




Biomolecules 2024, 14(1), 5; https://doi.org/10.3390/biom14010005 - 20 Dec 2023
Viewed by 1142
Abstract
►
Show Figures
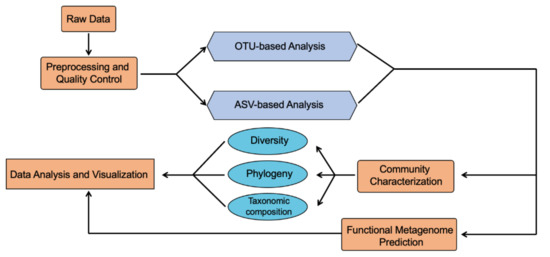
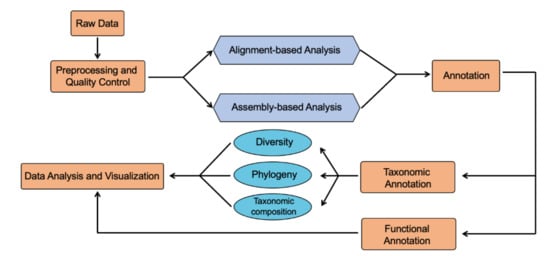
Continuous and significant progress in sequencing technologies and bioinformatics pipelines has revolutionized our comprehension of microbial communities, especially for human microbiomes. However, most studies have focused on studying the taxonomic composition of the microbiomes and we are still not able to characterize dysbiosis
[...] Read more.
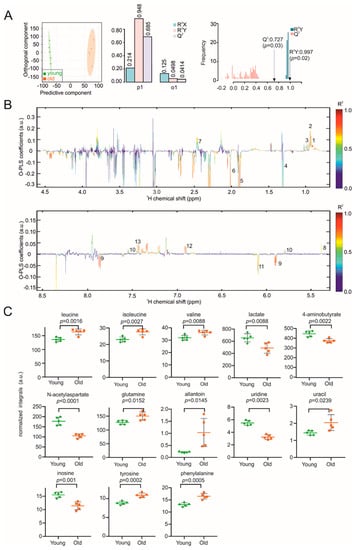
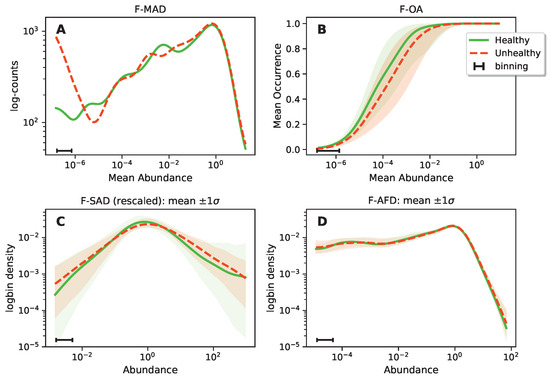
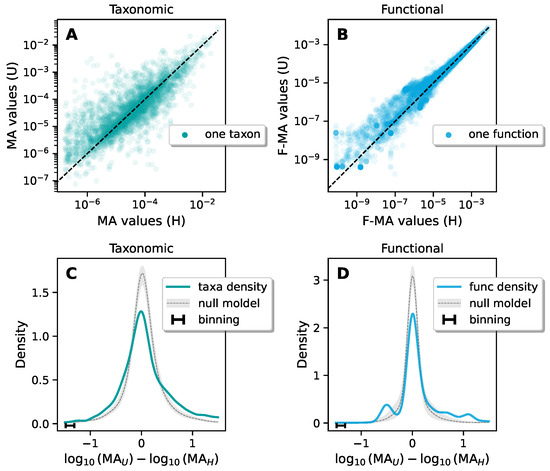
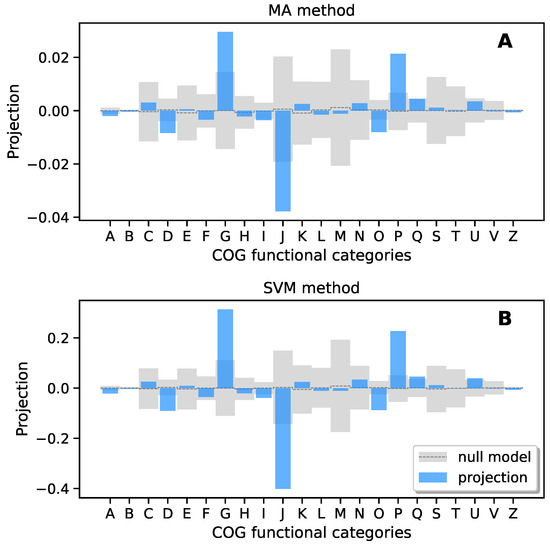
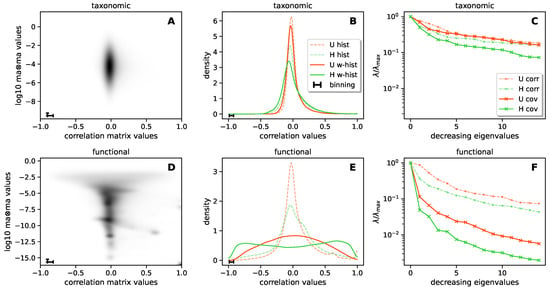
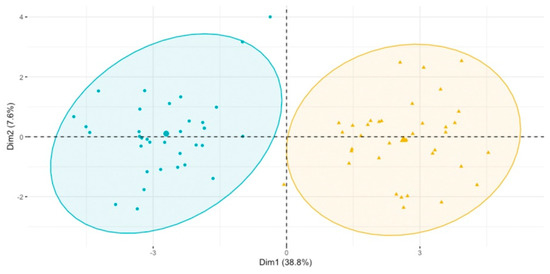
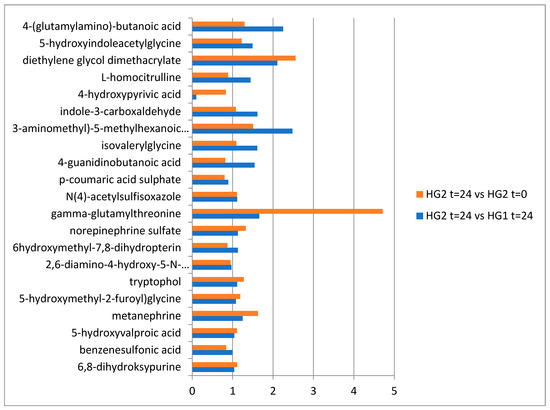
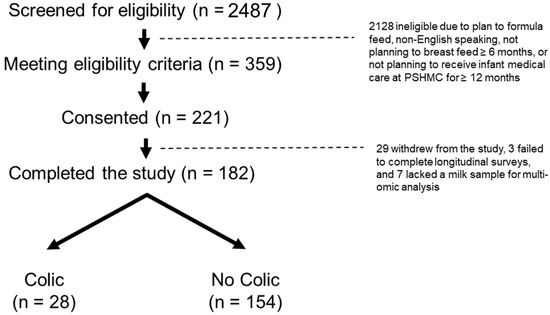
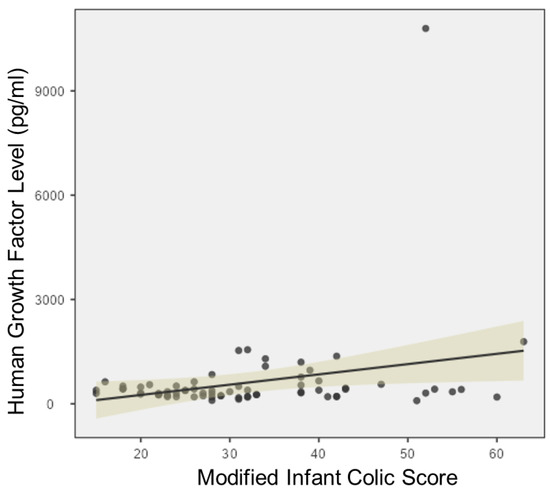
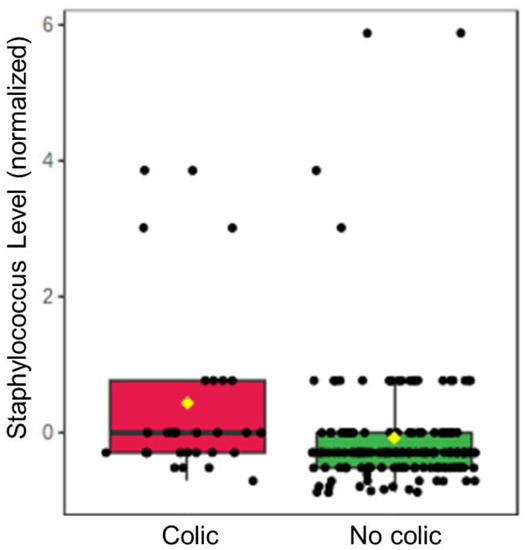
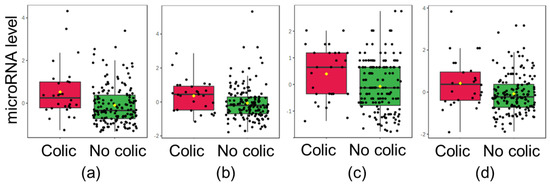
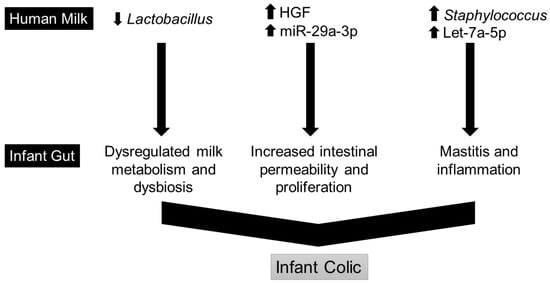
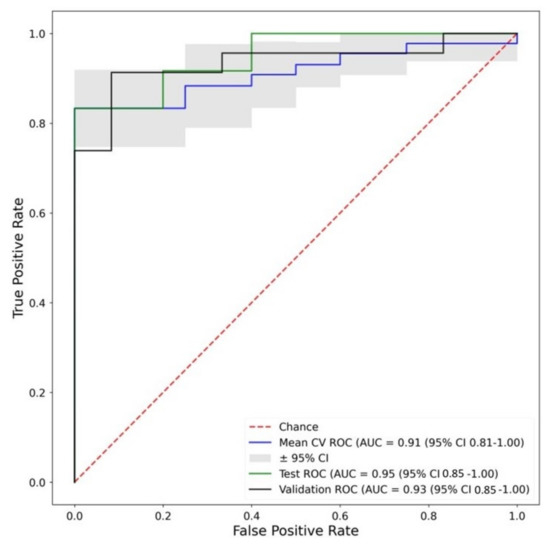
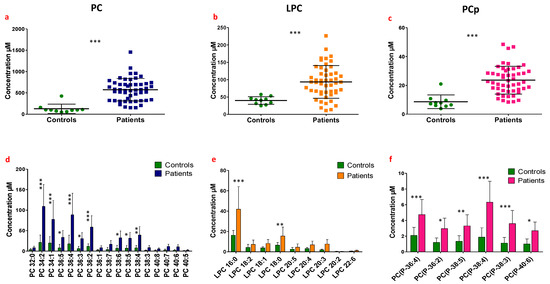
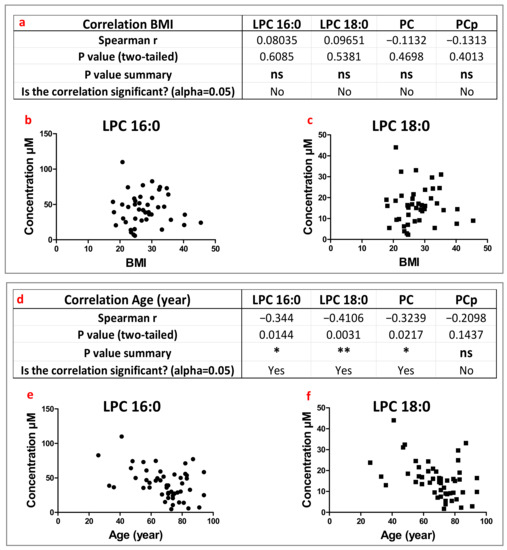
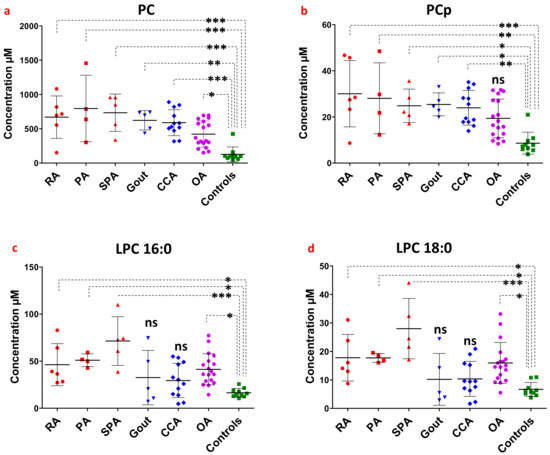
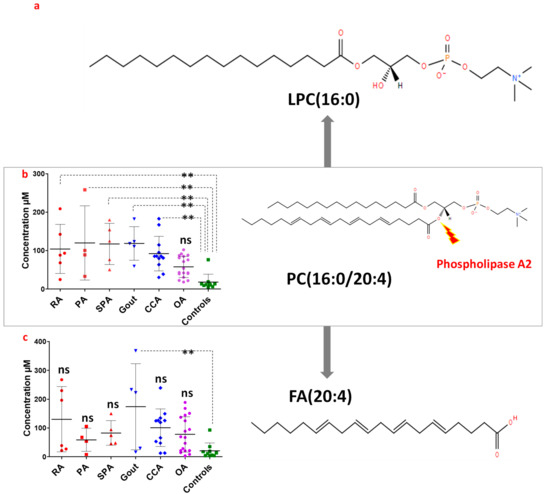
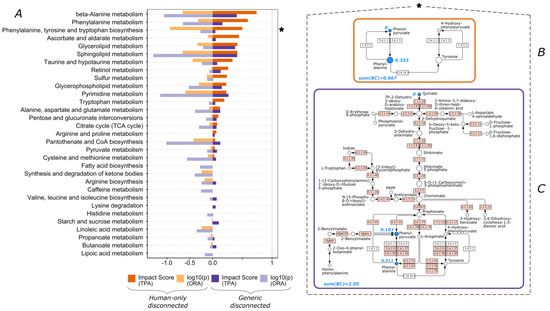
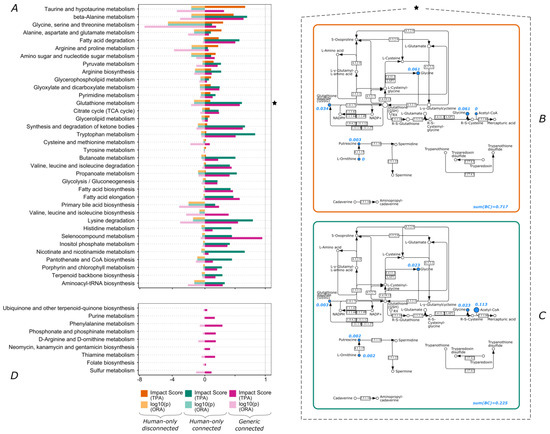
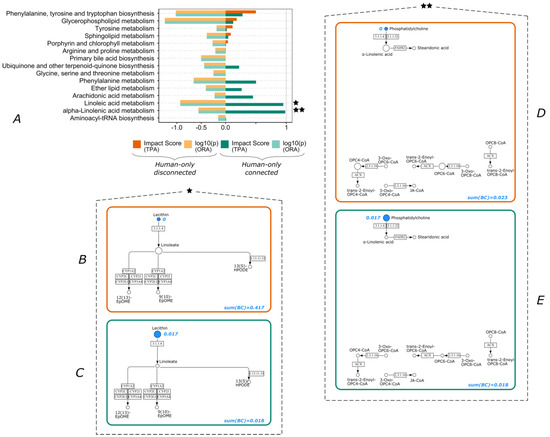
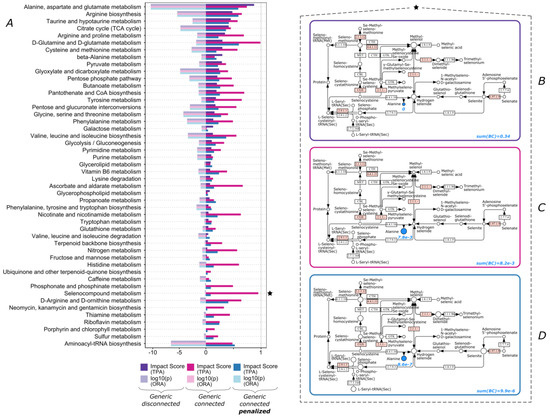
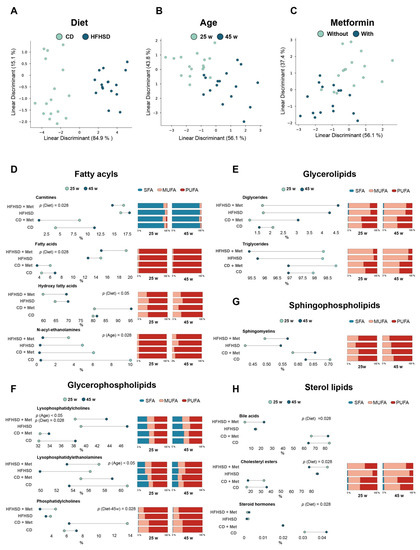
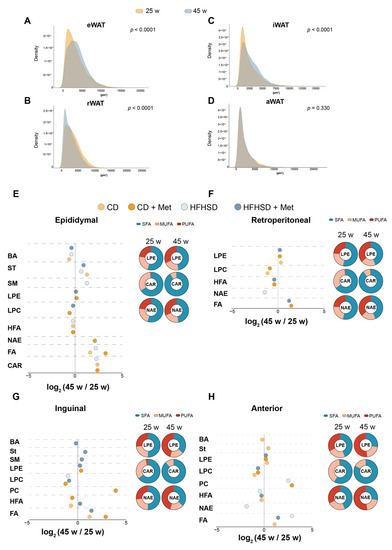
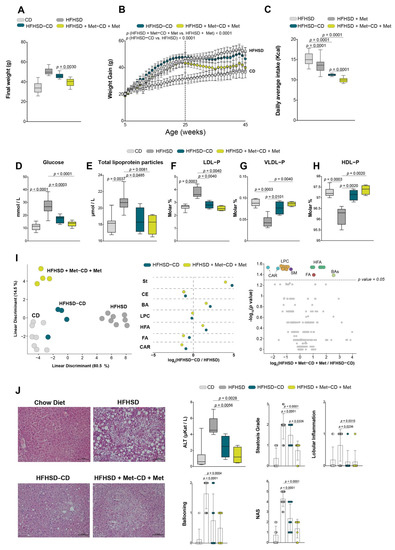
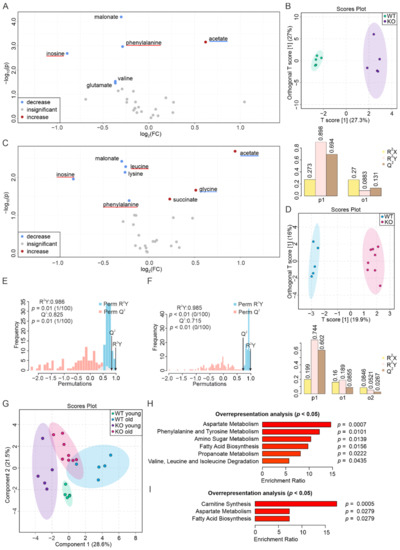
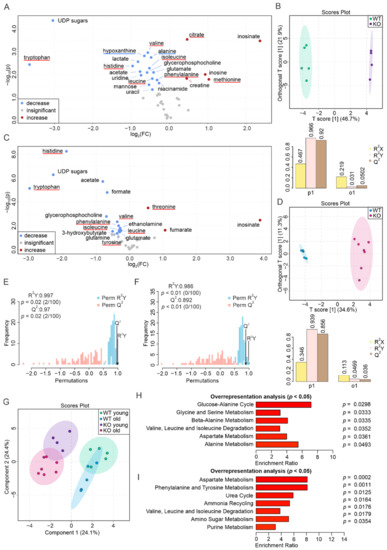
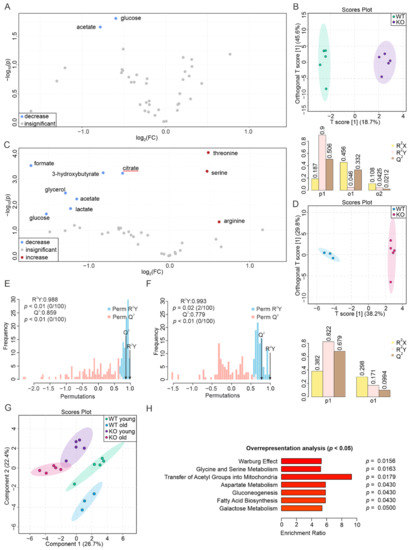
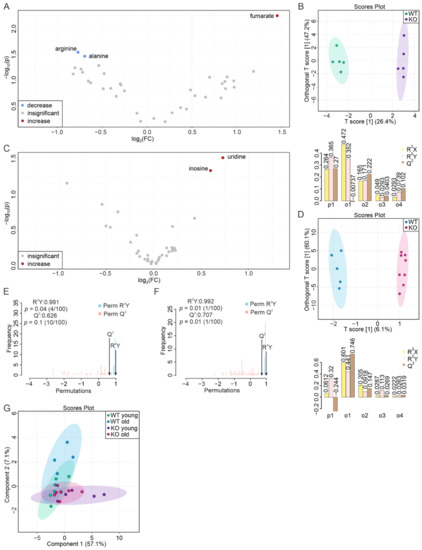
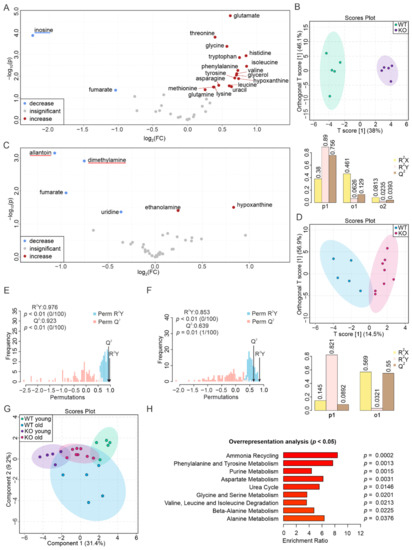
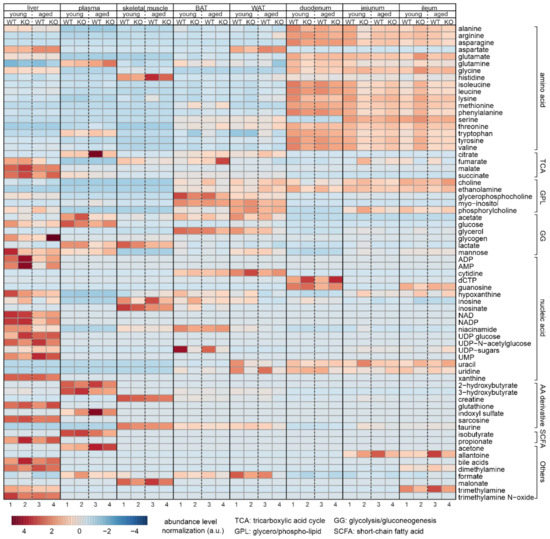
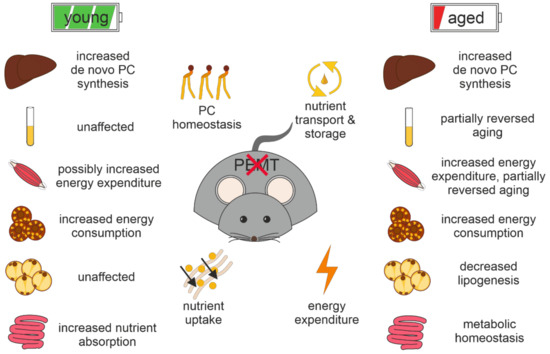
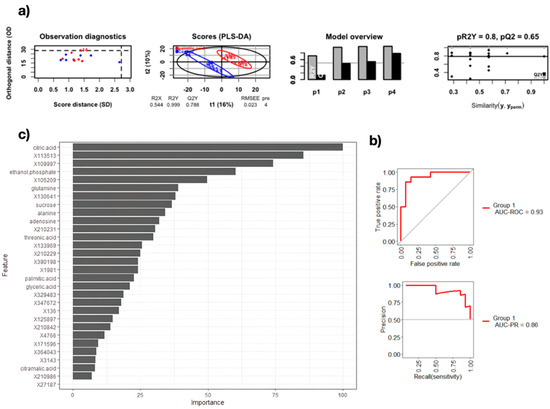
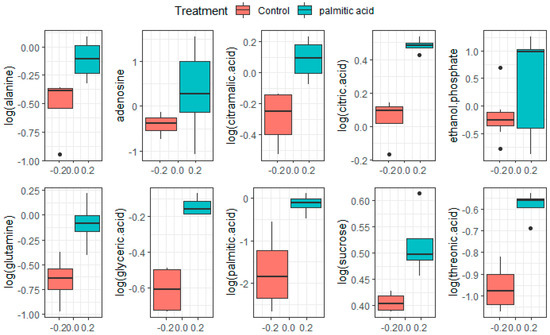
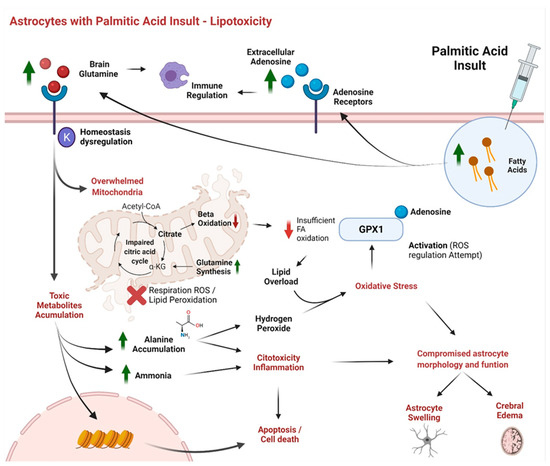
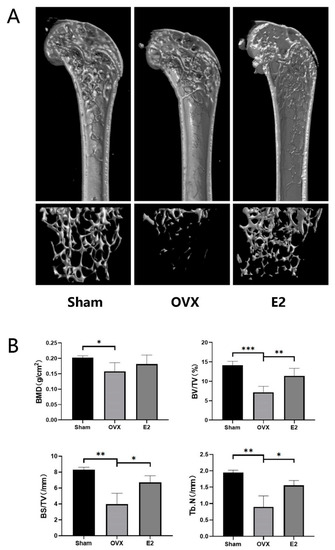
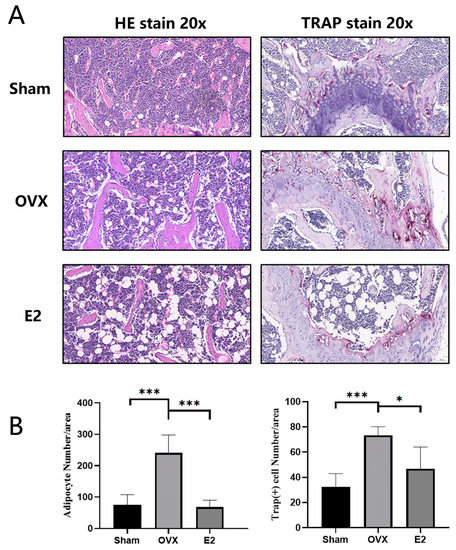
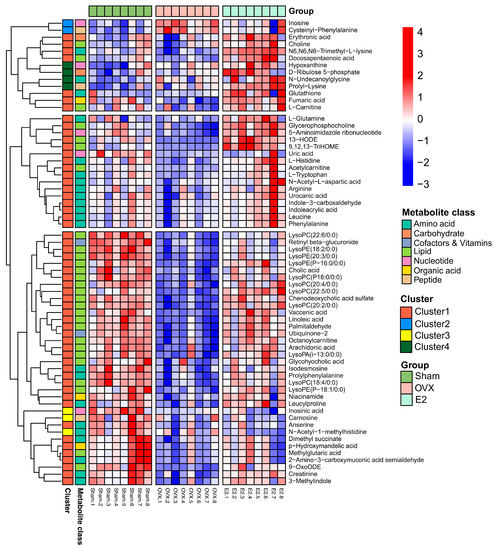
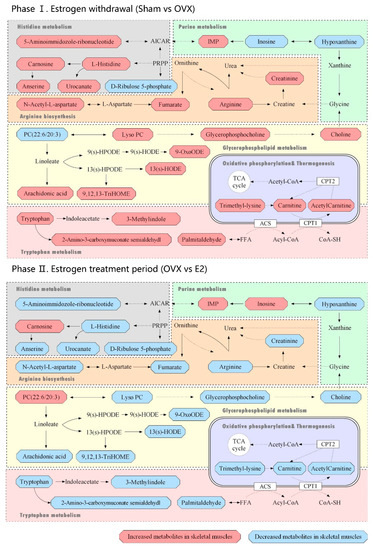
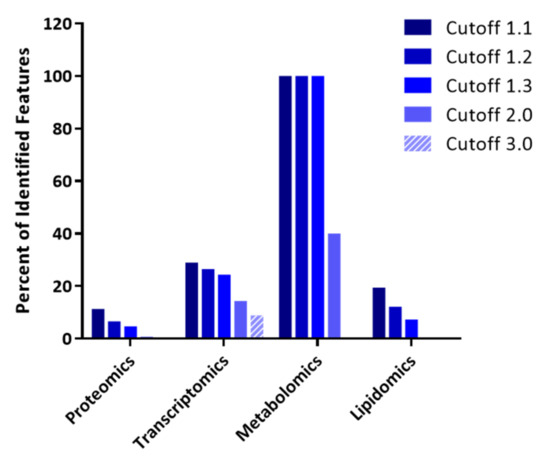
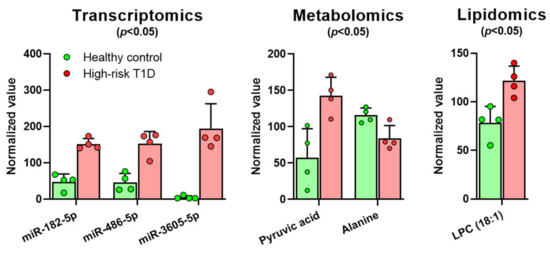
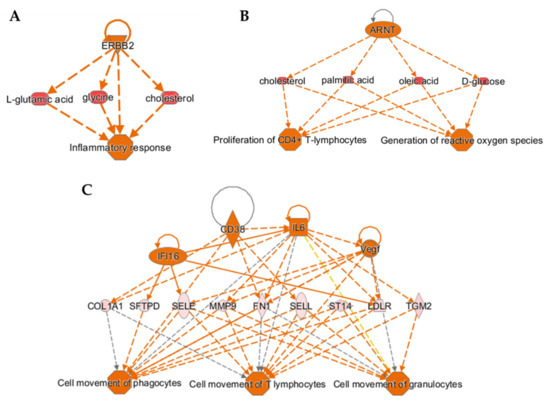
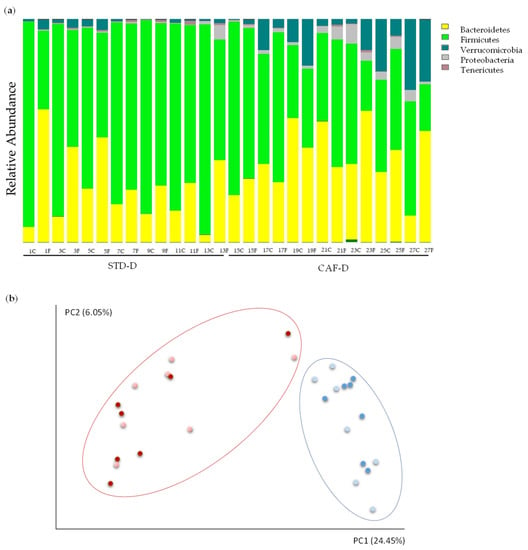
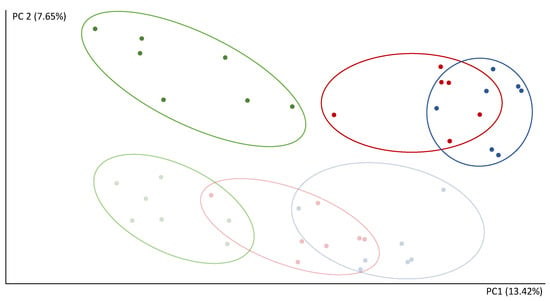
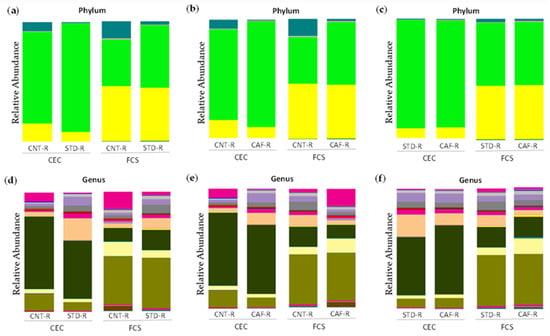
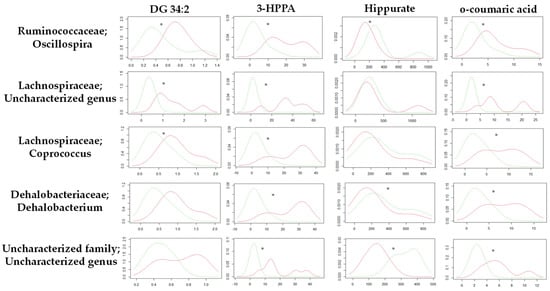
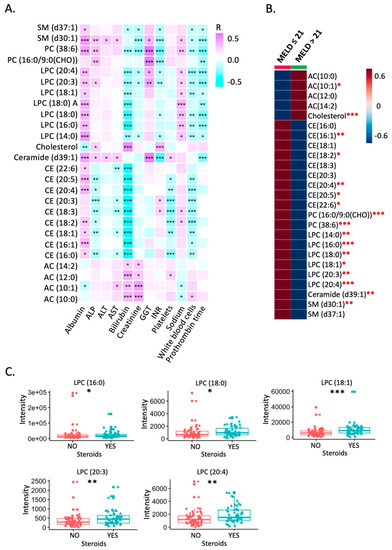
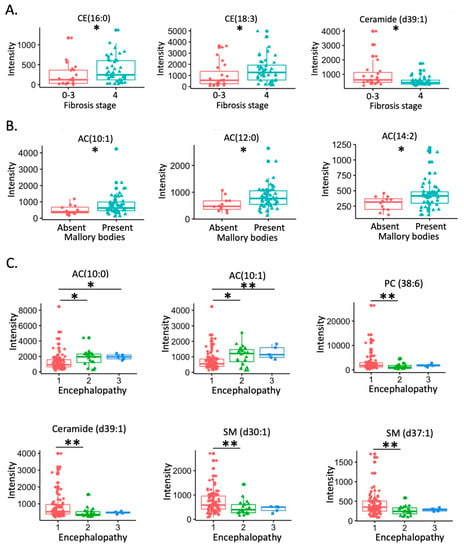
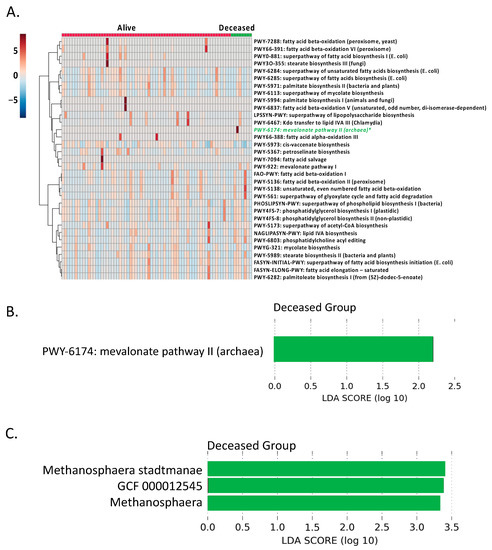
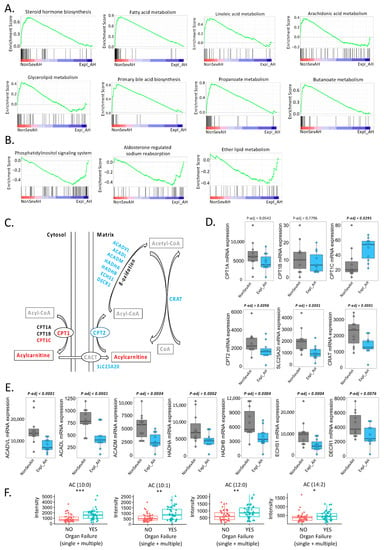
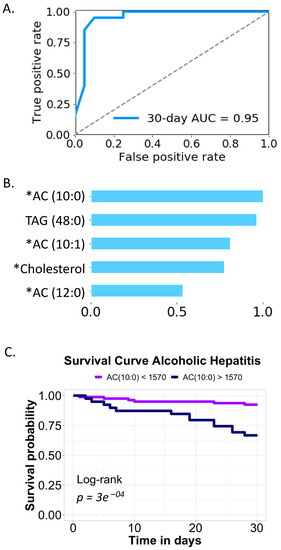
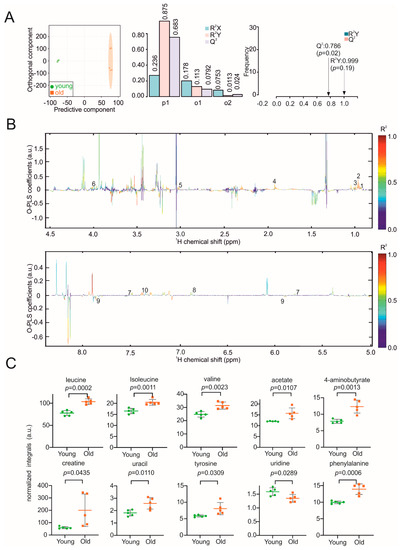
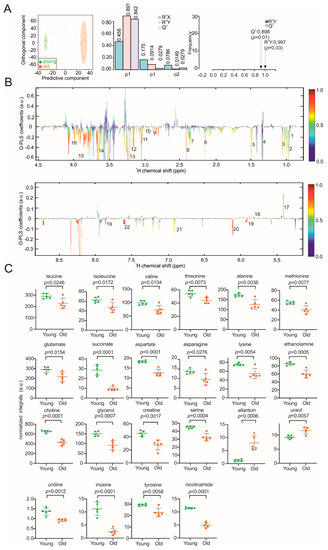
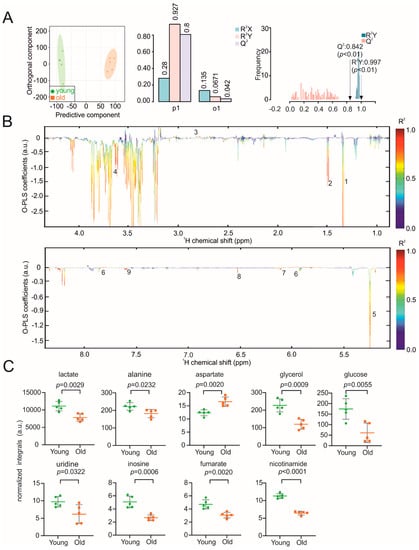
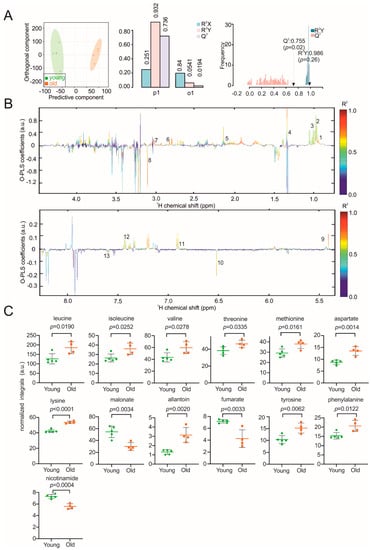
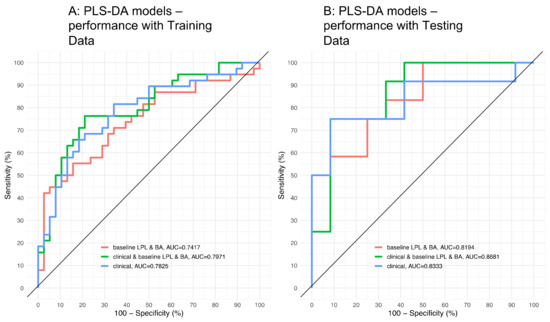
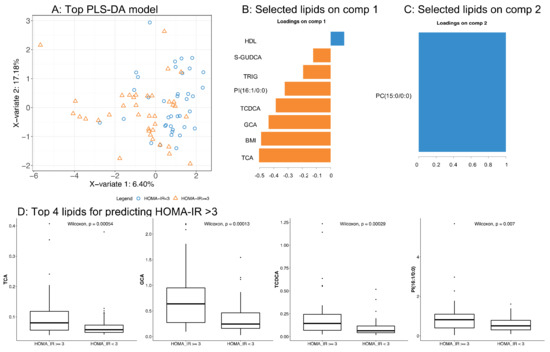
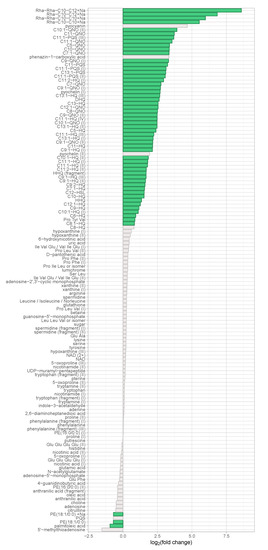
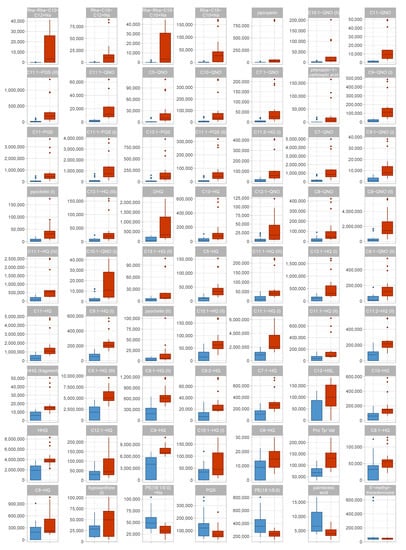
Continuous and significant progress in sequencing technologies and bioinformatics pipelines has revolutionized our comprehension of microbial communities, especially for human microbiomes. However, most studies have focused on studying the taxonomic composition of the microbiomes and we are still not able to characterize dysbiosis and unveil the underlying ecological consequences. This study explores the emergent organization of functional abundances and correlations of gut microbiomes in health and disease. Leveraging metagenomic sequences, taxonomic and functional tables are constructed, enabling comparative analysis. First, we show that emergent taxonomic and functional patterns are not useful to characterize dysbiosis. Then, through differential abundance analyses applied to functions, we reveal distinct functional compositions in healthy versus unhealthy microbiomes. In addition, we inquire into the functional correlation structure, revealing significant differences between the healthy and unhealthy groups, which may significantly contribute to understanding dysbiosis. Our study demonstrates that scrutinizing the functional organization in the microbiome provides novel insights into the underlying state of the microbiome. The shared data structure underlying the functional and taxonomic compositions allows for a comprehensive macroecological examination. Our findings not only shed light on dysbiosis, but also underscore the importance of studying functional interrelationships for a nuanced understanding of the dynamics of the microbial community. This research proposes a novel approach, bridging the gap between microbial ecology and functional analyses, promising a deeper understanding of the intricate world of the gut microbiota and its implications for human health.
Full article
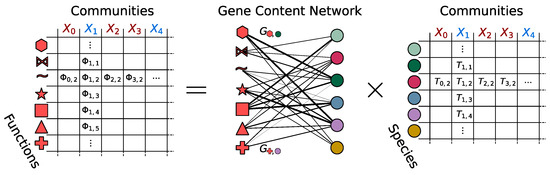
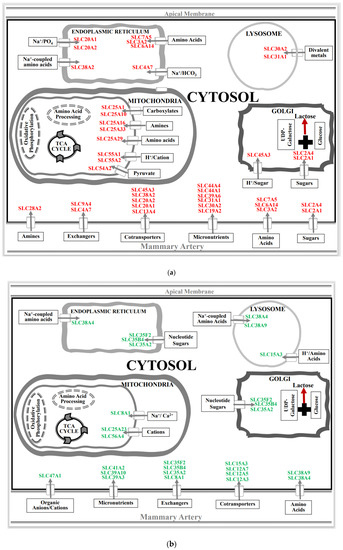
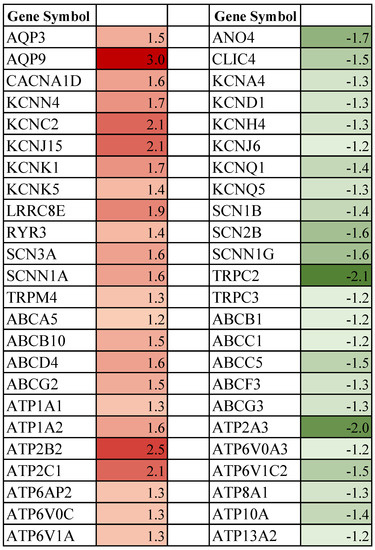
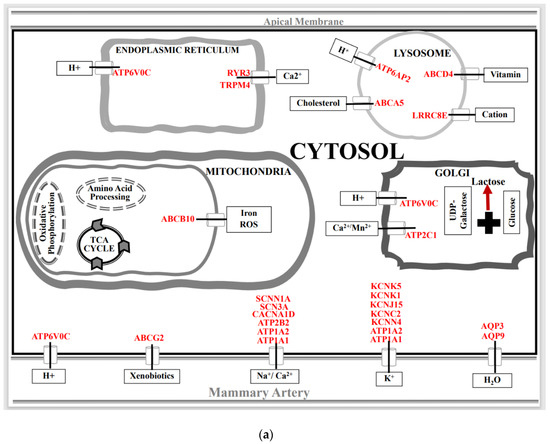
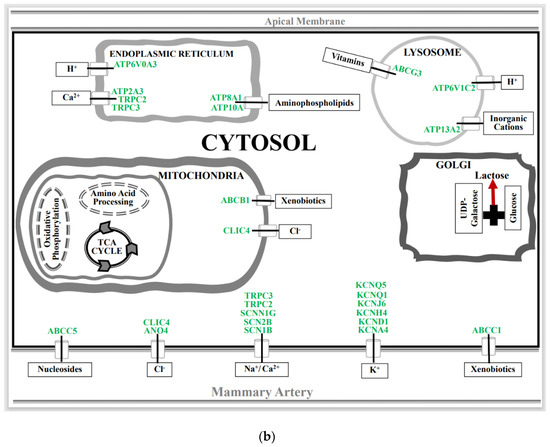
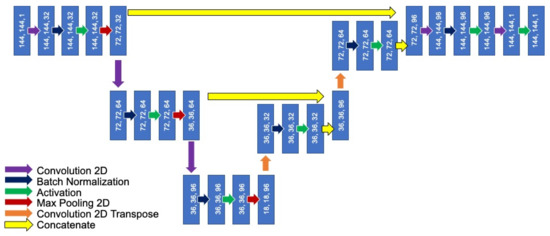
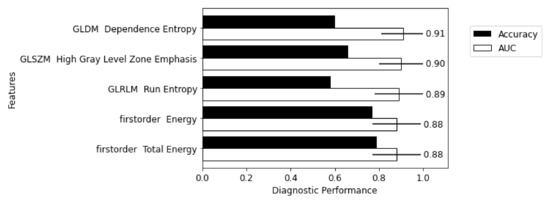
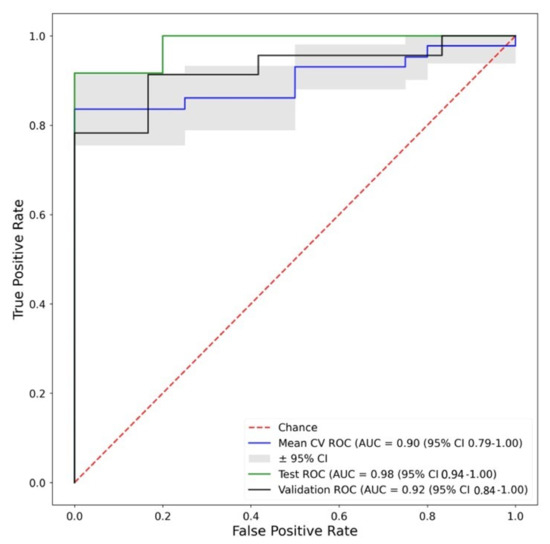
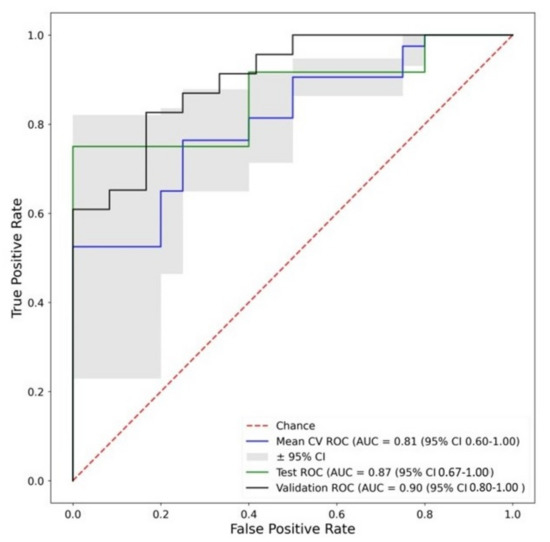
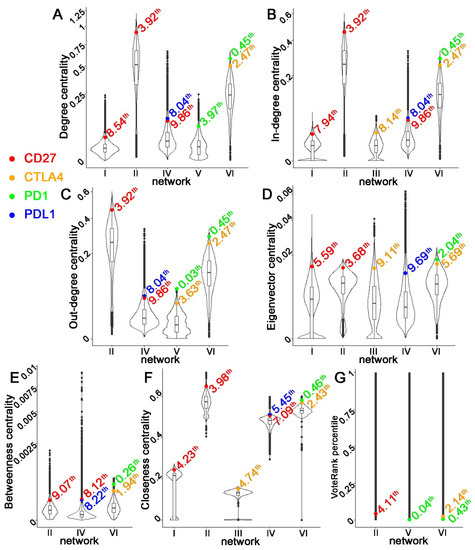
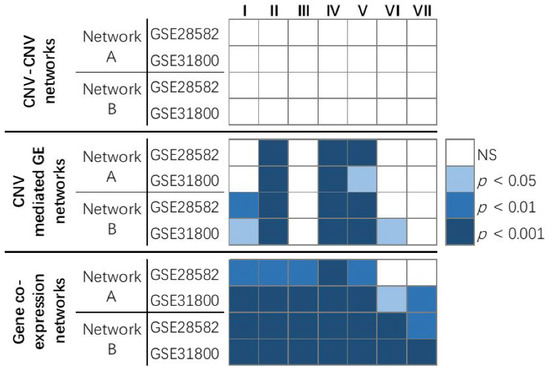
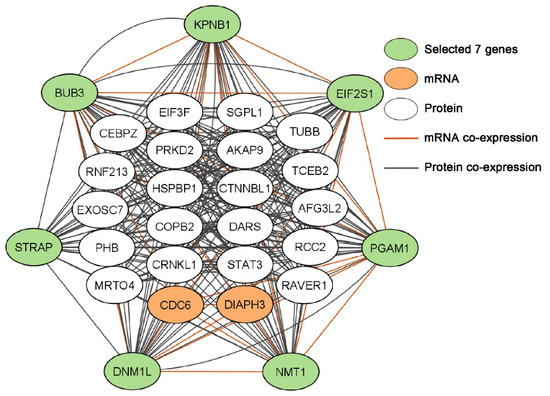
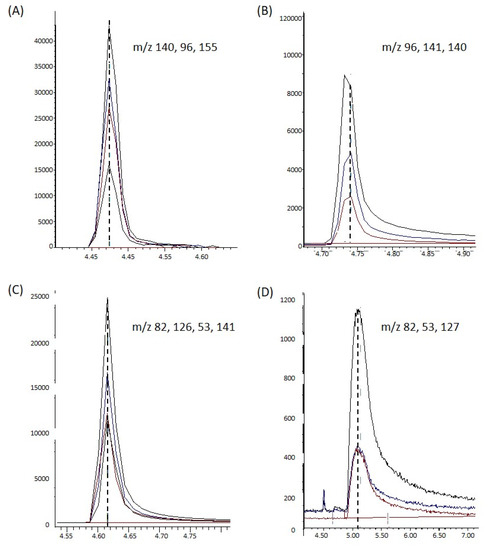
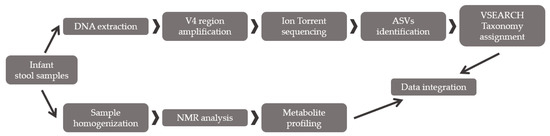
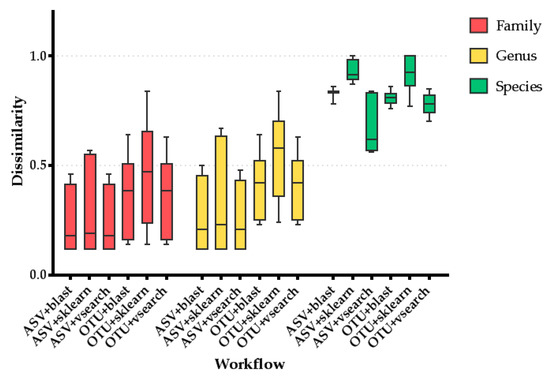
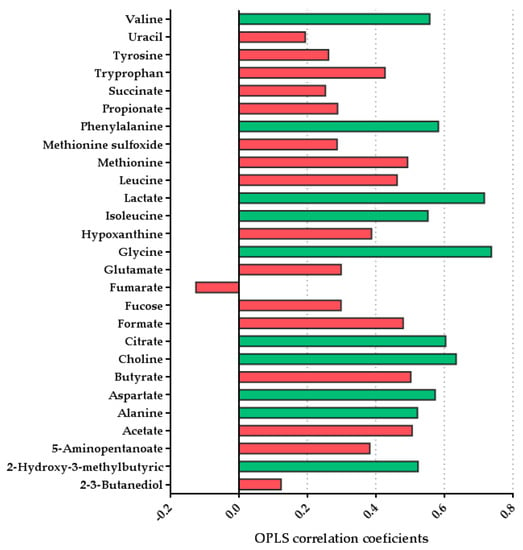
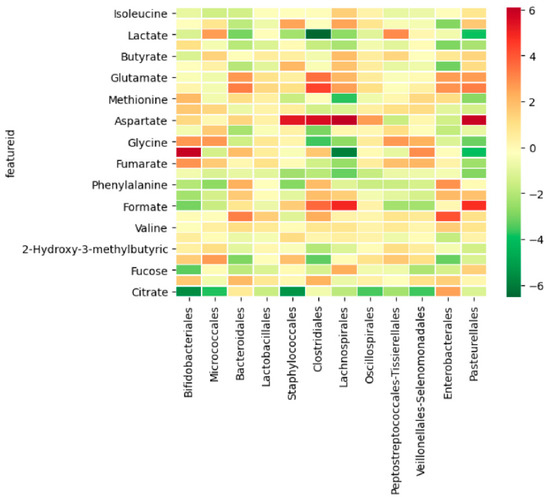
Figure 1



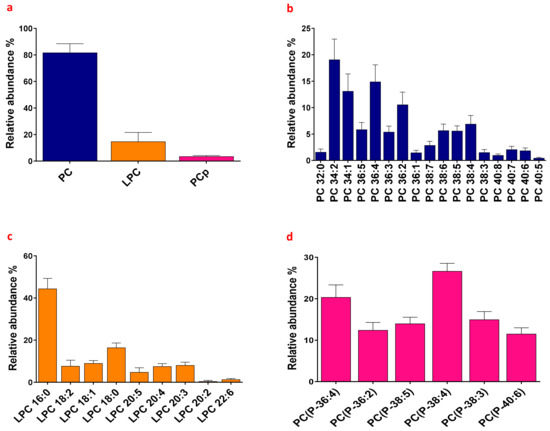


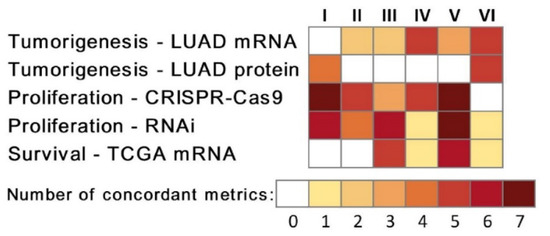

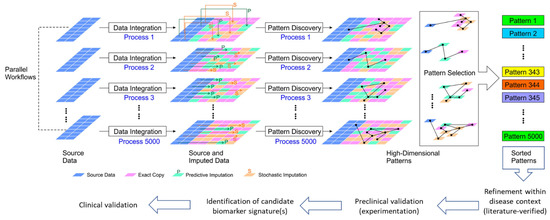
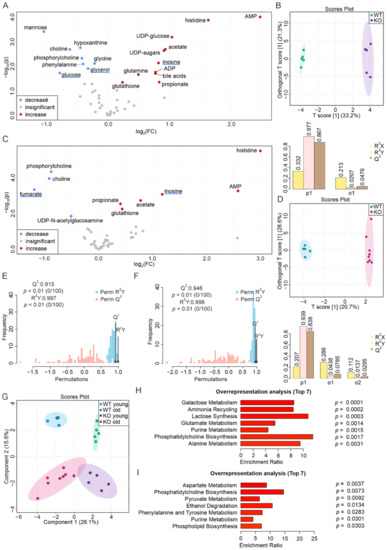




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}