
Exploring Computational Data Amplification and Imputation for the Discovery of Type 1 Diabetes (T1D) Biomarkers from Limited Human Datasets

 and
and
Abstract
:1. Introduction

2. Materials and Methods
2.1. Sample Collection
2.2. Multi-Omics Analyses
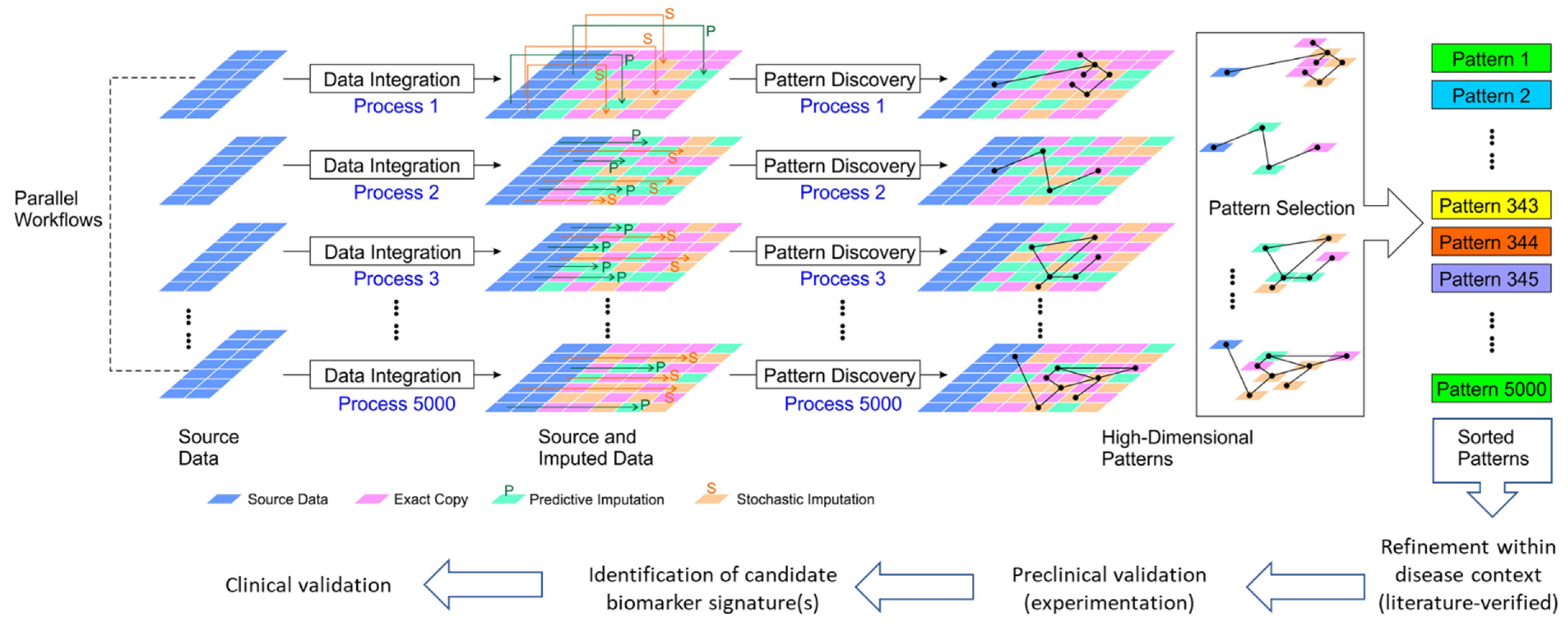
2.3. Algorithms for Multiple Imputation/Amplification of Multi-Omics
2.4. Data Analysis
3. Results
3.1. Comparative Enrichment Analyses for Canonical Pathways
3.2. Comparative Enrichment Analyses for Diseases and Functions
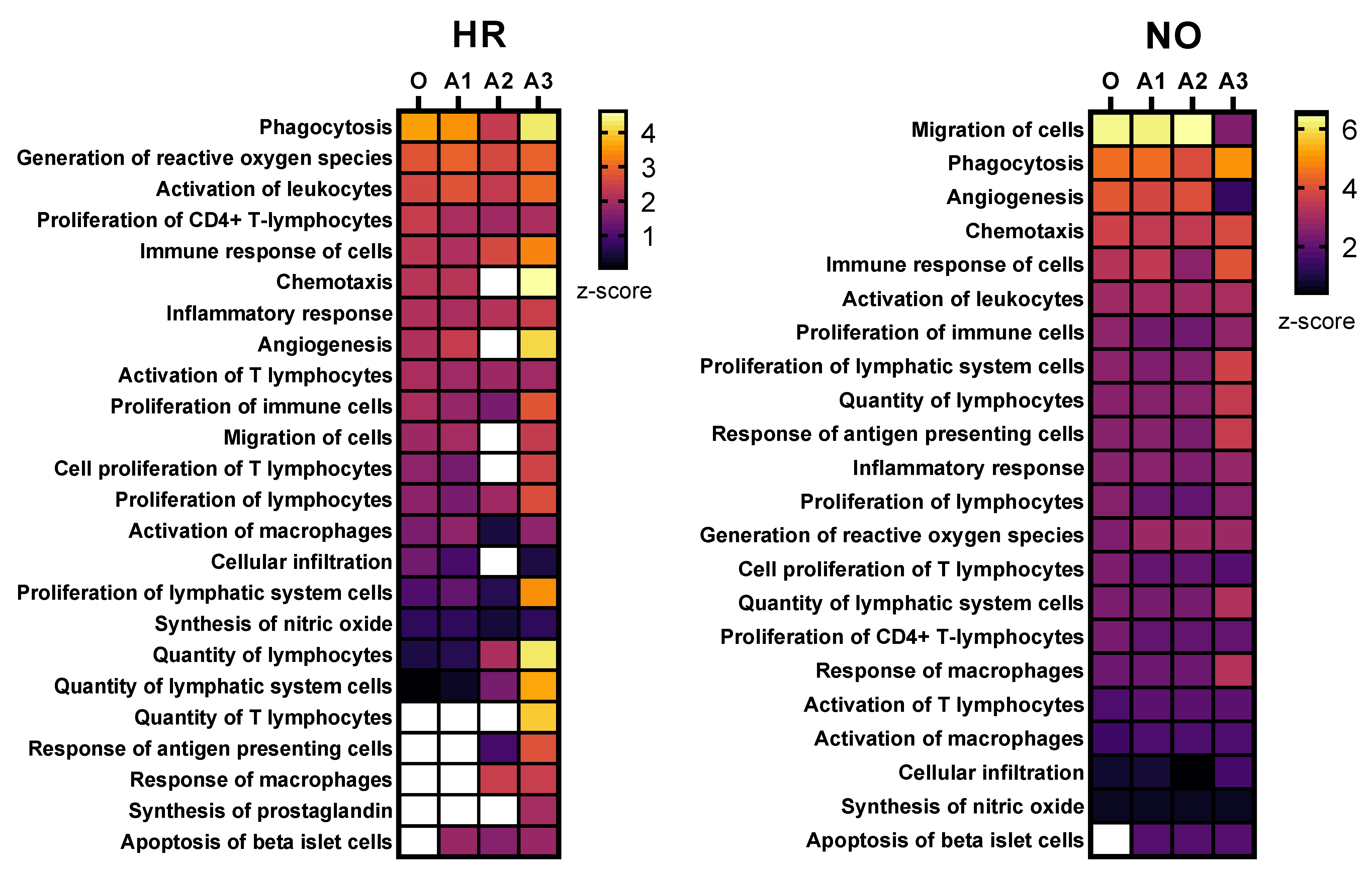
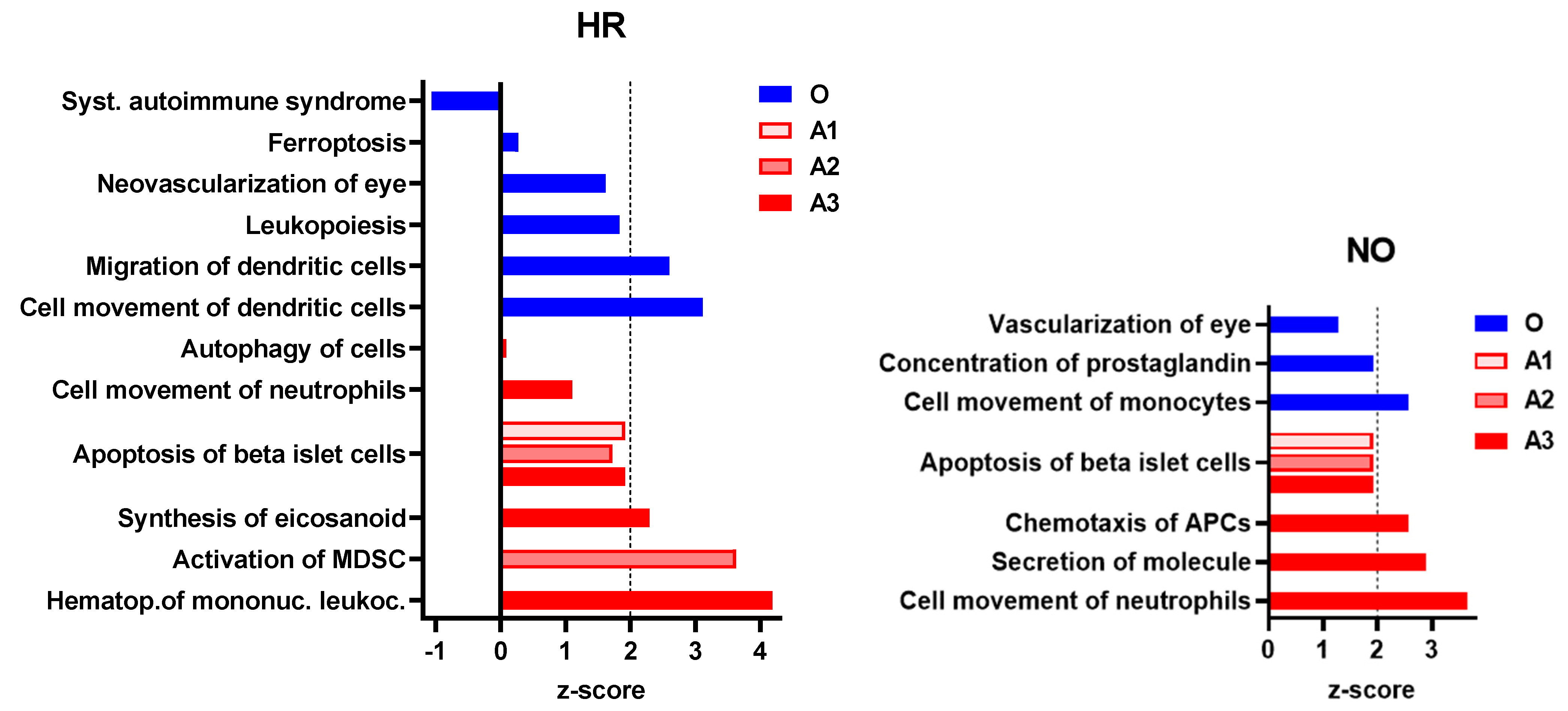
3.2.1. Enhanced Prediction of T1D-Relevant Immune Functions in Multi-Omics Datasets Independently
3.2.2. Enhanced Prediction of Immune and Inflammatory Diseases and Functions in Amplified Integrated Multi-Omics Datasets
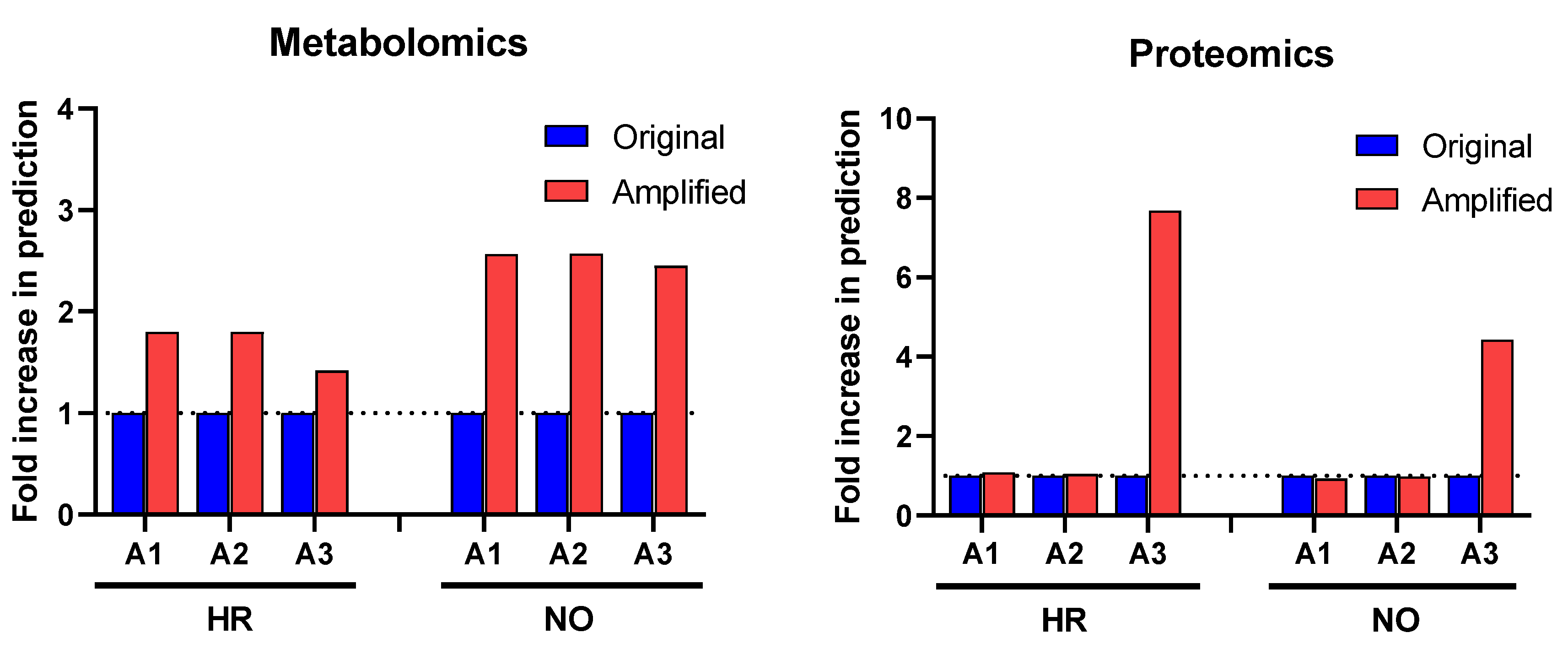
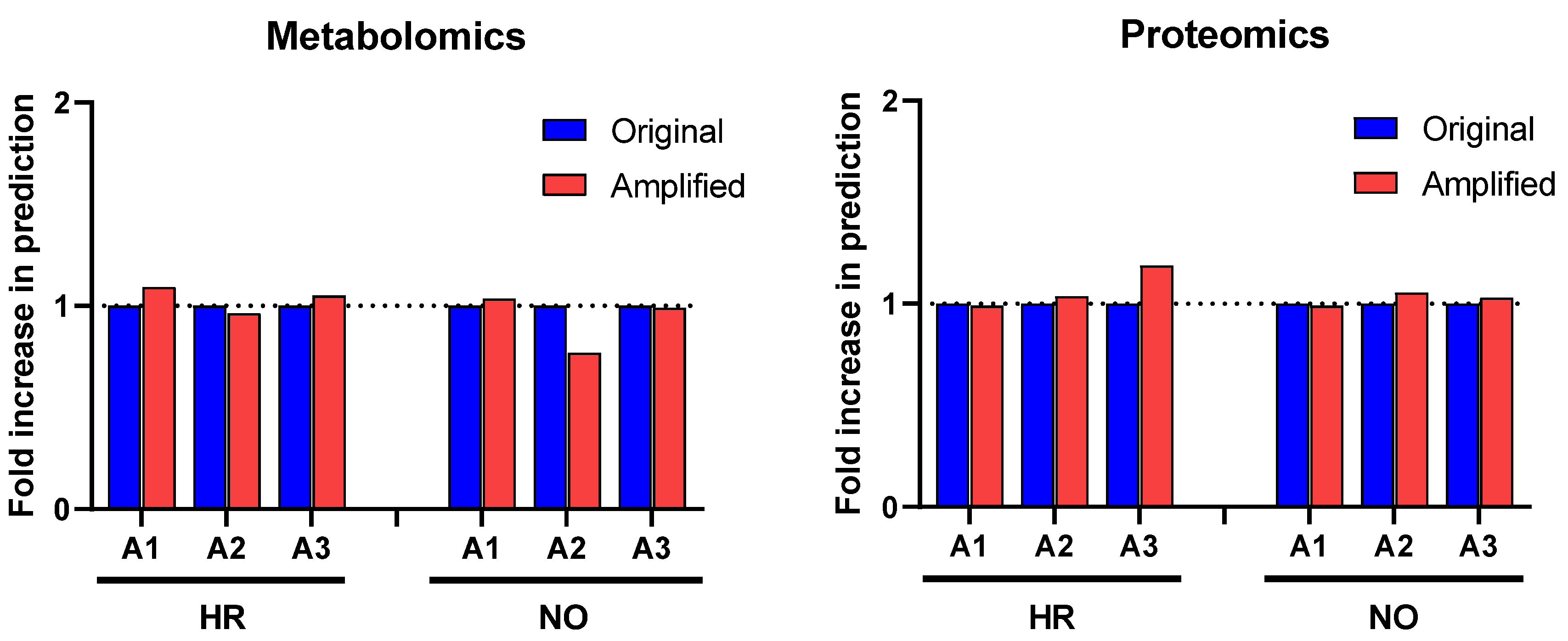
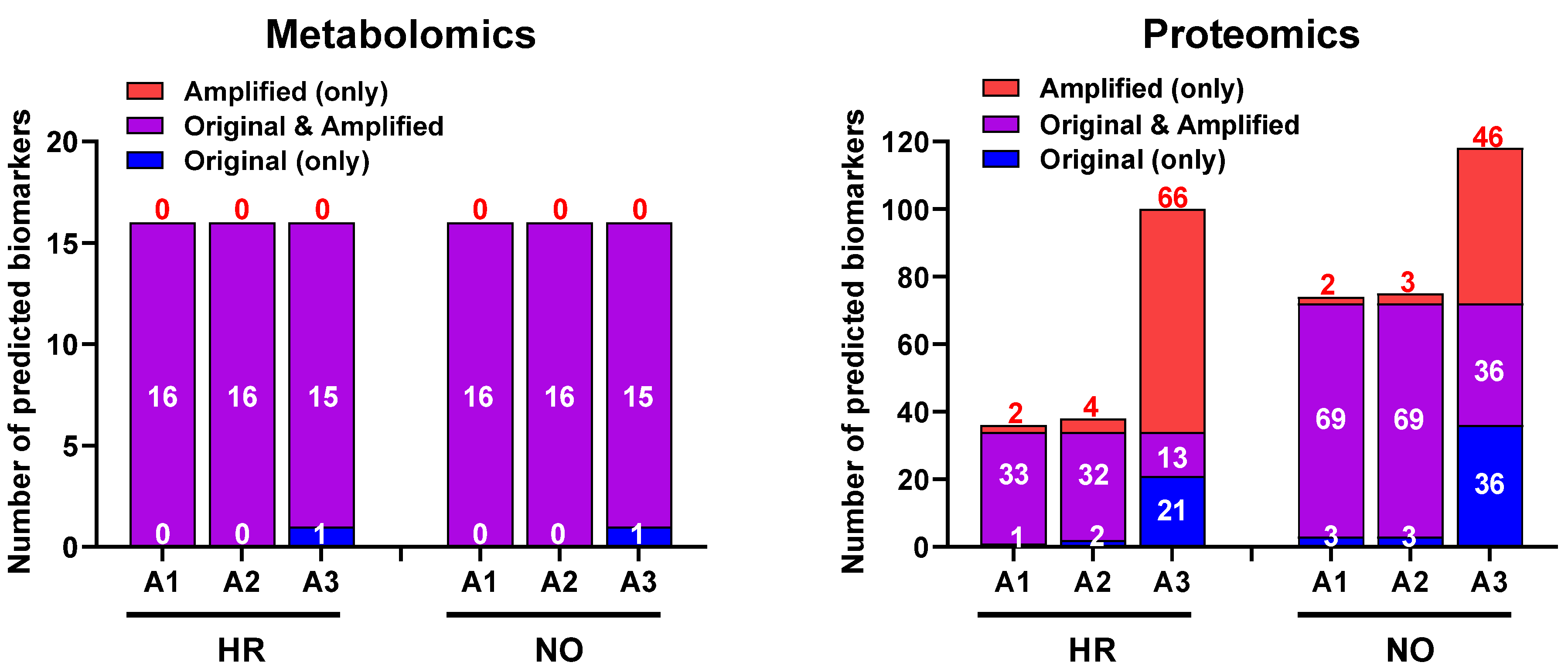
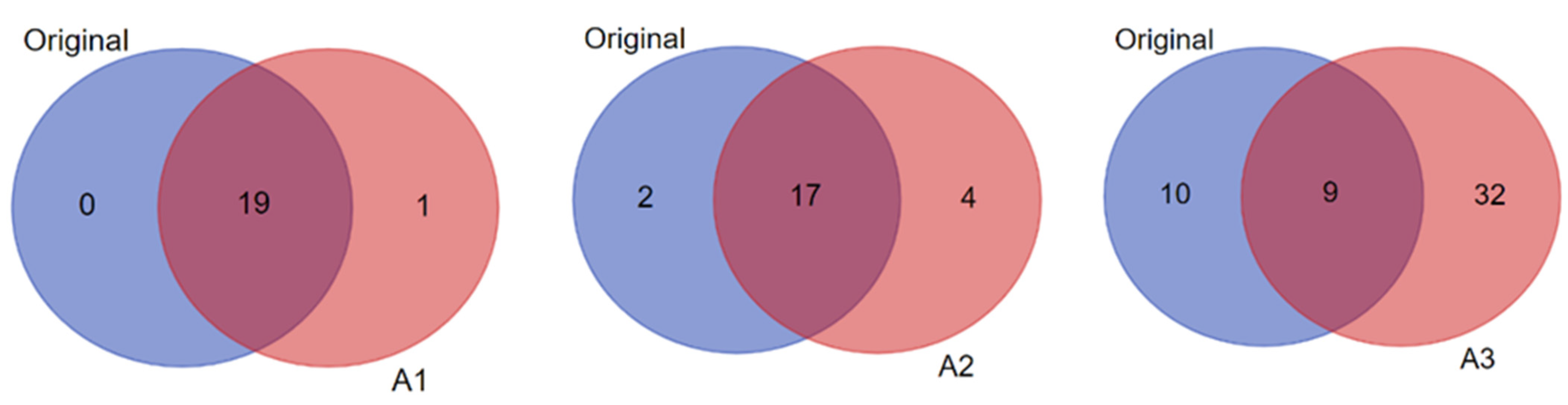
3.3. Biomarker Prediction in the Amplified Versus Original Proteomics and Metabolomics Datasets
4. Discussion
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- DiMeglio, L.A.; Evans-Molina, C.; A Oram, R. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Warshauer, J.T.; Bluestone, J.A.; Anderson, M.S. New Frontiers in the Treatment of Type 1 Diabetes. Cell Metab. 2020, 31, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, S.J.; Levin, D.; Looker, H.C.; Lindsay, R.; Wild, S.H.; Joss, N.; Leese, G.; Leslie, P.; McCrimmon, R.; Metcalfe, W.; et al. Estimated Life Expectancy in a Scottish Cohort With Type 1 Diabetes, 2008–2010. JAMA J. Am. Med. Assoc. 2015, 313, 37–44. [Google Scholar] [CrossRef]
- Rawshani, A.; Sattar, N.; Franzén, S.; Rawshani, A.; Hattersley, A.T.; Svensson, A.-M.; Eliasson, B.; Gudbjörnsdottir, S. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: A nationwide, register-based cohort study. Lancet 2018, 392, 477–486. [Google Scholar] [CrossRef] [Green Version]
- von Scholten, B.J.; Kreiner, F.F.; Gough, S.C.L.; von Herrath, M. Current and future therapies for type 1 diabetes. Diabetologia 2021, 64, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Lahesmaa, R.; Bonifacio, E.; Achenbach, P.; Tree, T. Immunological biomarkers for the development and progression of type 1 diabetes. Diabetologia 2018, 61, 2252–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, E.K.; Evans-Molina, C.; Tersey, S.A.; Eizirik, D.L.; Mirmira, R.G. Biomarkers of islet beta cell stress and death in type 1 diabetes. Diabetologia 2018, 61, 2259–2265. [Google Scholar] [CrossRef] [Green Version]
- Canzler, S.; Schor, J.; Busch, W.; Schubert, K.; Rolle-Kampczyk, U.E.; Seitz, H.; Kamp, H.; von Bergen, M.; Buesen, R.; Hackermüller, J. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 2020, 94, 371–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-X.; Wheelock, C.E.; Sköld, C.M.; Wheelock, A.M. Integration of multi-omics datasets enables molecular classification of COPD. Eur. Respir. J. 2018, 51, 1701930. [Google Scholar] [CrossRef]
- Alcazar, O.; Hernandez, L.; Nakayasu, E.; Nicora, C.; Ansong, C.; Muehlbauer, M.; Bain, J.; Myer, C.; Bhattacharya, S.; Buchwald, P.; et al. Parallel Multi-Omics in High-Risk Subjects for the Identification of Integrated Biomarker Signatures of Type 1 Diabetes. Biomolecules 2021, 11, 383. [Google Scholar] [CrossRef] [PubMed]
- Golub, G.H.; Loan, C.F.V. Matrix Computations, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 1996. [Google Scholar]
- Trefethen, L.N.; Bau, D., III. Numerical Linear Algebra; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 1997. [Google Scholar]
- Jakobsen, J.C.; Gluud, C.; Wetterslev, J.; Winkel, P.; Jakobsen, J.C.; Gluud, C.; Wetterslev, J.; Winkel, P. When and how should multiple imputation be used for handling missing data in randomised clinical trials—A practical guide with flowcharts. BMC Med. Res. Methodol. 2017, 17, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, W.; Sun, Y.; Milanovic, S.; Kon, M.; Castrillon-Candas, J.E. Multilevel Stochastic Optimization for Imputation in Massive Medical Data Records. arXiv 2021, arXiv:211009680. [Google Scholar]
- Miao, X.; Wu, Y.; Chen, L.; Gao, Y.; Wang, J.; Yin, J. Efficient and effective data imputation with influence functions. Proc. VLDB Endow. 2021, 15, 624–632. [Google Scholar] [CrossRef]
- Si, Y.A.; Heeringa, S.; Johnson, D.; Little, R.; Liu, W.; Pfeffer, F. Multiple Imputation with Massive Data: An Application to the Panel Study of Income Dynamics. arXiv 2021, arXiv:2007.03016. [Google Scholar] [CrossRef]
- Zhou, Z.; Mo, J.; Shi, Y. Data imputation and dimensionality reduction using deep learning in industrial data. In Proceedings of the 3rd IEEE International Conference on Computer and Communications (ICCC), Chengdu, China, 13–16 December 2017; pp. 2329–2333. [Google Scholar]
- Rubin, D.B. Multiple Imputation after 18+ Years. J. Am. Stat. Assoc. 1996, 91, 473–489. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Lin, W.-C.; Tsai, C.-F. Missing value imputation: A review and analysis of the literature (2006–2017). Artif. Intell. Rev. 2020, 53, 1487–1509. [Google Scholar] [CrossRef]
- Lohmann, T.; Laue, S.; Nietzschmann, U.; Kapellen, T.; Lehmann, I.; Schroeder, S.; Paschke, R.; Kiess, W. Reduced Expression of Th1-Associated Chemokine Receptors on Peripheral Blood Lymphocytes at Diagnosis of Type 1 Diabetes. Diabetes 2002, 51, 2474–2480. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Lu, Y.; Wang, B.; Jiao, P.; Ma, J. Analysis of immune cell components and immune-related gene expression profiles in peripheral blood of patients with type 1 diabetes mellitus. J. Transl. Med. 2021, 19, 319. [Google Scholar] [CrossRef]
- Ajjan, R.A.; Schroeder, V. Role of complement in diabetes. Mol. Immunol. 2019, 114, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.; Wasserfall, C.; Croker, B.; Campbell-Thompson, M.; Pugliese, A.; Atkinson, M.; Schatz, D. Increased Complement Activation in Human Type 1 Diabetes Pancreata. Diabetes Care 2013, 36, 3815–3817. [Google Scholar] [CrossRef] [Green Version]
- Leng, Q.; Nie, Y.; Zou, Y.; Chen, J. Elevated CXCL12 expression in the bone marrow of NOD mice is associated with altered T cell and stem cell trafficking and diabetes development. BMC Immunol. 2008, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Yuan, J.; Duncanson, S.; Hibert, M.L.; Kodish, B.C.; Mylavaganam, G.; Maker, M.; Li, H.; Sremac, M.; Santosuosso, M.; et al. Alginate Encapsulant Incorporating CXCL12 Supports Long-Term Allo- and Xenoislet Transplantation Without Systemic Immune Suppression. Am. J. Transplant. 2015, 15, 618–627. [Google Scholar] [CrossRef]
- Marée, A.F.; Kublik, R.; Finegood, D.T.; Edelstein-Keshet, L. Modelling the onset of Type 1 diabetes: Can impaired macrophage phagocytosis make the difference between health and disease? Philos. Trans. R. Soc. London. Ser. A Math. Phys. Eng. Sci. 2006, 364, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kaifu, T.; Sugahara-Tobinai, A.; Nakamura, A.; Miyazaki, J.-I.; Takai, T. Activating Fcγ Receptors Participate in the Development of Autoimmune Diabetes in NOD Mice. J. Immunol. 2007, 179, 764–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryba-Stanisławowska, M.; Rybarczyk-Kapturska, K.; Mysliwiec, M.; Myśliwska, J. Elevated Levels of Serum IL-12 and IL-18 are Associated with Lower Frequencies of CD4+CD25 high FOXP3+ Regulatory T cells in Young Patients with Type 1 Diabetes. Inflammation 2014, 37, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Adorini, L.; Magram, J.; Trembeau, S. The role of endogenous IL 12 in the induction of Th1-cell-mediated autoimmune diseases. Res. Immunol. 1995, 146, 645–651. [Google Scholar] [CrossRef]
- Kuczyński, S.; Winiarska, H.; Abramczyk, M.; Szczawińska, K.; Wierusz-Wysocka, B.; Dworacka, M. IL-15 is elevated in serum patients with type 1 diabetes mellitus. Diabetes Res. Clin. Pract. 2005, 69, 231–236. [Google Scholar] [CrossRef]
- Ramanathan, S.; Dubois, S.; Chen, X.-L.; Leblanc, C.; Ohashi, P.S.; Ilangumaran, S. Exposure to IL-15 and IL-21 Enables Autoreactive CD8 T Cells To Respond to Weak Antigens and Cause Disease in a Mouse Model of Autoimmune Diabetes. J. Immunol. 2011, 186, 5131–5141. [Google Scholar] [CrossRef] [Green Version]
- Monti, P.; Bonifacio, E. Interleukin-7 and Type 1 Diabetes. Curr. Diabetes Rep. 2014, 14, 518. [Google Scholar] [CrossRef] [PubMed]
- Penaranda, C.; Kuswanto, W.; Hofmann, J.; Kenefeck, R.; Narendran, P.; Walker, L.S.K.; Bluestone, J.A.; Abbas, A.K.; Dooms, H. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12668–12673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albiero, M.; Poncina, N.; Ciciliot, S.; Cappellari, R.; Menegazzo, L.; Ferraro, F.; Bolego, C.; Cignarella, A.; Avogaro, A.; Fadini, G.P. Bone Marrow Macrophages Contribute to Diabetic Stem Cell Mobilopathy by Producing Oncostatin M. Diabetes 2015, 64, 2957–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prashanth, G.; Vastrad, B.; Tengli, A.; Vastrad, C.; Kotturshetti, I. Identification of hub genes related to the progression of type 1 diabetes by computational analysis. BMC Endocr. Disord. 2021, 21, 61. [Google Scholar] [CrossRef]
- Kröncke, K.-D.; Kolb-Bachofen, V.; Berschick, B.; Burkart, V.; Kolb, H. Activated macrophages kill pancreatic syngeneic islet cells via arginine-dependent nitric oxide generation. Biochem. Biophys. Res. Commun. 1991, 175, 752–758. [Google Scholar] [CrossRef]
- Ysmail-Dahlouk, L.; Nouari, W.; Aribi, M. 1,25-dihydroxyvitamin D 3 down-modulates the production of proinflammatory cytokines and nitric oxide and enhances the phosphorylation of monocyte-expressed STAT6 at the recent-onset type 1 diabetes. Immunol. Lett. 2016, 179, 122–130. [Google Scholar] [CrossRef]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef] [Green Version]
- Tratsiakovich, Y.; Kiss, A.; Gonon, A.T.; Yang, J.; Sjöquist, P.-O.; Pernow, J. Inhibition of Rho kinase protects from ischaemia–reperfusion injury via regulation of arginase activity and nitric oxide synthase in type 1 diabetes. Diabetes Vasc. Dis. Res. 2017, 14, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating sphingolipid biomarkers in models of type 1 diabetes. J. Lipid Res. 2011, 52, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, L.F.; Buchwald, P.; Abdulreda, M.H. Effect of Arginase-1 Inhibition on the Incidence of Autoimmune Diabetes in NOD Mice. Curr. Res. Diabetes Obes. J. 2018, 5, 555661. [Google Scholar]
- Flodström, M.; Chen, M.-C.; Smismans, A.; Schuit, F.; Pipeleers, D.G.; Eizirik, D.L. Interleukin 1β increases arginine accumulation and activates the citrulline–no cycle in rat pancreatic βcells. Cytokine 1999, 11, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Hakola, L.; Erlund, I.; Cuthbertson, D.; Miettinen, M.E.; Autio, R.; Nucci, A.M.; Härkönen, T.; Honkanen, J.; Vaarala, O.; Hyöty, H.; et al. Serum fatty acids and risk of developing islet autoimmunity: A nested case–control study within the TRIGR birth cohort. Pediatr. Diabetes 2021, 22, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Cort, L.; Habib, M.; Eberwine, R.A.; Hessner, M.J.; Mordes, J.P.; Blankenhorn, E.P. Diubiquitin (Ubd) is a susceptibility gene for virus-triggered autoimmune diabetes in rats. Genes Immun. 2014, 15, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, K.; Yoshitomi, H.; Furu, M.; Ishikawa, M.; Shibuya, H.; Ito, H.; Matsuda, S. MicroRNA-451 Down-Regulates Neutrophil Chemotaxis via p38 MAPK. Arthritis Rheumatol. 2014, 66, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taurino, F.; Stanca, E.; Vonghia, L.; Siculella, L.; Sardanelli, A.M.; Papa, S.; Zanotti, F.; Gnoni, A. Short-term Type-1 diabetes differentially modulates 14-3-3 proteins in rat brain and liver. Eur. J. Clin. Investig. 2014, 44, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.; Chernoff, J. Group I p21-activated kinases: Emerging roles in immune function and viral pathogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 13–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; He, K.; Li, C.; Ni, Y.; Li, M.; Chen, L.; Hou, M.; Zhou, Z.; Xu, Z.; Ji, M. P21 activated kinase-1 (PAK1) in macrophages is required for promotion of Th17 cell response during helminth infection. J. Cell. Mol. Med. 2020, 24, 14325–14338. [Google Scholar] [CrossRef]
- Nelson, A.J.; Stephenson, D.J.; Bone, R.N.; Cardona, C.; Park, M.A.; Tusing, Y.G.; Lei, X.; Kokotos, G.; Graves, C.L.; Mathews, C.E.; et al. Lipid mediators and biomarkers associated with type 1 diabetes development. JCI Insight 2020, 5, 138034. [Google Scholar] [CrossRef]
- Deeg, M.A.; Bowen, R.F.; Williams, M.D.; Olson, L.K.; Kirk, E.A.; LeBoeuf, R.C. Increased expression of GPI-specific phospholipase D in mouse models of type 1 diabetes. Am. J. Physiol. Metab. 2001, 281, E147–E154. [Google Scholar] [CrossRef]
- Jayasimhan, A.; Mansour, K.P.; Slattery, R.M. Advances in our understanding of the pathophysiology of Type 1 diabetes: Lessons from the NOD mouse. Clin. Sci. 2014, 126, 1–18. [Google Scholar] [CrossRef]
- Wållberg, M.; Cooke, A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013, 34, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.H.; Jenkins, A.J.; Majewski, H. Elevated plasma prostaglandins and acetylated histone in monocytes in Type 1 diabetes patients. Diabet. Med. 2009, 26, 182–186. [Google Scholar] [CrossRef]
- Hänninen, A.; Jalkanen, S.; Salmi, M.; Toikkanen, S.; Nikolakaros, G.; Simell, O. Macrophages, T cell receptor usage, and endothelial cell activation in the pancreas at the onset of insulin-dependent diabetes mellitus. J. Clin. Investig. 1992, 90, 1901–1910. [Google Scholar] [CrossRef] [Green Version]
- Hwa, R.; Resnik, P.; Weinberg, A.; Cabezas, C.; Kolak, O. Bootstrapping parsers via syntactic projection across parallel texts. Nat. Lang. Eng. 2005, 11, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Jin, Z.; Hu, Z.; Vechtomova, O.; Mihalcea, R. Deep Learning for Text Style Transfer: A Survey. Comput. Linguist. 2022, 48, 155–205. [Google Scholar] [CrossRef]
- Savitski, M.M.; Mathieson, T.; Zinn, N.; Sweetman, G.; Doce, C.; Becher, I.; Pachl, F.; Kuster, B.; Bantscheff, M. Measuring and Managing Ratio Compression for Accurate iTRAQ/TMT Quantification. J. Proteome Res. 2013, 12, 3586–3598. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Nishiyama, S.; Nakamura, T.; Saku, K.; Maung, K.K.; Matsuda, I. Predominance of large low density lipoprotein particles and lower fractional esterification rate of cholesterol in high density lipoprotein in children with insulin-dependent diabetes mellitus. Eur. J. Pediatr. 1998, 157, 276–281. [Google Scholar] [CrossRef]
- Soggiu, A.; Piras, C.; Bonizzi, L.; Hussein, H.A.; Pisanu, S.; Roncada, P. A discovery-phase urine proteomics investigation in type 1 diabetes. Geol. Rundsch. 2012, 49, 453–464. [Google Scholar] [CrossRef]
- Assayag-Asherie, N.; Sever, D.; Bogdani, M.; Johnson, P.; Weiss, T.; Ginzberg, A.; Perles, S.; Weiss, L.; Sebban, L.E.; Turley, E.A.; et al. Can CD44 Be a Mediator of Cell Destruction? The Challenge of Type 1 Diabetes. PLoS ONE 2015, 10, e0143589. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-K.; Tso, T.; Snook, J.; Huang, Y.-S.; Lozano, R.; Zipf, W. Cholesteryl ester transfer and cholesterol esterification in type 1 diabetes: Relationships with plasma glucose. Geol. Rundsch. 2001, 38, 37–42. [Google Scholar] [CrossRef]
- Monteiro, M.B.; Pelaes, T.S.; Santos-Bezerra, D.P.; Thieme, K.; Lerario, A.M.; Oba-Shinjo, S.M.; Machado, U.F.; Passarelli, M.; Marie, S.K.N.; Correa-Giannella, M.L. Urinary Sediment Transcriptomic and Longitudinal Data to Investigate Renal Function Decline in Type 1 Diabetes. Front. Endocrinol. 2020, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.R.; Wasserfall, C.H.; McGrail, S.M.; Posgai, A.L.; Bacher, R.; Muir, A.; Haller, M.J.; Schatz, D.A.; Wesley, J.D.; von Herrath, M.; et al. Insulin-Like Growth Factor Dysregulation Both Preceding and Following Type 1 Diabetes Diagnosis. Diabetes 2020, 69, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Zhi, W.; Sharma, A.; Purohit, S.; Miller, E.; Bode, B.; Anderson, S.W.; Reed, J.C.; Steed, R.D.; Steed, L.; Hopkins, D.; et al. Discovery and Validation of Serum Protein Changes in Type 1 Diabetes Patients Using High Throughput Two Dimensional Liquid Chromatography-Mass Spectrometry and Immunoassays. Mol. Cell. Proteom. 2011, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planas, R.; Carrillo, J.; Sanchez, A.; de Villa, M.C.R.; Nuñez, F.; Verdaguer, J.; James, R.F.L.; Pujol-Borrell, R.; Vives-Pi, M. Gene expression profiles for the human pancreas and purified islets in Type 1 diabetes: New findings at clinical onset and in long-standing diabetes. Clin. Exp. Immunol. 2010, 159, 23–44. [Google Scholar] [CrossRef]
- Johansson, F.; Kramer, F.; Barnhart, S.; Kanter, J.E.; Vaisar, T.; Merrill, R.D.; Geng, L.; Oka, K.; Chan, L.; Chait, A.; et al. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 2082–2087. [Google Scholar] [CrossRef] [Green Version]
- Bylander, J.E.; Ahmed, F.; Conley, S.M.; Mwiza, J.-M.; Ongeri, E.M. Meprin Metalloprotease Deficiency Associated with Higher Mortality Rates and More Severe Diabetic Kidney Injury in Mice with STZ-Induced Type 1 Diabetes. J. Diabetes Res. 2017, 2017, 9035038. [Google Scholar] [CrossRef] [Green Version]
- Shiau, M.Y.; Tsai, S.T.; Tsai, K.J.; Haung, M.L.; Hsu, Y.T.; Chang, Y.H. Increased circulatory MMP-2 and MMP-9 levels and activities in patients with type 1 diabetes mellitus. Mt. Sinai J. Med. 2006, 73, 1024–1028. [Google Scholar]
- Åkerman, L.; Ludvigsson, J.; Casas, R. Low C-peptide levels and decreased expression of TNF and CD45 in children with high risk of type 1 diabetes. Clin. Immunol. 2013, 148, 4–15. [Google Scholar] [CrossRef]
- Mora, C.; Grewal, I.; Wong, F.S.; Flavell, R.A. Role of L-selectin in the development of autoimmune diabetes in non-obese diabetic mice. Int. Immunol. 2004, 16, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Triñanes, J.; Salido, E.; Fernández, J.; Rufino, M.; González-Posada, J.M.; Torres, A.; Hernández, D. Type 1 Diabetes Increases the Expression of Proinflammatory Cytokines and Adhesion Molecules in the Artery Wall of Candidate Patients for Kidney Transplantation. Diabetes Care 2012, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Rolim, I.; Duarte, N.; Barata, G.; Costa, J.; Gardete-Correia, L.; Boavida, J.; Duarte, R.; Raposo, J.; Peerally, Z.; Catarino, M.; et al. Immunoglobulin M gene association with autoantibody reactivity and type 1 diabetes. Immunogenetics 2017, 69, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, G.S.D.; Chiazza, F.; Chen, J.; Azevedo-Loiola, R.; Martin, L.; Kusters, D.H.M.; Reutelingsperger, C.; Fountoulakis, N.; Gnudi, L.; Yaqoob, M.M.; et al. Annexin A1 attenuates microvascular complications through restoration of Akt signalling in a murine model of type 1 diabetes. Diabetologia 2018, 61, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Lu, Y.-C.; Chen, Y.-W.; Lee, W.-L.; Lu, C.-H.; Chen, Y.-H.; Lee, Y.-C.; Lin, S.-T.; Timms, J.F.; Lee, Y.-R.; et al. Hemopexin is up-regulated in plasma from type 1 diabetes mellitus patients: Role of glucose-induced ROS. J. Proteom. 2012, 75, 3760–3777. [Google Scholar] [CrossRef] [PubMed]
- Tonon, F.; Candido, R.; Toffoli, B.; Tommasi, E.; Cortello, T.; Fabris, B.; Bernardi, S. Type 1 diabetes is associated with significant changes of ACE and ACE2 expression in peripheral blood mononuclear cells. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Al Muhtaseb, N.; Al Yousuf, A.; Bajaj, J.S. Apolipoprotein A-I, A-II, B, C-II, and C-III in children with insulin-dependent diabetes mellitus. Pediatrics 1992, 89, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Vendrame, F.; Santangelo, C.; Misasi, R.; Dionisi, S.; Gizzi, C.; Realacci, M.; Grassetti, D.; Di Mario, U.; Dotta, F. Defective lymphocyte caspase-3 expression in type 1 diabetes mellitus. Eur. J. Endocrinol. 2005, 152, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Jamali, Z.; Nazari, M.; Khorramdelazad, H.; Hakimizadeh, E.; Mahmoodi, M.; Karimabad, M.N.; Hassanshahi, G.; Rezaeaian, M.; Balaei, P.; Derakhshan, S.; et al. Expression of CC Chemokines CCL2, CCL5, and CCL11 is Associated with Duration of Disease and Complications in Type-1 Diabetes: A Study on Iranian Diabetic Patients. Clin. Lab. 2013, 59, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.-S.; Yoon, J.-W. A new look at viruses in type 1 diabetes. Diabetes/Metab. Res. Rev. 2003, 19, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Laforgue, D.; Hendel, H.; Caillat-Zucman, S.; Zagury, J.-F.; Winkler, C.; Boitard, C.; Timsit, J. A Common Stromal Cell–Derived Factor-1 Chemokine Gene Variant is Associated With the Early Onset of Type 1 Diabetes. Diabetes 2001, 50, 1211–1213. [Google Scholar] [CrossRef] [Green Version]
- Sanlioglu, A.D.; Griffith, T.S.; Omer, A.; Dirice, E.; Sari, R.; Altunbas, H.A.; Balci, M.K.; Sanlioglu, S. Molecular mechanisms of death ligand-mediated immune modulation: A gene therapy model to prolong islet survival in type 1 diabetes. J. Cell. Biochem. 2008, 104, 710–720. [Google Scholar] [CrossRef]
- Ou, D.; Wang, X.; Metzger, D.; Robbins, M.; Huang, J.; Jobin, C.; Chantler, J.; James, R.; Pozzilli, P.; Tingle, A. Regulation of TNF-Related Apoptosis-Inducing Ligand-Mediated Death-Signal Pathway in Human β Cells by Fas-Associated Death Domain and Nuclear Factor κB. Hum. Immunol. 2005, 66, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, I.; Hao, Z.; Rajewsky, K.; von Boehmer, H. Effective Destruction of Fas-deficient Insulin-producing β Cells in Type 1 Diabetes. J. Exp. Med. 2003, 198, 1103–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, S.; Tsui, H.; Lau, A.; Song, A.; Li, X.; Cheung, R.K.; Sampson, A.; Afifiyan, F.; Elford, A.; Jackowski, G.; et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not β-cell exclusive. Nat. Med. 2003, 9, 198–205. [Google Scholar] [CrossRef]
- Simeonovic, C.J.; Ziolkowski, A.F.; Wu, Z.; Choong, F.J.; Freeman, C.; Parish, C.R. Heparanase and Autoimmune Diabetes. Front. Immunol. 2013, 4, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folli, F.; Guzzi, V.; Perego, L.; Coletta, D.K.; Finzi, G.; Placidi, C.; La Rosa, S.; Capella, C.; Socci, C.; Lauro, D.; et al. Proteomics Reveals Novel Oxidative and Glycolytic Mechanisms in Type 1 Diabetic Patients’ Skin Which Are Normalized by Kidney-Pancreas Transplantation. PLoS ONE 2010, 5, e9923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marleau, A.M.; Sarvetnick, N.E. IL-18 is required for self-reactive T cell expansion in NOD mice. J. Autoimmun. 2011, 36, 263–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenny, L.; Ajjan, R.; King, R.; Thiel, S.; Schroeder, V. Plasma levels of mannan-binding lectin-associated serine proteases MASP-1 and MASP-2 are elevated in type 1 diabetes and correlate with glycaemic control. Clin. Exp. Immunol. 2015, 180, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cao, L.; Ye, J.; Zhu, D. Upregulation of myostatin gene expression in streptozotocin-induced type 1 diabetes mice is attenuated by insulin. Biochem. Biophys. Res. Commun. 2009, 388, 112–116. [Google Scholar] [CrossRef]
- Levenson, A.E.; Wadwa, R.P.; Shah, A.S.; Khoury, P.R.; Kimball, T.R.; Urbina, E.M.; de Ferranti, S.D.; Bishop, F.K.; Maahs, D.M.; Dolan, L.M.; et al. PCSK9 Is Increased in Youth With Type 1 Diabetes. Diabetes Care 2017, 40, e85–e87. [Google Scholar] [CrossRef] [Green Version]
- Crèvecoeur, I.; Gudmundsdottir, V.; Vig, S.; Sodré, F.M.C.; D’Hertog, W.; Fierro, A.C.; Van Lommel, L.; Gysemans, C.; Marchal, K.; Waelkens, E.; et al. Early differences in islets from prediabetic NOD mice: Combined microarray and proteomic analysis. Diabetologia 2017, 60, 475–489. [Google Scholar] [CrossRef] [Green Version]
- Mackness, B.; Durrington, P.N.; Boulton, A.J.M.; Hine, D.; Mackness, M.I. Serum paraoxonase activity in patients with type 1 diabetes compared to healthy controls. Eur. J. Clin. Investig. 2002, 32, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Iclal, G.; Mehmet, K.; Yılmaz, K.; Müslüm, A.; Geyikli, I.; Keskin, M.; Kör, Y.; Akan, M. Increased Resistin Serum Concentrations in Patients with Type 1 Diabetes Mellitus. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Safari-Alighiarloo, N.; Taghizadeh, M.; Tabatabaei, S.M.; Shahsavari, S.; Namaki, S.; Khodakarim, S.; Rezaei-Tavirani, M. Identification of new key genes for type 1 diabetes through construction and analysis of protein-protein interaction networks based on blood and pancreatic islet transcriptomes. J. Diabetes 2017, 9, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.L. Multiple imputation: A primer. Stat. Methods Med. Res. 1999, 8, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.R.; Kenward, M.G. Survival Data, Skips and Large Datasets. In Multiple Imputation and Its Application; Wiley: Chichester, UK, 2013; pp. 165–202. [Google Scholar]
- Kenward, M.G.; Carpenter, J. Multiple imputation: Current perspectives. Stat. Methods Med. Res. 2007, 16, 199–218. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| −log(p-Value) * | |||||||
|---|---|---|---|---|---|---|---|
| Canonical Pathways | Original | A1 | A2 | A3 | References | ||
| T1D High-Risk (HR) | Proteomics | CCR3 Signaling in Eosinophils | 0.206 | 0.201 | 0.201 | 4.03 | [22,23] |
| Complement System | 0.631 | 0.623 | 0.625 | 5.69 | [24,25] | ||
| CXCR4 Signaling | 5.53 | [26,27] | |||||
| Fcγ Receptor-Mediated Phagocytosis in Macrophages | 0.833 | 0.821 | 0.824 | 9.16 | [28] | ||
| FcγRIIB Signaling in B Lymphocytes | 0.333 | 0.333 | 2.39 | [29] | |||
| IL-12 Signaling and Production in Macrophages | 0.592 | 0.582 | 0.583 | 4.65 | [30,31] | ||
| IL-15 Production | 2.71 | 2.67 | 3.54 | 7.42 | [32,33] | ||
| IL-7 Signaling Pathway | 0.361 | 0.361 | 1.97 | [34,35] | |||
| Oncostatin M Signaling | 0.567 | 1.73 | [36] | ||||
| Paxillin Signaling | 0.266 | 0.261 | 0.262 | 7.39 | [37] | ||
| Production of Nitric Oxide and ROS in Macrophages | 0.398 | 0.389 | 0.39 | 2.67 | [38,39] | ||
| RHOA Signaling | 1.22 | 1.2 | 1.2 | 7.37 | [40,41] | ||
| Sphingosine-1-Phosphate Signaling | 0.237 | 0.232 | 0.233 | 3.23 | [42] | ||
| Metabolomics | Arginine Biosynthesis IV | 3.79 | 12.2 | 5.4 | 5.39 | [38,43] | |
| Citrulline–Nitric Oxide Cycle | 1.35 | 6.83 | 3.11 | 3.1 | [43,44] | ||
| Stearate Biosynthesis I (Animals) | 1.16 | 4.02 | 2.67 | 2.67 | [45] | ||
| FAT10 Signaling Pathway | 1.04 | 1.84 | 2.29 | 2.29 | [46] | ||
| T1D New-Onset (NO) | Proteomics | 14-3-3-Mediated Signaling | 3 | [47] | |||
| CCR3 Signaling in Eosinophils | 4.63 | [48] | |||||
| CXCR4 Signaling | 0.259 | 4.82 | [26,27] | ||||
| Fcγ Receptor-Mediated Phagocytosis in Macrophages | 0.666 | 0.664 | 0.325 | 3.36 | [28] | ||
| FcγRIIB Signaling in B Lymphocytes | 0.375 | 3 | [29] | ||||
| Oncostatin M Signaling | 0.282 | 0.281 | 0.284 | 2.45 | [36] | ||
| PAK Signaling | 6.01 | [49,50] | |||||
| Phospholipases | 0.975 | 1.56 | 0.983 | 3.7 | [51,52] | ||
| RHOA Signaling | 0.793 | 0.45 | 0.457 | 8.12 | [40,41] | ||
| Sphingosine-1-Phosphate Signaling | 0.475 | 0.474 | 4.48 | [42] | |||
| Metabolomics | Arginine Biosynthesis IV | 2.92 | 12.2 | 5.4 | 5.39 | [38,43] | |
| Citrulline–Nitric Oxide Cycle | 1.45 | 6.83 | 3.11 | 3.1 | [43,44] | ||
| Stearate Biosynthesis I (Animals) | 1.26 | 4.02 | 2.67 | 2.67 | [45] | ||
| FAT10 Signaling Pathway | 1.09 | 1.84 | 2.29 | 2.29 | [46] | ||
| High-Risk T1D (HR) | Original | A1 | A2 | A3 | ||||
|---|---|---|---|---|---|---|---|---|
| p | Biomarker Name * | p | Biomarker Name * | p | Biomarker Name * | p | Biomarker Name * | |
| Diabetes Mellitus | 3.53 × 10−13 | APOA2, APOE, CD44, CETP, GPNMB, IGF1, IGFBP2, JAG1, L1CAM, LDLR, MEP1B, MMP14, MMP2, MMP9, PTGDS, PTPRC, SELL, SFTPD, VCAM1 | 4.69 × 10−14 | APOA2, APOE, CD44, CETP, GPNMB, IGF1, IGFBP2, IGHM, JAG1, L1CAM, LDLR, MEP1B, MMP14, MMP2, MMP9, PTGDS, PTPRC, SELL, SFTPD, VCAM1 | 6.1 × 10−15 | ANXA1, APOA2, APOE, CD44, CETP, FGFR1, GPNMB, IGF1, IGFBP2, IGHM, JAG1, L1CAM, LDLR, MEP1B, MMP14, MMP2, MMP9, PTGDS, SELL, SFTPD, VCAM1 | 1.91 × 10−25 | ACE, ADK, AKT1, APOA1, APOA4, APOB, APOC1, CASP3, CCL5, CD36, CETP, CR2, CXCL12, EGFR, FADD, FAS, FGFR1, GFAP, GSTO1, GSTP1, HPSE, HSPB1, IGF1, IGF2, IGHM, IL18, L1CAM, MASP2, MMP14, MMP9, MSTN, PCSK9, PDE5A, PON1, PTGDS, PTPRC, RETN, SFTPD, SOD1, SRC, VCAM1 |
| T1D ** | n/a | APOA2 [60], APOE [61], CD44 [62], CETP [63], GPNMB [64], IGF1 [65], IGFBP2 [66], JAG1 [67], LDLR [68], MEP1B [69], MMP2 [70], MMP9 [70], PTPRC [71], SELL [72], VCAM1 [73] | n/a | APOA2 [60], APOE [61], CD44 [62], CETP [63], GPNMB [64], IGF1 [65], IGFBP2 [66], IGHM [74], JAG1 [67], LDLR [68], MEP1B [69], MMP2 [70], MMP9 [70], PTPRC [71], SELL [72], VCAM1 [73] | n/a | ANXA1 [75], APOA2 [60], APOE [61], CD44 [62], CETP [63], FGFR1 [76], GPNMB [64], IGF1 [65], IGFBP2 [66], IGHM [74], JAG1 [67], LDLR [68], MEP1B [69], MMP2 [70], MMP9 [70], SELL [72], VCAM1 [73] | n/a | ACE [77], APOA1 [60], APOB [78], CASP3 [79], CCL5 [80], CETP [63], CR2 [81], CXCL12 [82], FADD [83,84], FAS [83,85], FGFR1 [76], GFAP [86], HPSE [87], HSPB1 [88], IGF1 [65], IGF2 [65], IGHM [74], IL18 [89], MASP2 [90], MMP9 [70], MSTN [91], PCSK9 [92], PDE5A [93], PON1 [94], PTPRC [71], RETN [95], SRC [96], VCAM1 [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcazar, O.; Ogihara, M.; Ren, G.; Buchwald, P.; Abdulreda, M.H. Exploring Computational Data Amplification and Imputation for the Discovery of Type 1 Diabetes (T1D) Biomarkers from Limited Human Datasets. Biomolecules 2022, 12, 1444. https://doi.org/10.3390/biom12101444
Alcazar O, Ogihara M, Ren G, Buchwald P, Abdulreda MH. Exploring Computational Data Amplification and Imputation for the Discovery of Type 1 Diabetes (T1D) Biomarkers from Limited Human Datasets. Biomolecules. 2022; 12(10):1444. https://doi.org/10.3390/biom12101444
Chicago/Turabian StyleAlcazar, Oscar, Mitsunori Ogihara, Gang Ren, Peter Buchwald, and Midhat H. Abdulreda. 2022. "Exploring Computational Data Amplification and Imputation for the Discovery of Type 1 Diabetes (T1D) Biomarkers from Limited Human Datasets" Biomolecules 12, no. 10: 1444. https://doi.org/10.3390/biom12101444