
Comparative Transcriptomic Analysis Reveals the Effects of Drought on the Biosynthesis of Methyleugenol in Asarum sieboldii Miq.


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Drought-Stress Procedures
2.2. Volatile-Oils Extraction and GC-MS Analysis
2.3. Library Construction and Illumina Sequencing
2.4. Quality Control and De Novo Transcriptome Assembly
2.5. Gene Functional Annotation
2.6. Differential Expression Analysis
2.7. Real-Time qPCR Validation of RNA-Seq Data
3. Results
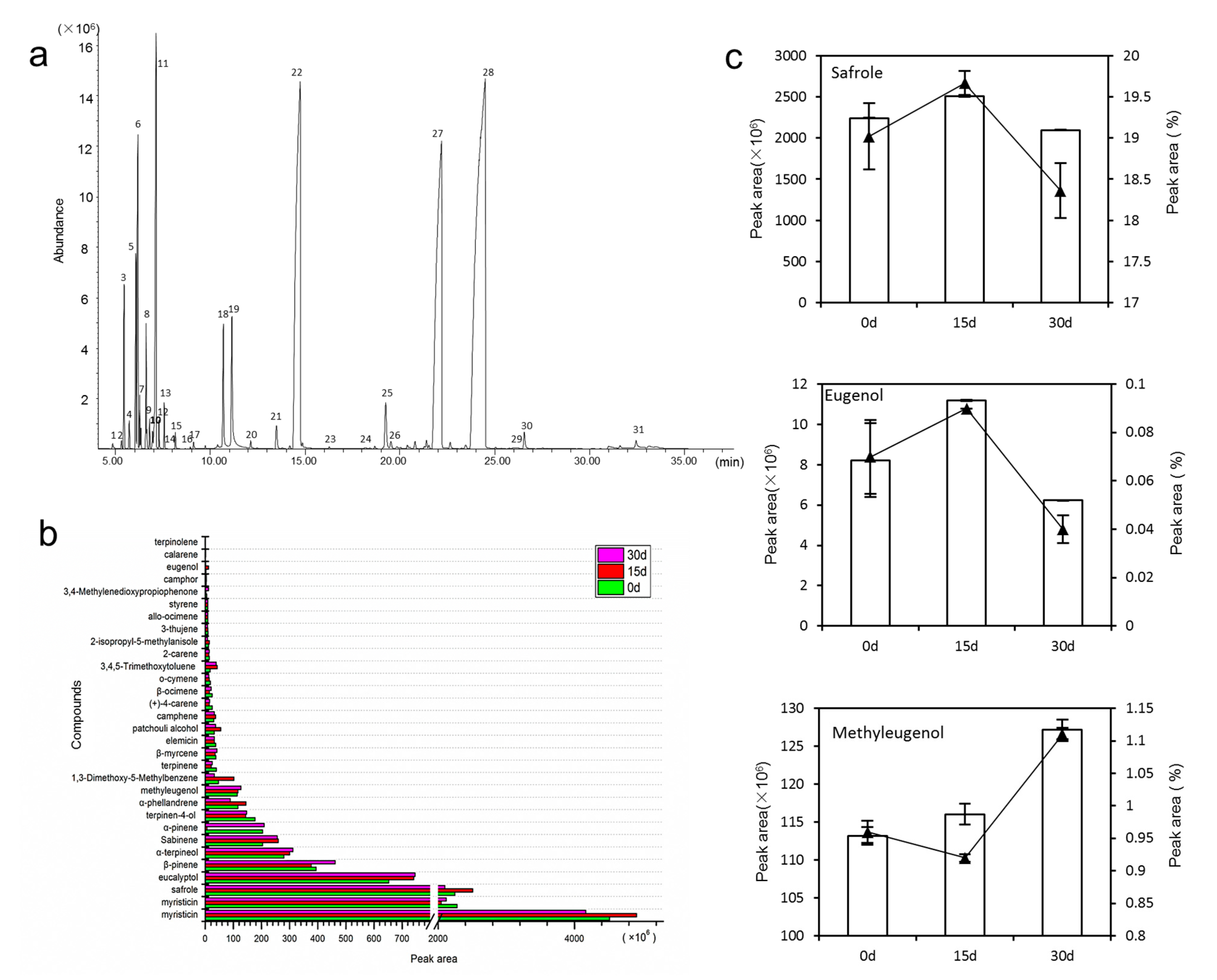
3.1. Volatile Oils Content and Composition
3.2. Sequencing Output and Assembly
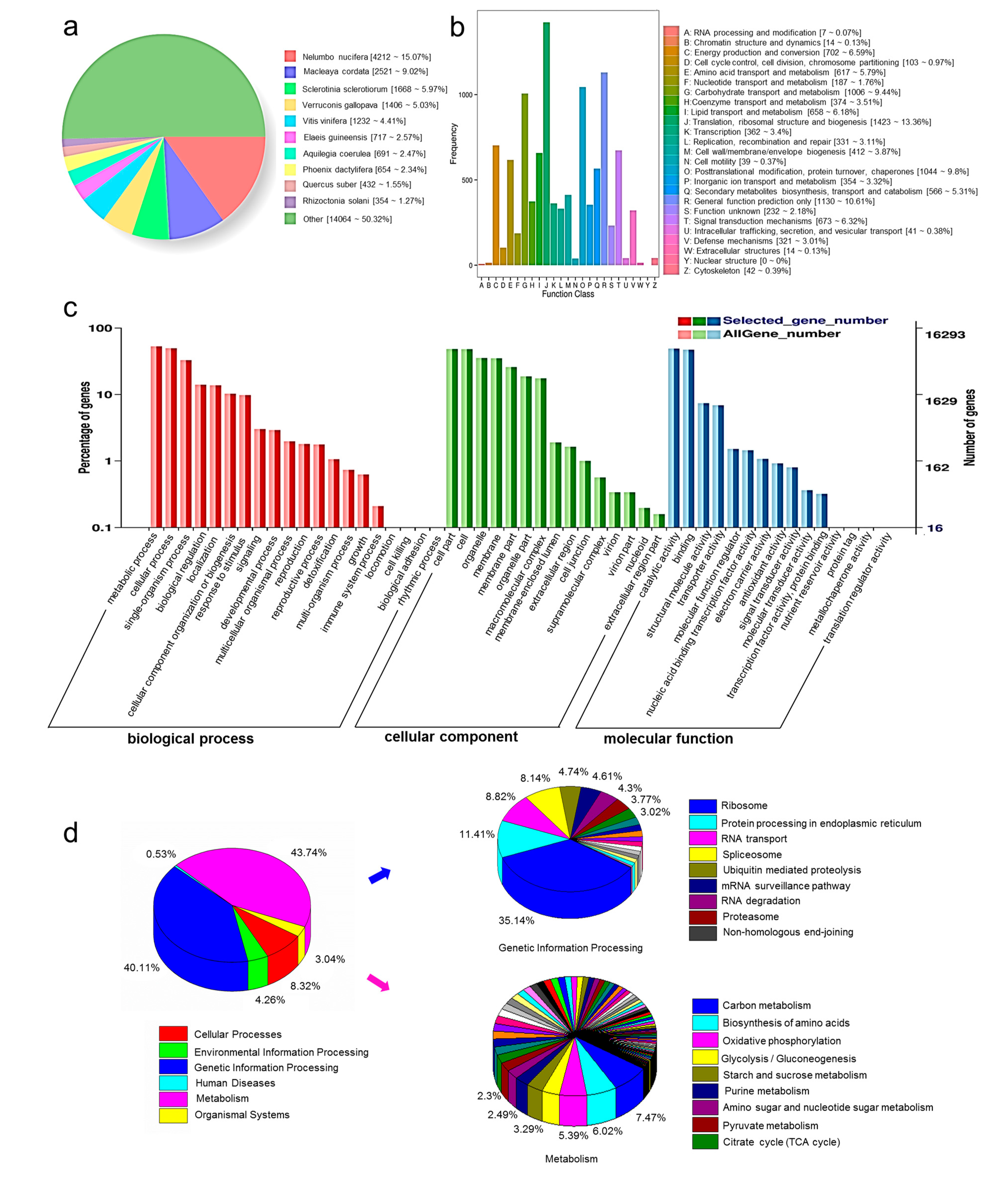
3.3. Functional Annotation and Classification
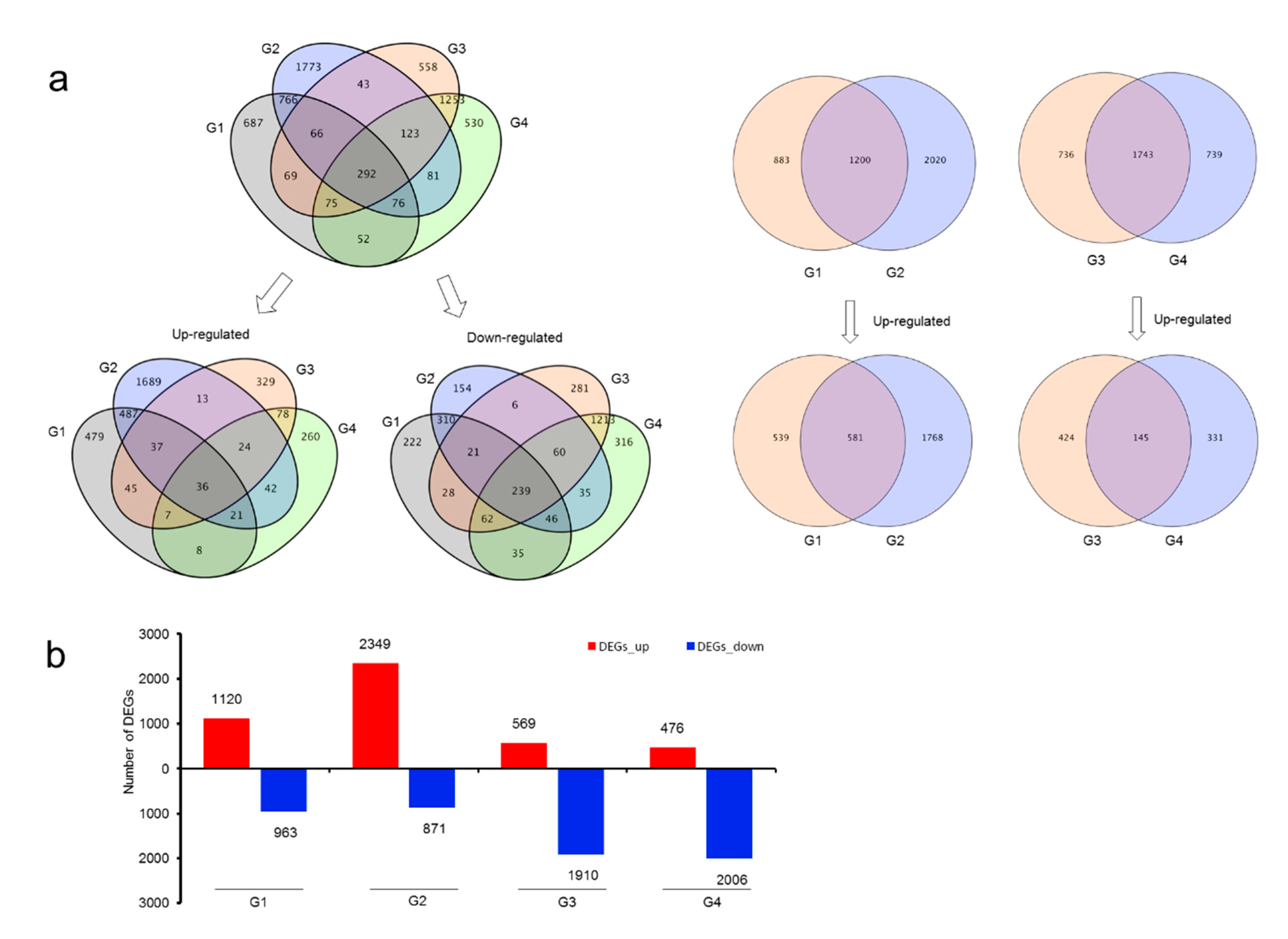
3.4. Differentially Expressed Genes in Root and Rhizome
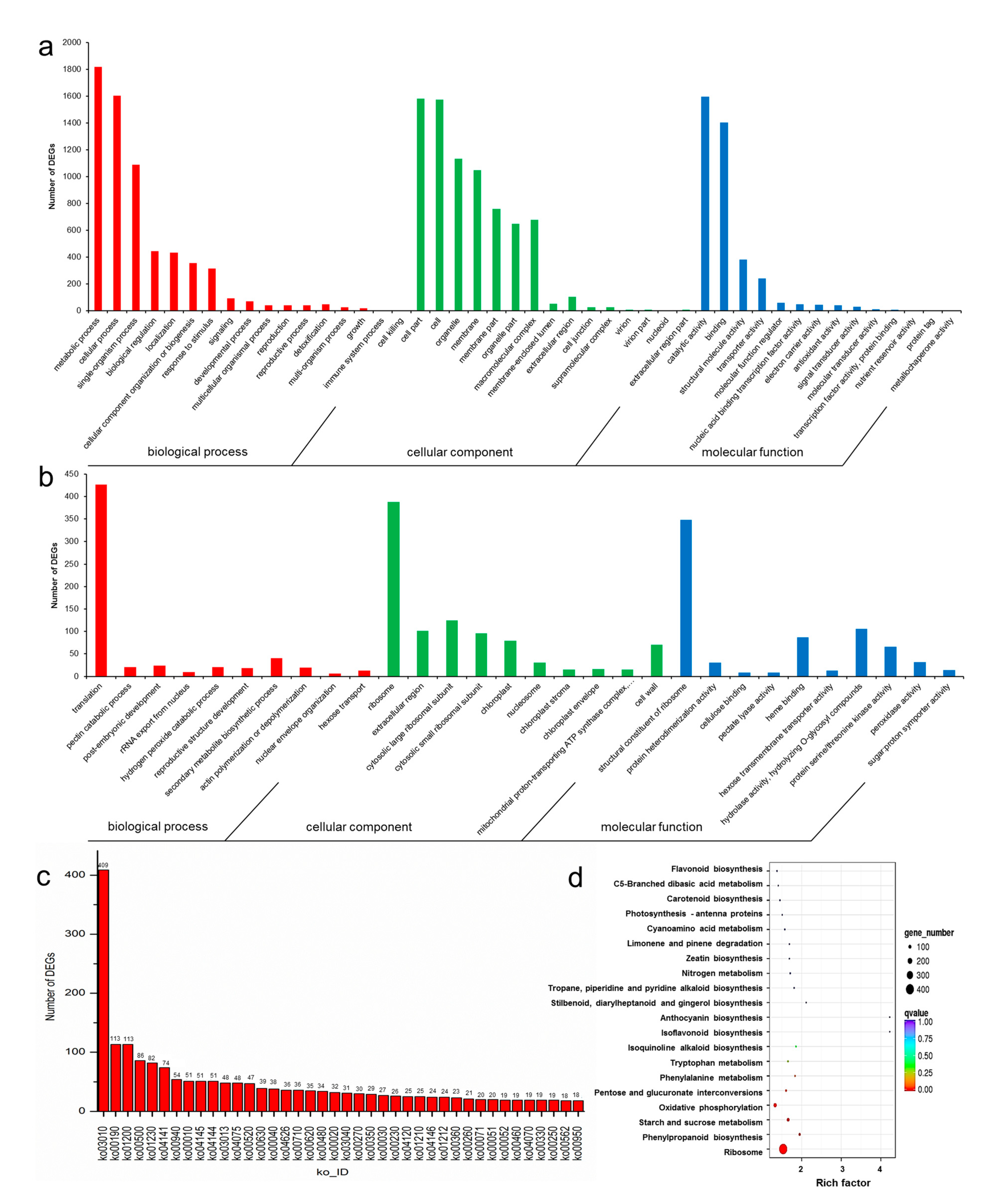
3.5. GO and KEGG Enrichment of DEGs
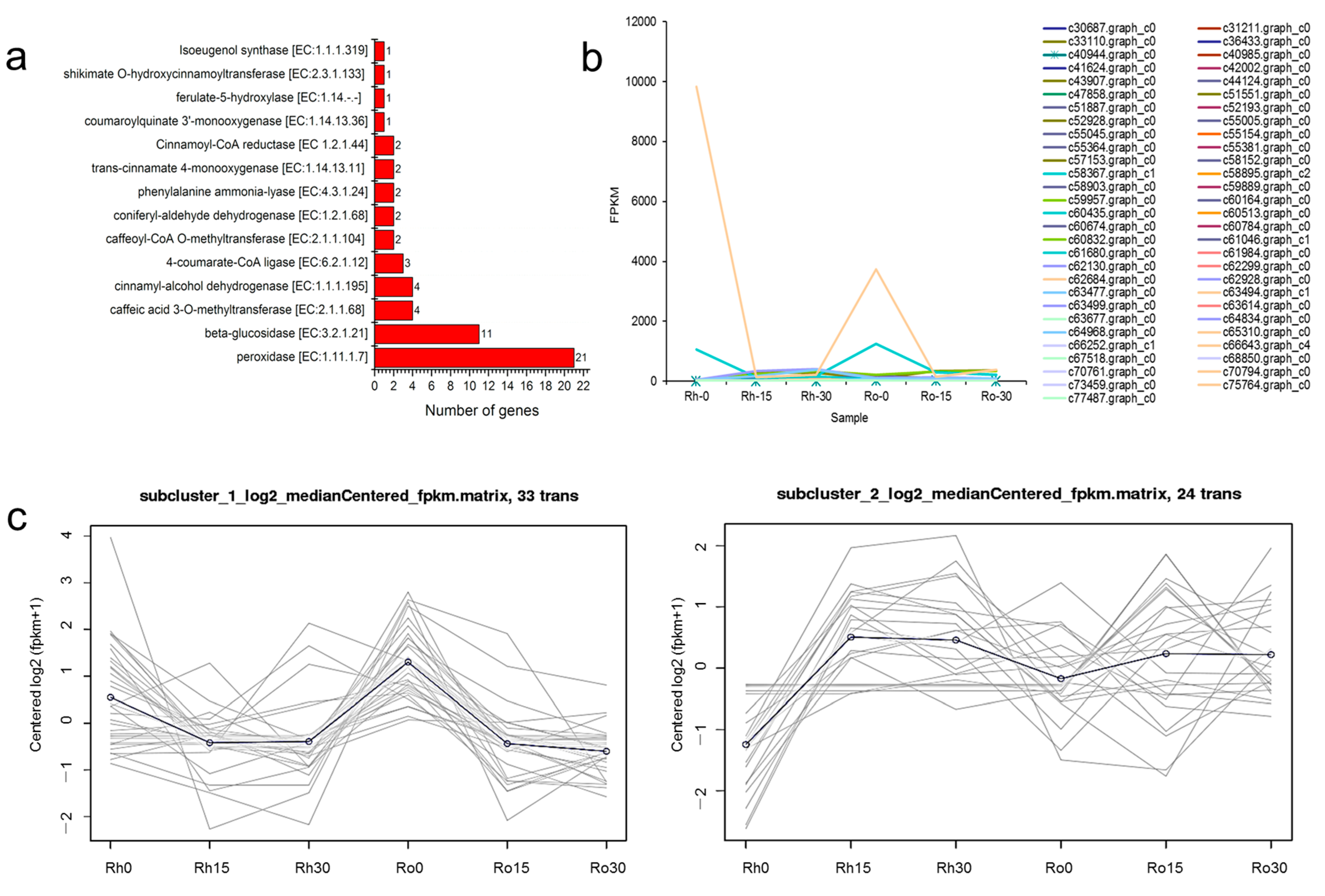
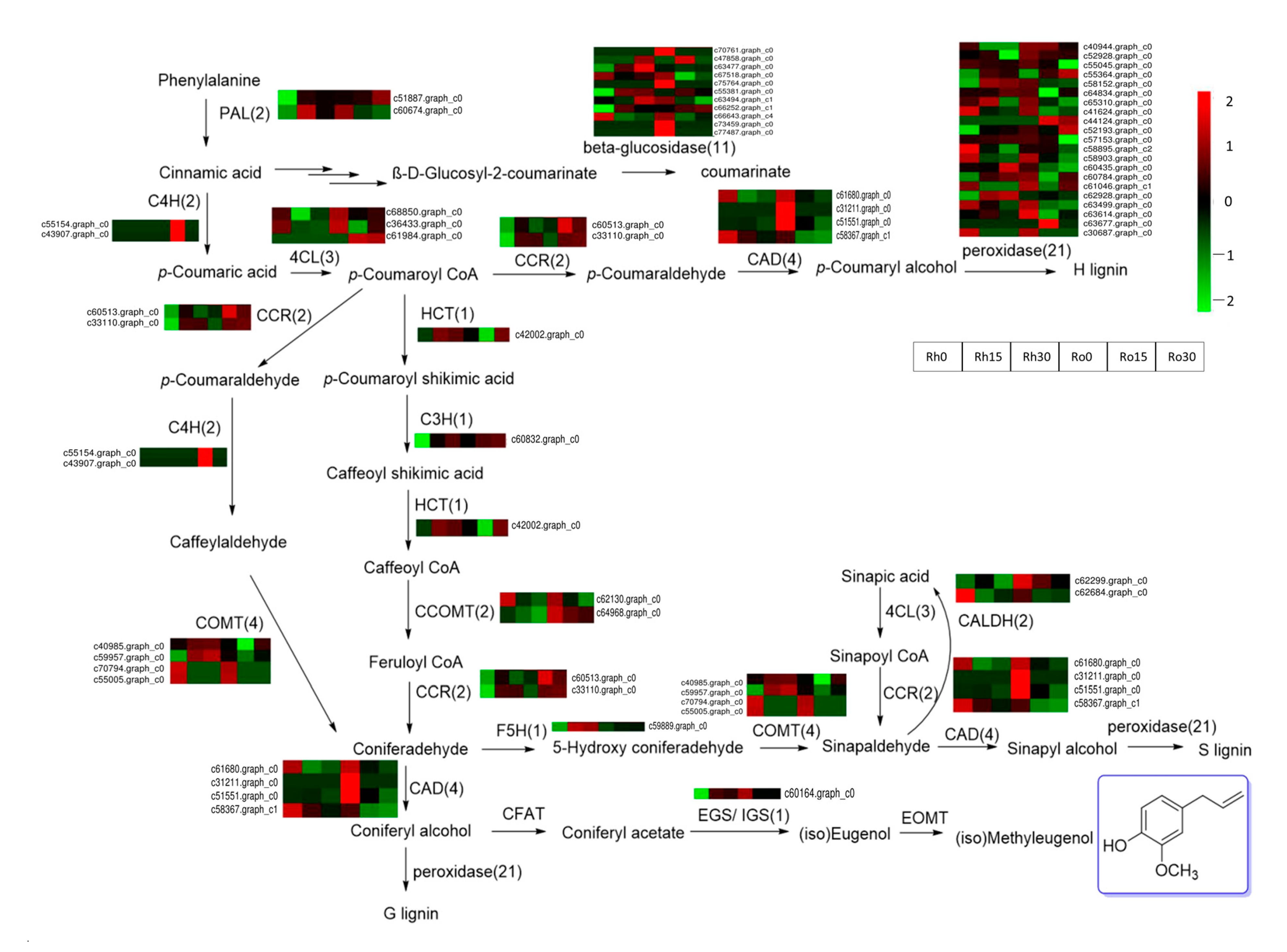
3.6. DEGs Related to Methyleugenol Biosynthesis
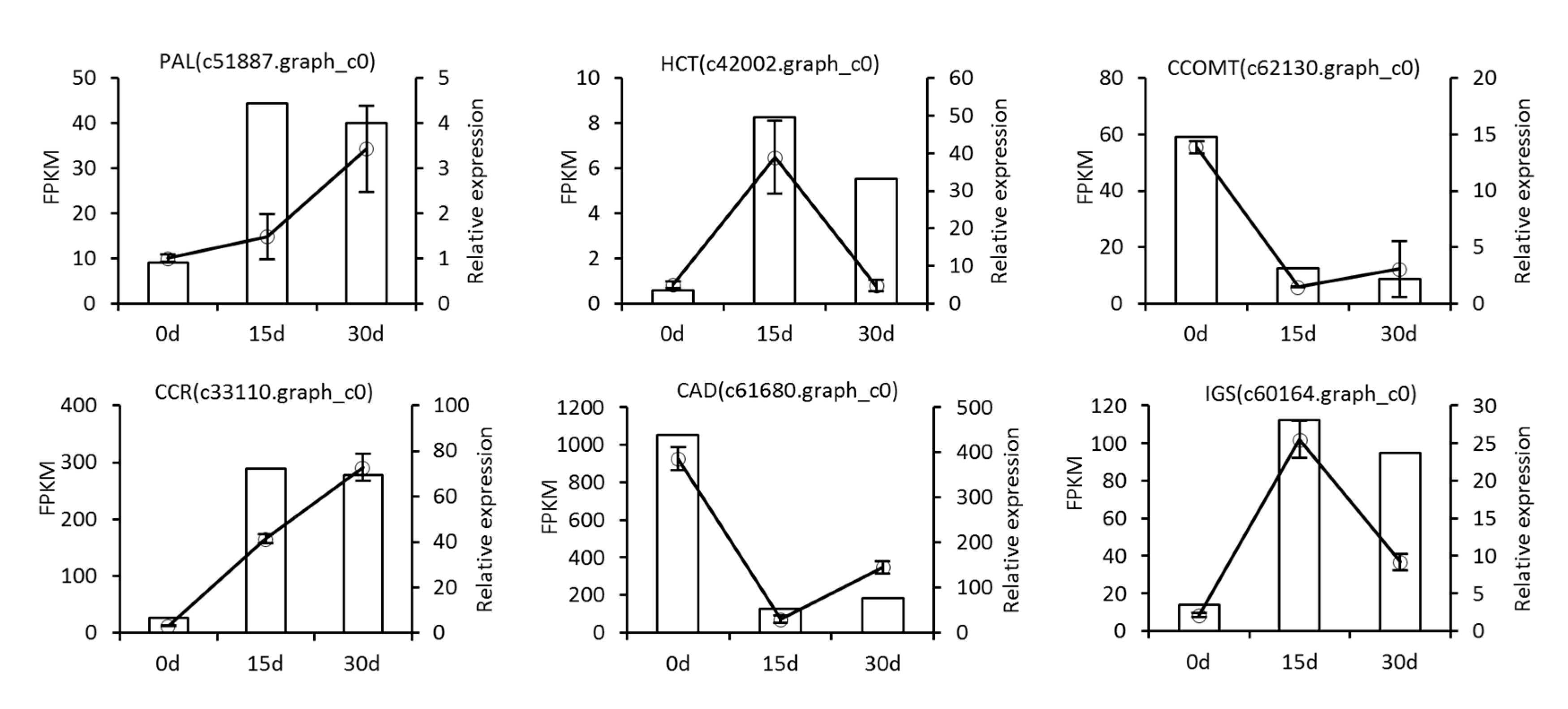
3.7. qRT-PCR Validation
4. Discussion
4.1. Drought Stress Reduce Volatile Oil Content
4.2. Drought Stress Induced the Gene Differentially Expressed in Methyleugenol Biosynthesis Pathway
4.3. Drought Stress Induced the Expression of Which Was Hypothetically Involved in the Biosynthesis of Safrole the Synthesis of Safrole
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mahajan, S.; Tuteja, N. Cold, salinity and drought stresses: An overview. Arch. Biochem. Biophys. 2005, 444, 139–158. [Google Scholar] [CrossRef]
- Yokota, A.; Kawasaki, S.; Iwano, M.; Nakamura, C.; Miyake, C.; Akashi, K. Citrulline and DRIP-1 protein (ArgE homologue) in drought tolerance of wild watermelon. Ann. Bot. 2002, 89, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Sourour, A. A review: Morphological, physiological, biochemical and molecular plant responses to water deficit stress. Int. J. Eng. Sci. 2017, 6, 1–4. [Google Scholar] [CrossRef]
- Chaves, M.M.; Maroco, J.P.; Pereira, J.S. Understanding plant responses to drought from genes to the whole plant. Funct. Plant Biol. 2003, 30, 239–264. [Google Scholar] [CrossRef]
- Rao, S.; Tian, Y.; Xia, X.; Li, Y.; Chen, J. Chromosome doubling mediates superior drought tolerance in Lycium ruthenicum via abscisic acid signaling. Hortic. Res. 2020, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Wang, Y.; Huang, L.; Du, F.; Zhao, X.; Li, Z.; Wang, W.; Fu, B. A U-box E3 ubiquitin ligase OsPUB67 is positively involved in drought tolerance in rice. Plant Mol. Biol. 2020, 102, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Kamei, A.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Molecular responses to drought, salinity and frost: Common and different paths for plant protection. Curr. Opin. Biotechnol. 2003, 14, 194–199. [Google Scholar] [CrossRef]
- Lee, J.Y.; Moon, S.S.; Hwang, B.K. Isolation and antifungal activity of kakuol, a propiophenone derivative from Asarum sieboldii rhizome. Pest Manag. Sci. 2005, 61, 821–825. [Google Scholar] [CrossRef]
- Chen, C.; Spriano, D.; Lehmann, T.; Meier, B. Reduction of safrole and methyleugenol in Asari radix et rhizoma by decoction. Forsch. Komplementmed. 2009, 16, 162–166. [Google Scholar] [CrossRef]
- Wu, H.; Li, J.; Zhang, F.; Li, L.; Liu, Z.; He, Z. Essential oil components from Asarum sieboldii Miquel are toxic to the house dust mite Dermatophagoides farinae. Parasitol. Res. 2012, 111, 1895–1899. [Google Scholar] [CrossRef]
- Kim, S.-J.; Gao Zhang, C.; Taek Lim, J. Mechanism of anti-nociceptive effects of Asarum sieboldii Miq. Radix: Potential role of bradykinin, histamine and opioid receptor-mediated pathways. J. Ethnopharmacol. 2003, 88, 5–9. [Google Scholar] [CrossRef]
- Jeong, M.; Kim, H.M.; Lee, J.S.; Choi, J.H.; Jang, D.S. (-)-Asarinin from the Roots of Asarum sieboldii Induces Apoptotic Cell Death via Caspase Activation in Human Ovarian Cancer Cells. Molecules 2018, 23, 1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, A.K.; Whyte, I.M.; Bensoussan, A.; Dawson, A.H.; Zhu, X.; Myers, S.P. Chinese Herbal Medicine Toxicology Database: Monograph on Herba Asari, “Xi Xin”. Clin. Toxicol. 2002, 40, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Kim, S.J. Phytochemical, toxicological and pharmacological studies of Asiasari Radix et Rhizoma: A Review. Trop. J. Pharm. Res. 2015, 14, 545. [Google Scholar] [CrossRef] [Green Version]
- De Vincenzi, M.; Silano, M.; Stacchini, P.; Scazzocchio, B. Constituents of aromatic plants: I. Methyleugenol. Fitoterapia 2000, 71, 216–221. [Google Scholar] [CrossRef]
- Liu, J.; Xu, C.; Zhang, H.; Liu, F.; Ma, D.; Liu, Z. Comparative Transcriptomics Analysis for Gene Mining and Identification of a Cinnamyl Alcohol Dehydrogenase Involved in Methyleugenol Biosynthesis from Asarum sieboldii Miq. Molecules 2018, 23, 3184. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi Mandoulakani, B.; Eyvazpour, E.; Ghadimzadeh, M. The effect of drought stress on the expression of key genes involved in the biosynthesis of phenylpropanoids and essential oil components in basil (Ocimum basilicum L.). Phytochemistry 2017, 139, 1–7. [Google Scholar] [CrossRef]
- Khakdan, F.; Nasiri, J.; Ranjbar, M.; Alizadeh, H. Water deficit stress fluctuates expression profiles of 4Cl, C3H, COMT, CVOMT and EOMT genes involved in the biosynthetic pathway of volatile phenylpropanoids alongside accumulation of methylchavicol and methyleugenol in different Iranian cultivars of basil. J. Plant Physiol. 2017, 218, 74–83. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Kim, H.C.; Shin, S.; Kim, K.-H.; Moon, J.-C.; Kim, J.Y.; Lee, B.-M. Transcriptome Analysis of Flowering Time Genes under Drought Stress in Maize Leaves. Front. Plant Sci. 2017, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Ma, Y.; Shukla, V.; Merewitz, E.B. Transcriptome analysis of creeping bentgrass exposed to drought stress and polyamine treatment. PLoS ONE 2017, 12, e0175848. [Google Scholar]
- Liu, C.Y.; Liu, X.Q.; Long, D.P.; Cao, B.N.; Xiang, Z.H.; Zhao, A.C. De novo assembly of mulberry (Morus alba L.) transcriptome and identification of candidate unigenes related to salt stress responses. Russ. J. Plant Physiol. 2017, 64, 738–748. [Google Scholar] [CrossRef]
- Han, X.; Zhang, L.; Miao, X.; Hu, X.; Nan, S.; Fu, H. Transcriptome analysis reveals the molecular mechanisms of mucilage biosynthesis during Artemisia sphaerocephala seed development. Ind. Crops Prod. 2020, 145, 111991. [Google Scholar] [CrossRef]
- Lin, M.-Y.; Niu, H.; Liu, J.-J.; Liu, W.; Liu, Z. Identification of genes related to biosynthesis of methyleugenol in Asarum sieboldii by transcriptome sequence analysis. Chin. Tradit. Herb. Drugs 2017, 48, 3160–3167. [Google Scholar]
- National Pharmacopoeia Committee. Pharmacopoeia of People’s Republic of China [M]. Part 4; China Medical Science Press: Beijing, China, 2015; Appendix 2204. [Google Scholar]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.; Chen, Y.W.; He, F.C. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Tatusov, R.; Galperin, M.; Natale, D.; Koonin, E. The COG Database: A Tool for Genome-Scale Analysis of Protein Functions and Evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.; Abrams, N.; Jackson, J.; Jacobs, A.; Krylov, D.; Makarova, K.; Mazumder, R.; Mekhedov, S.; Nikolskaya, A.; Bachoti, S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.; Barker, W.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, S.; Cook, H.; Heller, D.; Walter, M.; Rattei, T.; Mende, D.; Sunagawa, S.; Kuhn, M.; et al. EGGNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nastou, K.C.; Tsaousis, G.N.; Papandreou, N.C.; Hamodrakas, S.J. MBPpred: Proteome-wide detection of membrane lipid-binding proteins using profile Hidden Markov Models. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Qu, C.; Yang, J.; Zhang, W.; Ji, Y. Transcriptome changes in the phenylpropanoid pathway in senescing leaves of Toona sinensis. Acta Physiol. Plant. 2019, 41, 126. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Shen, C.; Chen, J.X.; He, C.T.; Zhou, Q.; Tan, X.; Yuan, J.G.; Yang, Z.Y. Comparative Transcriptome Analysis of Two Ipomoea aquatica Forsk. Cultivars Targeted to Explore Possible Mechanism of Genotype-Dependent Accumulation of Cadmium. J. Agric. Food Chem. 2016, 64, 5241–5250. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing Under Dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Pan, L.; Liu, W.; Lin, M.; Hongqing, L.I.; Liu, Z. Screening of Reference Genes for Quantitative Real-time PCR Analysis in Asarum sieboldii. Biotechnol. Bull. 2017, 33, 174–179. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Manzi, M.; Pitarch-Bielsa, M.; Arbona, V.; Gómez-Cadenas, A. Leaf dehydration is needed to induce abscisic acid accumulation in roots of citrus plants. Environ. Exp. Bot. 2017, 139, 116–126. [Google Scholar] [CrossRef]
- Smith, S.; De Smet, I. Root system architecture: Insights from Arabidopsis and cereal crops. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1441–1452. [Google Scholar] [CrossRef] [Green Version]
- Borchert, P.; Wislocki, A.P.; Miller, J.A.; Miller, E.C. The metabolism of it naturally occurring hepatocarcinogen safrole to 1’-hydroxysafrole and the electronphilic reactivity of 1-acetoxysafrole. Cancer Res. 1973, 33, 575–589. [Google Scholar] [PubMed]
- Miller, E.C.; Swanson, A.B.; Phillips, D.H.; Fletcher, T.L.; Miller, J.A. Structure-Activity Studies of the Carcinogenicities in the Mouse and Rat of Some Naturally Occurring and Synthetic Alkenylbenzene Derivatives Related to Safrole and Estragole. Cancer Res. 1983, 43, 1124–1134. [Google Scholar] [PubMed]
- Daimon, H.; Sawada, S.; Asakura, S.; Sagami, F. In vivo genotoxicity and DNA adduct levels in the liver of rats treated with safrole. Carcinogenesis 1998, 19, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.-H.; Chai, Y.-R.; Li, J.-N.; Chen, L. Molecular cloning of two genes encoding cinnamate 4-hydroxylase (C4H) from oilseed rape (Brassica napus). J. Biochem. Mol. Biol. 2007, 40, 247–260. [Google Scholar] [CrossRef]
- Ikezawa, N.; Iwasa, K.; Sato, F. CYP719A subfamily of cytochrome P450 oxygenases and isoquinoline alkaloid biosynthesis in Eschscholzia californica. Plant Cell Rep. 2009, 28, 123–133. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range | Transcript | Unigene |
|---|---|---|
| 300–500 | 38,585 (16.88%) | 23,455 (43.97%) |
| 500–1000 | 48,103 (21.04%) | 15,156 (28.41%) |
| 1000–2000 | 67,681 (29.60%) | 8559 (16.04%) |
| 2000+ | 74,249 (32.48%) | 6174 (11.57%) |
| Total Number | 228,618 | 53,344 |
| Total Length | 387,891,699 | 51,209,482 |
| N50 Length | 2423 | 1469 |
| Mean Length | 1696.68 | 959.99 |
| Database | Annotated_Number | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| COG | 9620 | 4634 | 4986 |
| GO | 16,293 | 9510 | 6783 |
| KEGG | 10,789 | 5899 | 4890 |
| KOG | 16,792 | 9060 | 7732 |
| Pfam | 19,739 | 9586 | 10,153 |
| Swiss-Prot | 17,548 | 8390 | 9158 |
| eggNOG | 26,155 | 14,173 | 11,982 |
| NR | 27,995 | 15,693 | 12,302 |
| All_Annotated | 28,715 | 16,313 | 12,402 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Ali, T.; Liu, Z. Comparative Transcriptomic Analysis Reveals the Effects of Drought on the Biosynthesis of Methyleugenol in Asarum sieboldii Miq. Biomolecules 2021, 11, 1233. https://doi.org/10.3390/biom11081233
Liu F, Ali T, Liu Z. Comparative Transcriptomic Analysis Reveals the Effects of Drought on the Biosynthesis of Methyleugenol in Asarum sieboldii Miq. Biomolecules. 2021; 11(8):1233. https://doi.org/10.3390/biom11081233
Chicago/Turabian StyleLiu, Fawang, Tahir Ali, and Zhong Liu. 2021. "Comparative Transcriptomic Analysis Reveals the Effects of Drought on the Biosynthesis of Methyleugenol in Asarum sieboldii Miq." Biomolecules 11, no. 8: 1233. https://doi.org/10.3390/biom11081233