
Computational Insights into the Structural Dynamics of MDA5 Variants Associated with Aicardi–Goutières Syndrome and Singleton–Merten Syndrome


 ,
, 
Abstract
:1. Introduction
- I.
- Analysis of MDA5 sequence conservation, and stability of mutated sequences (MDA5L372F, MDA5A452T, MDA5R779H, and MDA5R822Q);
- II.
- Preparation and MD simulation of MDA5WT, MDA5L372F, MDA5A452T, MDA5R779H, and MDA5R822Q;
- III.
- Trajectory analysis of MDA5 wild-type and mutant complexes: RMSD, RMSF, SASA, Rg, inter H-bonds, intra-H-bonds, interaction energies, PCA and residue network analysis.
2. Materials and Methods
2.1. Conservation and Mutant Stability Analyses
2.2. Preparation of dsRNA-Bound MDA5 Wild Type and Mutant Complexes
2.3. Molecular Dynamic Simulations
2.4. PCA and Free Energy Landscape (FEL) Analyses
2.5. Residue Network Analysis
3. Results
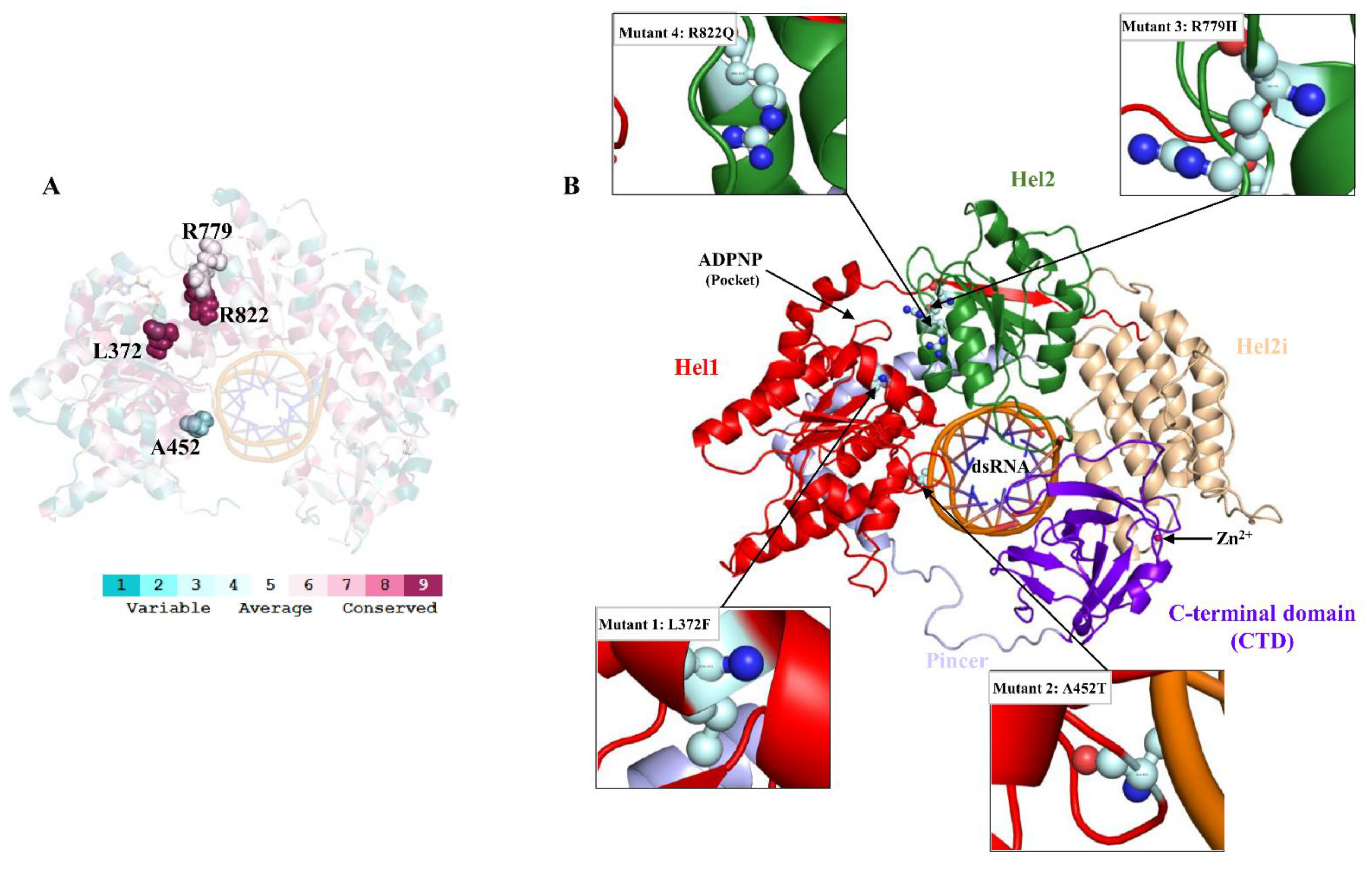
3.1. Conservation, Mutant Stability and Functional Analyses
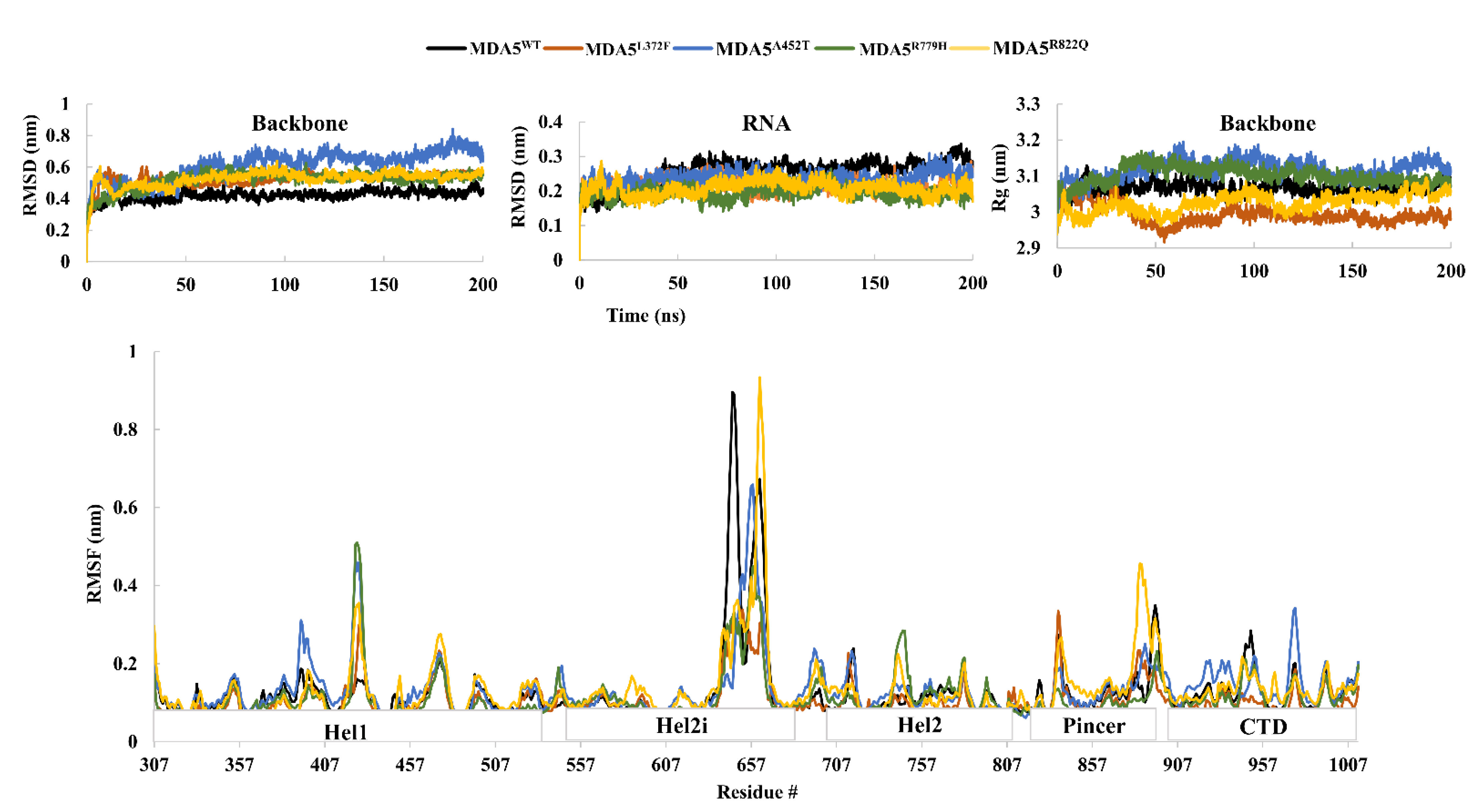
3.2. Structural Dynamics of Wild-Type and Mutant MDA5-dsRNA Bound Complexes
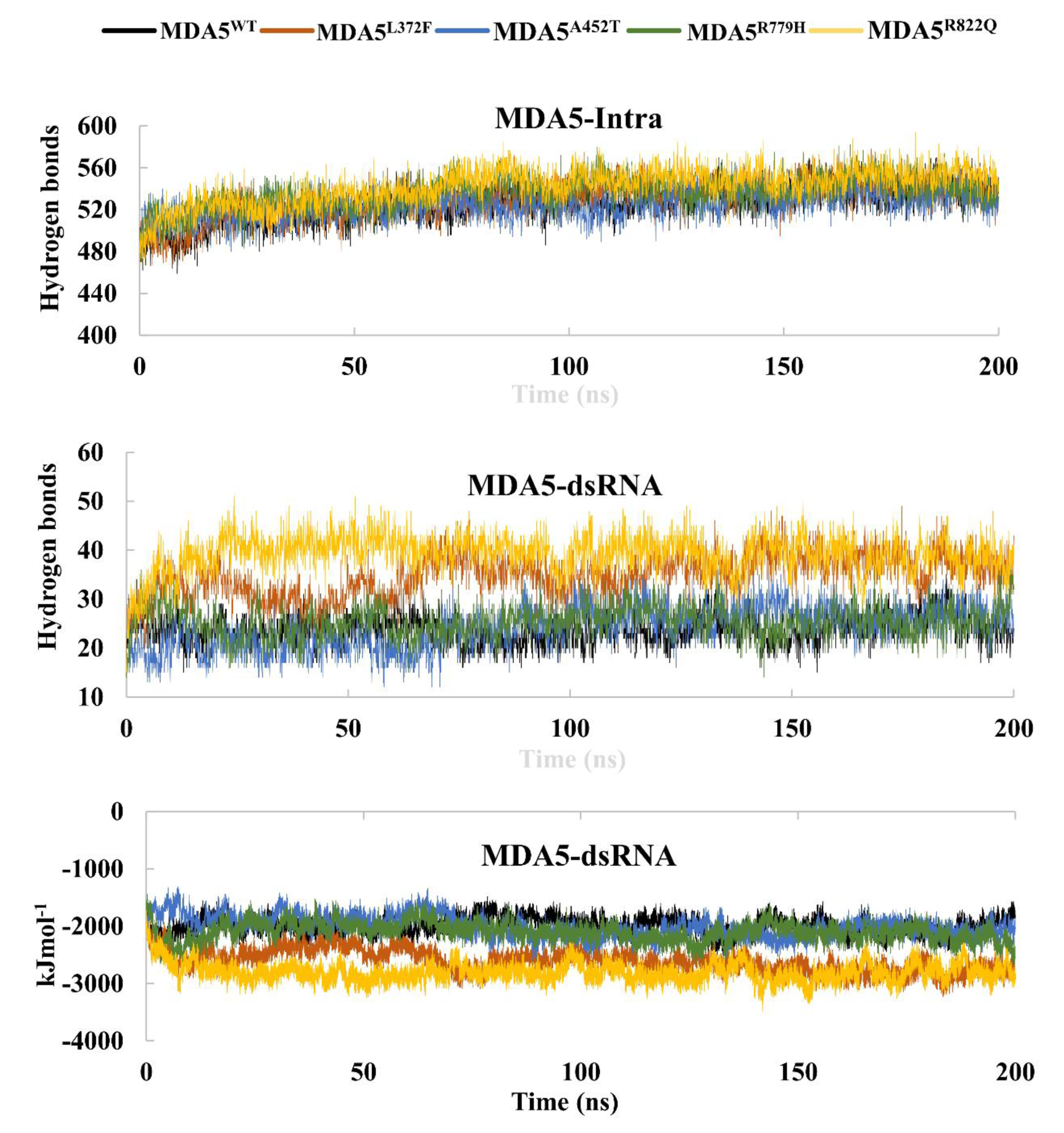
3.3. Hydrogen Bonds and Interaction Energies
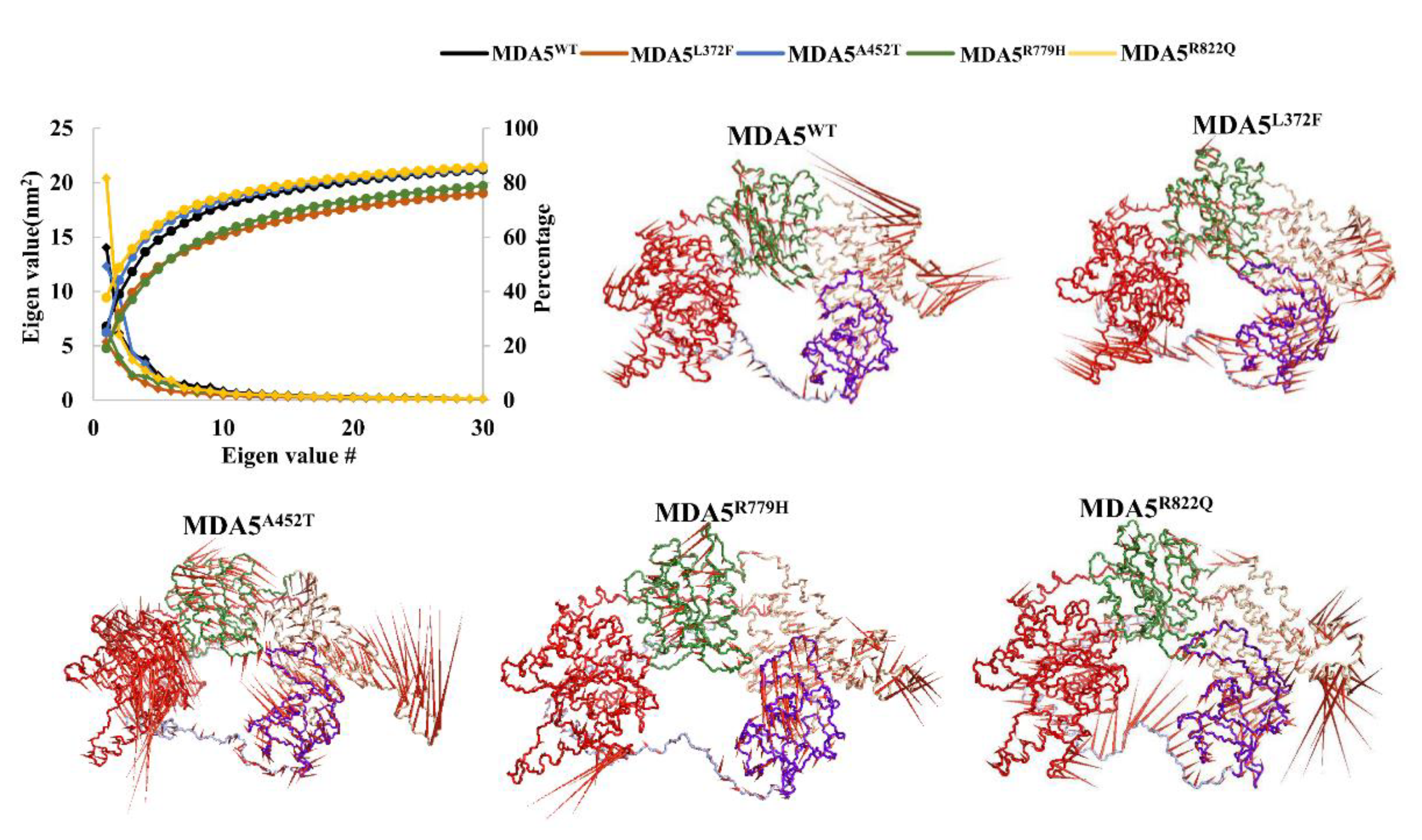
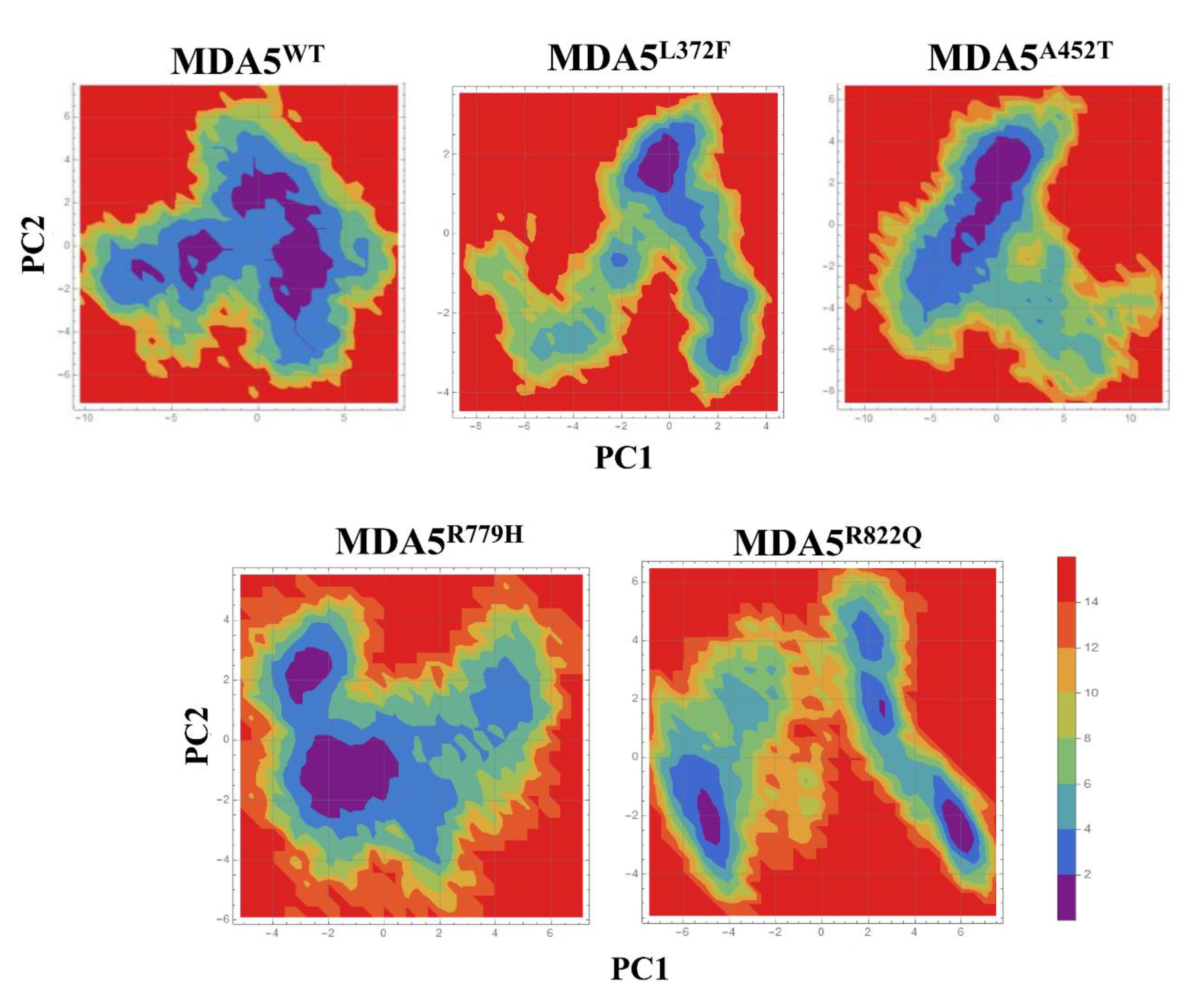
3.4. Principal Component Analysis
3.5. RNA Interactions with MDA5
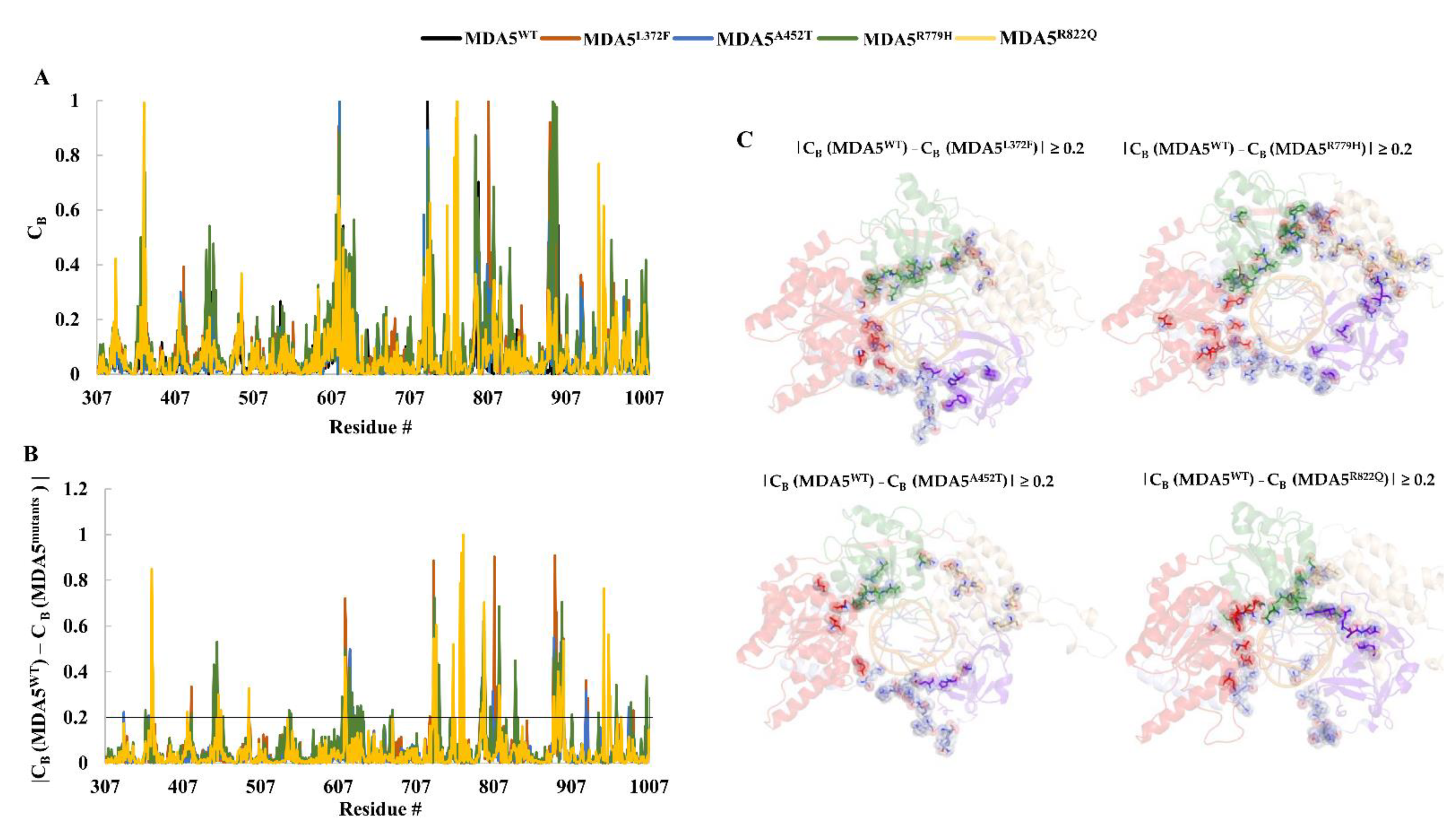
3.6. Residue Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Brahma, B.; Maharana, J.; Patra, M.C.; Kumar, S.; Mishra, P.; Saini, M.; De, B.C.; Mahanty, S.; Datta, T.K.; et al. Insight into buffalo (Bubalus bubalis) RIG1 and MDA5 receptors: A comparative study on dsRNA recognition and in-vitro antiviral response. PLoS ONE 2014, 9, e89788. [Google Scholar] [CrossRef] [Green Version]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Takahasi, K.; Kumeta, H.; Tsuduki, N.; Narita, R.; Shigemoto, T.; Hirai, R.; Yoneyama, M.; Horiuchi, M.; Ogura, K.; Fujita, T.; et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: Identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 2009, 284, 17465–17474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.; Eisenächer, K.; Kirchhofer, A.; Brzózka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias Junior, A.G.; Sampaio, N.G.; Rehwinkel, J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 2019, 27, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-T.; Verma, A.; Hodgkinson, C.P. Toll-like receptors and human disease: Lessons from single nucleotide polymorphisms. Curr. Genom. 2012, 13, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Han, M.X.; Ding, C.; Kyung, H.M. Genetic polymorphisms in pattern recognition receptors and risk of periodontitis: Evidence based on 12,793 subjects. Hum. Immunol. 2015, 76, 496–504. [Google Scholar] [CrossRef]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Ferrara, T.M.; Ben, S.; Riccardi, S.L.; Cole, J.B.; Gowan, K.; Holland, P.J.; Bennett, D.C.; et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012, 44, 676–680. [Google Scholar] [CrossRef]
- Cunninghame Graham, D.S.; Morris, D.L.; Bhangale, T.R.; Criswell, L.A.; Syvanen, A.C.; Ronnblom, L.; Behrens, T.W.; Graham, R.R.; Vyse, T.J. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 2011, 7, e1002341. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Jin, X.; Xu, J.; Gao, J.; Du, X.; Duan, D.; Li, B.; Zhao, J.; Zhan, W.; Tang, H.; et al. Sequencing-based approach identified three new susceptibility loci for psoriasis. Nat. Commun. 2014, 5, 4331. [Google Scholar] [CrossRef] [Green Version]
- Funabiki, M.; Kato, H.; Miyachi, Y.; Toki, H.; Motegi, H.; Inoue, M.; Minowa, O.; Yoshida, A.; Deguchi, K.; Sato, H.; et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 2014, 40, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Rutsch, F.; MacDougall, M.; Lu, C.; Buers, I.; Mamaeva, O.; Nitschke, Y.; Rice, G.I.; Erlandsen, H.; Kehl, H.G.; Thiele, H.; et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef]
- Oda, H.; Nakagawa, K.; Abe, J.; Awaya, T.; Funabiki, M.; Hijikata, A.; Nishikomori, R.; Funatsuka, M.; Ohshima, Y.; Sugawara, Y.; et al. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet. 2014, 95, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Qu, K.; Modis, Y. Cryo-EM Structures of MDA5-dsRNA Filaments at Different Stages of ATP Hydrolysis. Mol. Cell 2018, 72, 999–1012.e1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchikawa, E.; Lethier, M.; Malet, H.; Brunel, J.; Gerlier, D.; Cusack, S. Structural Analysis of dsRNA Binding to Anti-viral Pattern Recognition Receptors LGP2 and MDA5. Mol. Cell 2016, 62, 586–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger. Available online: https://pymol.org/2/support.html (accessed on 15 August 2021).
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systèmes BIOVIA. Available online: https://www.3ds.com/products-services/biovia/(accessed on 15 August 2021).
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Gosu, V.; Son, S.; Shin, D.; Song, K.-D. Insights into the dynamic nature of the dsRNA-bound TLR3 complex. Sci. Rep. 2019, 9, 3652. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, B.; Parekh, N. NAPS: Network Analysis of Protein Structures. Nucleic Acids Res. 2016, 44, W375–W382. [Google Scholar] [CrossRef] [Green Version]
- Mahita, J.; Sowdhamini, R. Probing subtle conformational changes induced by phosphorylation and point mutations in the TIR domains of TLR2 and TLR3. Proteins 2018, 86, 524–535. [Google Scholar] [CrossRef]
- Gosu, V.; Shin, D.; Song, K.D.; Heo, J.; Oh, J.D. Molecular modeling and dynamic simulation of chicken Mx protein with the S631N polymorphism. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Gosu, V.; Won, K.; Oh, J.D.; Shin, D. Conformational Changes Induced by S34Y and R98C Variants in the Death Domain of Myd88. Front. Mol. Biosci. 2020, 7, 27. [Google Scholar] [CrossRef]
- Anwar, M.A.; Choi, S. Structure-Activity Relationship in TLR4 Mutations: Atomistic Molecular Dynamics Simulations and Residue Interaction Network Analysis. Sci. Rep. 2017, 7, 43807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehury, B.; Raina, V.; Misra, N.; Suar, M. Effect of mutation on structure, function and dynamics of receptor binding domain of human SARS-CoV-2 with host cell receptor ACE2: A molecular dynamics simulations study. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trajectory Analysis | MDA5WT | MDA5L372F | MDA5A452T | MDA5R779H | MDA5R822Q |
|---|---|---|---|---|---|
| RMSD (nm) | 0.4206 (0.03) | 0.5222 (0.04) | 0.6089 (0.09) | 0.5186 (0.05) | 0.5255 (0.04) |
| RMSD(dsRNA) (nm) | 0.2527 (0.03) | 0.216 (0.02) | 0.2357 (0.02) | 0.1938 (0.02) | 0.2078 (0.02) |
| Rg (nm) | 3.0658 (0.01) | 2.9921 (0.02) | 3.1180 (0.02) | 3.1015 (0.02) | 3.0246 (0.02) |
| SASA (nm2) | 385 (7) | 377 (10) | 384 (9) | 383 (7) | 383 (7) |
| Intra H-bonds | 525 (16) | 529 (17) | 524 (13) | 536 (14) | 541 (27) |
| Interaction energies (kJ/mol) | −1997 (143) | −2612 (194) | −2003 (186) | −2102 (165) | −2803 (162) |
| Domains | MDA5WT | MDA5L372F | MDA5A452T | MDA5R779H | MDA5R822Q |
|---|---|---|---|---|---|
| Hel1 | K365 | K365 | K365 | K365 | N364 |
| K397 | V366 | H447 | T394 | V366 | |
| Q415 | G392 | N449 | Q395 | G392 | |
| K450 | T394 | H447 | T413 | ||
| A452 | Q395 | N449 | Q415 | ||
| T413 | |||||
| Q415 | |||||
| H447 | |||||
| N449 | |||||
| Hel2i | Q576 | Q576 | Q576 | Q580 | Q576 |
| Q580 | E579 | Q580 | Q584 | Q580 | |
| Q584 | Q580 | Q584 | K587 | ||
| K587 | K587 | K587 | |||
| R605 | |||||
| Hel2 | K726 | K726 | K726 | K726 | K726 |
| R728 | R728 | R728 | R728 | R728 | |
| G756 | A757 | G756 | G756 | A757 | |
| A757 | H759 | A757 | A757 | S761 | |
| Q771 | S760 | Q768 | V791 | K764 | |
| V791 | S760 | Q771 | Q768 | ||
| Q771 | I814 | Q771 | |||
| Pincer | K889 | K889 | K885 | K889 | K885 |
| R890 | R890 | K889 | N891 | T888 | |
| A893 | K894 | R890 | |||
| C-terminal domain | M926 | E924 | K949 | D953 | E924 |
| H927 | L947 | R985 | Y954 | M926 | |
| N944 | D953 | K1001 | K975 | K949 | |
| K949 | K975 | K983 | D953 | ||
| K983 | K1001 | I956 | |||
| R985 | K1002 | V973 | |||
| K1001 | K975 | ||||
| K1002 | K983 | ||||
| R985 | |||||
| K1001 | |||||
| K1002 |
| |CB (MDA5WT) − CB (MDA5L372F)| ≥ 0.2 |
| 418, 447, 449, 451, 453, 492, 616, 617, 618, 621, 622, 625, 725, 730, 733, 790, 791, 792, 793, 794, 795, 808, 810, 812, 814, 884, 886, 887, 888, 890, 894, 896, 897, 926, 928, 987, 1010 |
| |CB (MDA5WT) − CB (MDA5A452T)| ≥ 0.2 |
| 331, 363, 368, 451, 615, 621, 622, 625, 626, 634, 636, 733, 791, 792, 793, 794, 795, 806, 812, 885, 887, 888, 889, 891, 892, 894, 896, 897, 926, 928, 981 |
| |CB (MDA5WT) − CB (MDA5R779H)| ≥ 0.2 |
| 359, 418, 447, 449, 451, 455, 459, 492, 544, 546, 612, 616, 618, 621, 625, 628, 632, 636, 639, 674, 677, 730, 731, 733, 734, 735, 737, 790, 791, 792, 794, 795, 796, 803, 812, 814, 815, 835, 836, 885, 887, 888, 889, 890, 892, 893, 895, 897, 908, 942, 965, 984, 1004, 1009 |
| |CB (MDA5WT) − CB (MDA5R822Q)| ≥ 0.2 |
| 367, 368, 369, 413, 453, 456, 492, 615, 616, 617, 730, 731, 733, 755, 764, 766, 768, 791, 793, 795, 814, 815, 884, 885, 890, 892, 894, 895, 896, 897, 949, 955, 956, 957, 971 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gosu, V.; Sasidharan, S.; Saudagar, P.; Lee, H.-K.; Shin, D. Computational Insights into the Structural Dynamics of MDA5 Variants Associated with Aicardi–Goutières Syndrome and Singleton–Merten Syndrome. Biomolecules 2021, 11, 1251. https://doi.org/10.3390/biom11081251
Gosu V, Sasidharan S, Saudagar P, Lee H-K, Shin D. Computational Insights into the Structural Dynamics of MDA5 Variants Associated with Aicardi–Goutières Syndrome and Singleton–Merten Syndrome. Biomolecules. 2021; 11(8):1251. https://doi.org/10.3390/biom11081251
Chicago/Turabian StyleGosu, Vijayakumar, Santanu Sasidharan, Prakash Saudagar, Hak-Kyo Lee, and Donghyun Shin. 2021. "Computational Insights into the Structural Dynamics of MDA5 Variants Associated with Aicardi–Goutières Syndrome and Singleton–Merten Syndrome" Biomolecules 11, no. 8: 1251. https://doi.org/10.3390/biom11081251