Jeffries, N.; Litvak, S.; Nagle, J.W.; Schiffmann, R. Identification of fifteen novel mutations and genotype-phenotype relationship in Fabry disease. Clin. Genet. 2001, 60, 46–51. [Google Scholar] [CrossRef]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Ortiz, A.; Abiose, A.; Bichet, D.-G.; Cabrera, G.; Charrow, J.; Germain, D.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: Data from the Fabry Registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef]
- Schaefer, E.; Mehta, A.; Gal, A. Genotype and phenotype in Fabry disease: Analysis of the Fabry Outcome Survey. Acta Paediatr. Suppl. 2005, 94, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Blanch, L.C.; Meaney, C.; Morris, C.P. A sensitive mutation screening strategy for Fabry disease: Detection of nine mutations in the alpha-galactosidase A gene. Hum. Mutat. 1996, 8, 38–43. [Google Scholar] [CrossRef]
- Kase, R.; Bierfreund, U.; Klein, A.; Kolter, T.; Itoh, K.; Suzuki, M.; Hashimoto, Y.; Sandhoff, K.; Sakuraba, H. Only sphingolipid activator protein B (SAP-B or saposin B) stimulates the degradation of globotriaosylceramide by recombinant human lysosomal α-galactosidase in a detergent-free liposomal system. FEBS Lett. 1996, 393, 74–76. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, B.; Hu, J.; Madduri, A.; Lundberg, D.; Miller, B.; Gill, J.; Meiyappan, M.; Pan, C.; Miller, T.; Zhang, B. Affinity purification of human alpha galactosidase utilizing a novel small molecule biomimetic of alpha-D-galactose. Protein Expr. Purif. 2021, 177, 105752. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The Molecular Defect Leading to Fabry Disease: Structure of Human α-Galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Varela-Calais, P.; Nicolicht, P.; Martin, R.P.; Yamamoto, J.; D’Almeida, V.; Martins, A.M.; Pesquero, J.B. Functional characterization of novel variants found in patients with suspected Fabry disease. Clin. Chim. Acta 2022, 534, 156–160. [Google Scholar] [CrossRef]
- Perry, R.; Shah, R.; Saiedi, M.; Patil, S.; Ganesan, A.; Linhart, A.; Selvanayagam, J.B. The Role of Cardiac Imaging in the Diagnosis and Management of Anderson-Fabry Disease. JACC Cardiovasc. Imaging 2019, 12, 1230–1242. [Google Scholar] [CrossRef]
- Nair, V.; Belanger, E.C.; Veinot, J.P. Lysosomal storage disorders affecting the heart: A review. Cardiovasc. Pathol. 2019, 39, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Knott, K.D.; Augusto, J.B.; Nordin, S.; Kozor, R.; Camaioni, C.; Xue, H.; Hughes, R.K.; Manisty, C.; Brown, L.A.E.; Kellman, P.; et al. Quantitative Myocardial Perfusion in Fabry Disease. Circ. Cardiovasc. Imaging 2019, 12, e008872. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, M. Altered Sphingolipids Metabolism Damaged Mitochondrial Functions: Lessons Learned From Gaucher and Fabry Diseases. J. Clin. Med. 2020, 9, 1116. [Google Scholar] [CrossRef] [Green Version]
- Chimenti, C.; Hamdani, N.; Boontje, N.M.; DE Cobelli, F.; Esposito, A.; Bronzwaer, J.G.; Stienen, G.; Russo, M.A.; Paulus, W.J.; Frustaci, A.; et al. Myofilament Degradation and Dysfunction of Human Cardiomyocytes in Fabry Disease. Am. J. Pathol. 2008, 172, 1482–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A Human Stem Cell Model of Fabry Disease Implicates LIMP-2 Accumulation in Cardiomyocyte Pathology. Stem Cell Rep. 2019, 13, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M. Electrocardiographic Changes and Arrhythmia in Fabry Disease. Front. Cardiovasc. Med. 2016, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Publisher Correction: Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2019, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [Green Version]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients With Fabry Disease: A Critical Link to Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Beck, M.; Sunder-Plassmann, G. Fabry Disease: Perspectives from 5 Years of FOS; Oxford Pharma Genesis: Oxford, UK, 2006. [Google Scholar]
- Viggiano, E.; Politano, L. X Chromosome Inactivation in Carriers of Fabry Disease: Review and Meta-Analysis. Int. J. Mol. Sci. 2021, 22, 7663. [Google Scholar] [CrossRef] [PubMed]
- Makoto, Y.; Koji, H.; Masaaki, M.; Sean, D.; Andre, M.; Sandra, S.; Jeffrey, M.; Chuwa, T.; Toshihiro, T. Identification of novel mutations in the α-galactosidase A gene in patients with Fabry disease: Pitfalls of mutation analyses in patients with low α-galactosidase A activity. J. Cardiol. 2011, 57, 345–353. [Google Scholar]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.; Benistan, K.; Angelova, L. X-linked inheritance and its implication in the diagnosis and management of female patients in Fabry disease. Rev. Med. Interne 2010, 31 (Suppl. 2), S209–S213. [Google Scholar] [CrossRef]
- Kampine, J.P.; Brady, R.O.; Kanfer, J.N.; Feld, M.; Shapiro, D. Diagnosis of gaucher’s disease and niemann-pick disease with small samples of venous blood. Science 1967, 155, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Johnson, W.G.; Uhlendorf, B. Identification of heterozygous carriers of lipid storage diseases. Current status and clinical applications. Am. J. Med. 1971, 51, 423–431. [Google Scholar] [CrossRef]
- Kılavuz, S.; Basaranoglu, M.; Epcacan, S.; Bako, D.; Ozer, A.; Donmez, Y.N.; Ceylan, E.I.; Tukun, A.; Ceylaner, S.; Geylani, H.; et al. A rare cause of hydrops fetalis in two Gaucher disease type 2 patients with a novel mutation. Metab. Brain Dis. 2022, 37, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Grünfeld, J.P. How to improve the early diagnosis of Fabry’s disease? Kidney Int. 2003, 64, 1136–1137. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A.; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Jehn, U.; Bayraktar, S.; Pollmann, S.; Van Marck, V.; Weide, T.; Pavenstädt, H.; Brand, E.; Lenders, M. α-Galactosidase a Defi-ciency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes. Int. J. Mol. Sci. 2021, 22, 11339. [Google Scholar] [CrossRef]
- Froissart, R.; Guffon, N.; Vanier, M.T.; Desnick, R.J.; Maire, I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef]
- Daitx, V.V.; Mezzalira, J.; Goldim, M.P.D.S.; Coelho, J.C. Comparison between alpha-galactosidase A activity in blood samples collected on filter paper, leukocytes and plasma. Clin. Biochem. 2012, 45, 1233–1238. [Google Scholar] [CrossRef]
- Muto, R.; Suzuki, Y.; Shimizu, H.; Yasuda, K.; Ishimoto, T.; Maruyama, S.; Ito, Y.; Mizuno, M. Recurrent Cerebrovascular Complications under Enzyme Replacement Therapy in a Patient with Fabry Disease on Peritoneal Dialysis. Intern. Med. 2022, 10, 0185-22. [Google Scholar] [CrossRef] [PubMed]
- Mayes, J.S.; Scheerer, J.B.; Sifers, R.N.; Donaldson, M.L. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin. Chim. Acta 1981, 112, 247–251. [Google Scholar] [CrossRef]
- Hoffmann, B.; Koch, H.G.; Schweitzer-Krantz, S.; Wendel, U.; Mayatepek, E. Deficient α-galactosidase A activity in plasma but no Fabry disease—A pitfall in diagnosis. Clin. Chem. Lab. Med. 2005, 43, 1276–1277. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef]
- Hoffmann, B. Fabry disease: Recent advances in pathology, diagnosis, treatment and monitoring. Orphanet J. Rare Dis. 2009, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukacs, Z.; Keil, A.; Kohlschutter, A.; Beck, M.; Mengel, E. The ratio of alpha-galactosidase to beta-glucuronidase activities in dried blood for the identification of female Fabry disease patients. J. Inherit. Metab. Dis. 2005, 28, 803–805. [Google Scholar] [CrossRef]
- Zhang, X.K.; Elbin, C.S.; Chuang, W.-L.; Cooper, S.K.; Marashio, C.A.; Beauregard, C.; Keutzer, J.M. Multiplex Enzyme Assay Screening of Dried Blood Spots for Lysosomal Storage Disorders by Using Tandem Mass Spectrometry. Clin. Chem. 2008, 54, 1725–1728. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.; Lovejoy, M.; Brooks, D.; Harkin, M.L.; Hopwood, J.J.; Meikle, P. Immunoquantification of α-Galactosidase: Evaluation for the Diagnosis of Fabry Disease. Clin. Chem. 2004, 50, 1979–1985. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Scott, C.R.; Chamoles, N.A.; Ghavami, A.; Pinto, B.M.; Turecek, F.; Gelb, M.H. Direct Multiplex Assay of Lysosomal Enzymes in Dried Blood Spots for Newborn Screening. Clin. Chem. 2004, 50, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Linthorst, G.E.; Vedder, A.C.; Aerts, J.; Hollak, C.E. Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clin. Chim. Acta 2005, 353, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Ranieri, E.; Simonsen, H.; Rozaklis, T.; Ramsay, S.L.; Whitfield, P.D.; Fuller, M.; Christensen, E.; Skovby, F.; Hopwood, J.J. Newborn Screening for Lysosomal Storage Disorders: Clinical Evaluation of a Two-Tier Strategy. Pediatrics 2004, 114, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Berna, L.; Asfaw, B.; Conzelmann, E.; Černý, B.; Ledvinova, J. Determination of Urinary Sulfatides and Other Lipids by Combination of Reversed-Phase and Thin-Layer Chromatographies. Anal. Biochem. 1999, 269, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Vance, D.E.; Sweeley, C.C. Quantitative determination of the neutral glycosyl ceramides in human blood. J. Lipid Res. 1967, 8, 621–630. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, Y.; Wang, P. Separation and Determination of Phospholipids in Biological Samples by High-Performance Liquid Chromatography. J. Chromatogr. Sci. 2003, 41, 142–144. [Google Scholar] [CrossRef] [Green Version]
- Thurberg, B.L.; Fallon, J.T.; Mitchell, R.; Aretz, T.; Gordon, R.E.; O’Callaghan, M.W. Cardiac microvascular pathology in Fabry disease: Evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation 2009, 119, 2561–2567. [Google Scholar] [CrossRef] [Green Version]
- Monda, E.; Limongelli, G. Is There a Role for Genetic Testing in Patients With Myocarditis? Circ. Genom. Precis. Med. 2022. In Press. [Google Scholar]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020, 28, 1081–1090. [Google Scholar] [CrossRef]
- Monda, E.; Rubino, M.; Lioncino, M.; Di Fraia, F.; Pacileo, R.; Verrillo, F.; Cirillo, A.; Caiazza, M.; Fusco, A.; Esposito, A.; et al. Hypertrophic Cardiomyopathy in Children: Pathophysiology, Diagnosis, and Treatment of Non-sarcomeric Causes. Front. Pediatr. 2021, 9, 632293. [Google Scholar] [CrossRef]
- Monda, E.; Sarubbi, B.; Russo, M.G.; Caiazza, M.; Mazzaccara, C.; Magrelli, J.; Rubino, M.; Esposito, A.; Perna, A.; Passariello, A.; et al. Unexplained sudden cardiac arrest in children: Clinical and genetic characteristics of survivors. Eur. J. Prev. Cardiol. 2020, 28, 1134–1137. [Google Scholar] [CrossRef]
- Eng, C.M.; Resnick-Silverman, L.A.; Niehaus, D.J.; Astrin, K.H.; Desnick, R.J. Nature and frequency of mutations in the al-pha-galactosidase A gene that cause Fabry disease. Am. J. Hum. Genet. 1993, 53, 1186–1197. [Google Scholar]
- Mogensen, J.; van Tintelen, J.P.; Fokstuen, S.; Elliott, P.; van Langen, I.M.; Meder, B.; Richard, P.; Syrris, P.; Caforio, A.L.; Adler, Y.; et al. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: A viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur. Heart J. 2015, 36, 1367–1370. [Google Scholar] [CrossRef]
- Smid, B.E.; van der Tol, L.; Cecchi, F.; Elliott, P.M.; Hughes, D.A.; Linthorst, G.E.; Timmermans, J.; Weidemann, F.; West, M.L.; Biegstraaten, M.; et al. Uncertain diagnosis of Fabry disease: Consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int. J. Cardiol. 2014, 177, 400–408. [Google Scholar] [CrossRef]
- Germain, D.P.; Fouilhoux, A.; Decramer, S.; Tardieu, M.; Pillet, P.; Fila, M.; Rivera, S.; Deschênes, G.; Lacombe, D. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin. Genet. 2019, 96, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Nunes, J.P.; Costa, O.; Faria Mdo, S.; Almeida, P.B.; Lacerda, L. Cardiac Fabry’s disease: An unusual cause of left ventricular hypertrophy. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 630–633. [Google Scholar] [CrossRef]
- Ishii, S.; Nakao, S.; Minamikawa-Tachino, R.; Desnick, R.J.; Fan, J.-Q. Alternative Splicing in the α-Galactosidase A Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am. J. Hum. Genet. 2002, 70, 994–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornreich, R.; Bishop, D.F.; Desnick, R.J. Alpha-galactosidase A gene rearrangements causing Fabry disease. Identification of short direct repeats at breakpoints in an Alu-rich gene. J. Biol. Chem. 1990, 265, 9319–9326. [Google Scholar] [CrossRef]
- Shabbeer, J.; Robinson, M.; Desnick, R.J. Detection of α-galactosidase a mutations causing Fabry disease by denaturing high performance liquid chromatography. Hum. Mutat. 2005, 25, 299–305. [Google Scholar] [CrossRef]
- Seydelmann, N.; Liu, D.; Krämer, J.; Drechsler, C.; Hu, K.; Nordbeck, P.; Schneider, A.; Störk, S.; Bijnens, B.; Ertl, G.; et al. High-Sensitivity Troponin: A Clinical Blood Biomarker for Staging Cardiomyopathy in Fabry Disease. J. Am. Heart Assoc. 2016, 5, e002839. [Google Scholar] [CrossRef] [Green Version]
- Torralba-Cabeza, M.Á.; Olivera, S.; Hughes, D.A.; Pastores, G.M.; Mateo, R.N.; Pérez-Calvo, J.I. Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Mol. Genet. Metab. 2011, 104, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, A.; Beuschlein, F.; Sivasubramaniam, V.; Kasper, D.; Warnock, D.G. Lyso-Gb3 associates with adverse long-term outcome in patients with Fabry diseas. J. Med. Genet. 2022, 59, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Hughes, D.A.; Winchester, B. Toward a consensus in the laboratory diagnostics of Fabry disease—Recommendations of a European expert group. J. Inherit. Metab. Dis. 2011, 34, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Stiles, A.R.; Zhang, H.; Dai, J.; McCaw, P.; Beasley, J.; Rehder, C.; Koeberl, D.D.; McDonald, M.; Bali, D.S.; Young, S.P. A comprehensive testing algorithm for the diagnosis of Fabry disease in males and females. Mol. Genet. Metab. 2020, 130, 209–214, Mitochondrial microRNAs are dysregulated in patients with Fabry Disease. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Kasper, D.; Pagliardini, V.; Biamino, E.; Giachero, S.; Porta, F. Metabolic progression to clinical phenotype in classic Fabry disease. Ital. J. Pediatr. 2017, 43, 1. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, J.; Fiordelisi, A.; Sorriento, D.; Cerasuolo, F.; Buonaiuto, A.; Avvisato, R.; Pisani, A.; Varzideh, F.; Riccio, E.; Santulli, G.; et al. Mitochondrial microRNAs are dysregulated in patients with Fabry Disease. J. Pharmacol. Exp. Ther. 2022. In Press. [Google Scholar] [CrossRef]
- Fernández-Pereira, C.; Millán-Tejado, B.S.; Gallardo-Gómez, M.; Pérez-Márquez, T.; Alves-Villar, M.; Melcón-Crespo, C.; Fernández-Martín, J.; Ortolano, S. Therapeutic Approaches in Lysosomal Storage Diseases. Biomolecules 2021, 11, 1775. [Google Scholar] [CrossRef]
- Hughes, D.A.; Elliott, P.M.; Shah, J.; Zuckerman, J.; Coghlan, G.; Brookes, J.; Mehta, A.B. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: A randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 2008, 94, 153–158. [Google Scholar] [CrossRef]
- Banikazemi, M.; Bultas, J.; Waldek, S.; Wilcox, W.R.; Whitley, C.B.; McDonald, M.; Finkel, R.; Packman, S.; Bichet, D.G.; Warnock, D.G.; et al. Agalsidase-beta therapy for advanced Fabry disease: A randomized trial. Ann. Intern. Med. 2007, 146, 77–86. [Google Scholar] [CrossRef]
- Germain, D.P.; Elliott, P.M.; Falissard, B.; Fomin, V.V.; Hilz, M.J.; Jovanovic, A.; Kantola, I.; Linhart, A.; Mignani, R.; Namdar, M.; et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol. Genet. Metab. Rep. 2019, 19, 100454. [Google Scholar] [CrossRef]
- Germain, D.P.; Charrow, J.; Desnick, R.J.; Guffon, N.; Kempf, J.; Lachmann, R.; Lemay, R.; Linthorst, G.E.; Packman, S.; Scott, C.R.; et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J. Med. Genet. 2015, 52, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Bénichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Fan, J.-Q. Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: In vitro and preclinical studies. Int. J. Clin. Pharmacol. Ther. 2009, 47, S111–S117. [Google Scholar] [PubMed]
- Patel, V.; O’Mahony, C.; Hughes, D.; Rahman, M.S.; Coats, C.; Murphy, E.; Lachmann, R.; Mehta, A.; Elliott, P.M. Clinical and genetic predictors of major cardiac events in patients with Anderson–Fabry Disease. Heart 2015, 101, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Limongelli, G. A Roadmap to Predict Adverse Outcome in Fabry Disease. J. Am. Coll. Cardiol. 2022, 80, 995–997. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amodio, F.; Caiazza, M.; Monda, E.; Rubino, M.; Capodicasa, L.; Chiosi, F.; Simonelli, V.; Dongiglio, F.; Fimiani, F.; Pepe, N.; et al. An Overview of Molecular Mechanisms in Fabry Disease. Biomolecules 2022, 12, 1460. https://doi.org/10.3390/biom12101460
Amodio F, Caiazza M, Monda E, Rubino M, Capodicasa L, Chiosi F, Simonelli V, Dongiglio F, Fimiani F, Pepe N, et al. An Overview of Molecular Mechanisms in Fabry Disease. Biomolecules. 2022; 12(10):1460. https://doi.org/10.3390/biom12101460
Chicago/Turabian StyleAmodio, Federica, Martina Caiazza, Emanuele Monda, Marta Rubino, Laura Capodicasa, Flavia Chiosi, Vincenzo Simonelli, Francesca Dongiglio, Fabio Fimiani, Nicola Pepe, and et al. 2022. "An Overview of Molecular Mechanisms in Fabry Disease" Biomolecules 12, no. 10: 1460. https://doi.org/10.3390/biom12101460