
RET Regulates Human Medullary Thyroid Cancer Cell Proliferation through CDK5 and STAT3 Activation

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Cell Viability and Proliferation Assay
2.3. Western Blotting
2.4. Immunoprecipitation
2.5. Immunocytochemistry
2.6. siRNA Transfection
2.7. Statistical Analysis
3. Results
3.1. Downregulation of CDK5 Decreases GDNF-Induced Human Medullary Thyroid Cancer Cell Viability
3.2. CDK5 Physically Interacts with RET Protein in Human Medullary Thyroid Cancer Cells
3.3. Downregulation of CDK5 Inhibits GDNF-Induced STAT3 Activation in Human Medullary Thyroid Cancer Cells
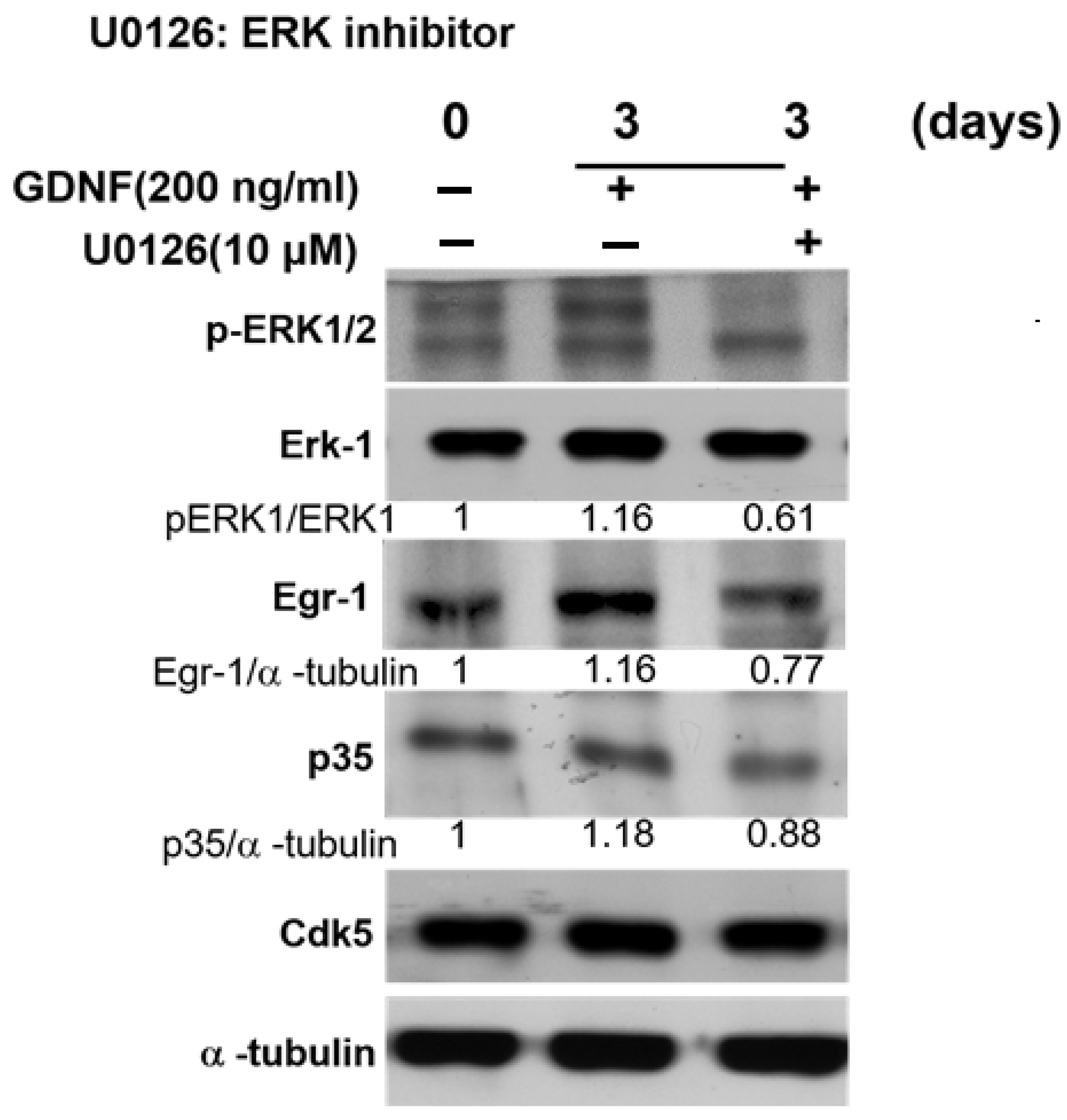
3.4. GDNF-Induced p35/CDK5 Activity Is ERK-Egr1-Dependent in Human Medullary Thyroid Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gambardella, C.; Offi, C.; Patrone, R.; Clarizia, G.; Mauriello, C.; Tartaglia, E.; Di Capua, F.; Di Martino, S.; Romano, R.M.; Fiore, L.; et al. Calcitonin negative Medullary Thyroid Carcinoma: A challenging diagnosis or a medical dilemma? BMC Endocr. Disord. 2019, 19, 45. [Google Scholar] [CrossRef]
- Ceolin, L.; Duval, M.; Benini, A.F.; Ferreira, C.V.; Maia, A.L. Medullary thyroid carcinoma beyond surgery: Advances, challenges, and perspectives. Endocr. Relat. Cancer 2019, 26, R499–R518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, N.; Shi, X.; Zhao, J.J.; Guan, H.; Zhang, T.T.; Wen, S.S.; Liao, T.; Hu, J.Q.; Liu, W.Y.; Wang, Y.L.; et al. Genomic and Transcriptomic Characterization of Sporadic Medullary Thyroid Carcinoma. Thyroid 2020. [Google Scholar] [CrossRef]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET fusions in solid tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef]
- Plaza Menacho, I.; Koster, R.; van der Sloot, A.M.; Quax, W.J.; Osinga, J.; van der Sluis, T.; Hollema, H.; Burzynski, G.M.; Gimm, O.; Buys, C.H.; et al. RET-familial medullary thyroid carcinoma mutants Y791F and S891A activate a Src/JAK/STAT3 pathway, independent of glial cell line-derived neurotrophic factor. Cancer Res. 2005, 65, 1729–1737. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Chakravarti, A. A gene regulatory network explains RET-EDNRB epistasis in Hirschsprung disease. Hum. Mol. Genet. 2019, 28, 3137–3147. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, J.M.; Ling, A.Y.; Turner, T.N.; Sosa, M.X.; Krumm, N.; Chatterjee, S.; Kapoor, A.; Coe, B.P.; Nguyen, K.H.; Gupta, N.; et al. Molecular Genetic Anatomy and Risk Profile of Hirschsprung’s Disease. N. Engl. J. Med. 2019, 380, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Tomuschat, C.; Puri, P. RET gene is a major risk factor for Hirschsprung’s disease: A meta-analysis. Pediatr. Surg. Int. 2015, 31, 701–710. [Google Scholar] [CrossRef]
- Asai, N.; Jijiwa, M.; Enomoto, A.; Kawai, K.; Maeda, K.; Ichiahara, M.; Murakumo, Y.; Takahashi, M. RET receptor signaling: Dysfunction in thyroid cancer and Hirschsprung’s disease. Pathol. Int. 2006, 56, 164–172. [Google Scholar] [CrossRef] [PubMed]
- De Falco, V.; Carlomagno, F.; Li, H.Y.; Santoro, M. The molecular basis for RET tyrosine-kinase inhibitors in thyroid cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 307–318. [Google Scholar] [CrossRef]
- Hadoux, J.; Schlumberger, M. Chemotherapy and tyrosine-kinase inhibitors for medullary thyroid cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 335–347. [Google Scholar] [CrossRef]
- Bagherzadeh, M.; Aminorroaya, A.; Vafaeimanesh, J.; Mohajeri-Tehrani, M.R. Coexistence of medullary thyroid carcinoma and recurrent non-functional pituitary adenoma: A case report. J. Med. Case Rep. 2018, 12, 220. [Google Scholar] [CrossRef]
- Hedayati, M.; Zarif Yeganeh, M.; Sheikholeslami, S.; Afsari, F. Diversity of mutations in the RET proto-oncogene and its oncogenic mechanism in medullary thyroid cancer. Crit. Rev. Clin. Lab. Sci. 2016, 53, 217–227. [Google Scholar] [CrossRef]
- FDA Approves Selpercatinib; Pralsetinib May Soon Follow. Cancer Discov. 2020. [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Oo, T.F.; Kholodilov, N.; Burke, R.E. Regulation of natural cell death in dopaminergic neurons of the substantia nigra by striatal glial cell line-derived neurotrophic factor in vivo. J. Neurosci. 2003, 23, 5141–5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisk, T.; Farnebo, F.; Zedenius, J.; Grimelius, L.; Hoog, A.; Wallin, G.; Larsson, C. Expression of RET and its ligand complexes, GDNF/GFRalpha-1 and NTN/GFRalpha-2, in medullary thyroid carcinomas. Eur. J. Endocrinol. 2000, 142, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Moon, A. Glial cell-derived neurotrophic factor (GDNF) promotes low-grade Hs683 glioma cell migration through JNK, ERK-1/2 and p38 MAPK signaling pathways. Neurosci. Res. 2006, 56, 29–38. [Google Scholar] [CrossRef]
- Subramaniam, S.; Strelau, J.; Unsicker, K. GDNF prevents TGF-beta-induced damage of the plasma membrane in cerebellar granule neurons by suppressing activation of p38-MAPK via the phosphatidylinositol 3-kinase pathway. Cell. Tissue Res. 2008, 331, 373–383. [Google Scholar] [CrossRef]
- Zhou, L.; Too, H.P. GDNF family ligand dependent STAT3 activation is mediated by specific alternatively spliced isoforms of GFRalpha2 and RET. Biochim. Biophys. Acta 2013, 1833, 2789–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Namgung, U. Cdk5 Phosphorylation of STAT3 in Dorsal Root Ganglion Neurons Is Involved in Promoting Axonal Regeneration After Peripheral Nerve Injury. Int. Neurourol. J. 2020, 24, S19–S27. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.N.; Chen, M.C.; Lin, K.C.; Peng, Y.T.; Li, P.C.; Lin, E.; Chiang, M.C.; Hsieh, J.T.; Lin, H. Cyclin-dependent kinase 5 modulates STAT3 and androgen receptor activation through phosphorylation of Ser(7)(2)(7) on STAT3 in prostate cancer cells. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E975–E986. [Google Scholar] [CrossRef]
- Huang, P.H.; Chen, M.C.; Peng, Y.T.; Kao, W.H.; Chang, C.H.; Wang, Y.C.; Lai, C.H.; Hsieh, J.T.; Wang, J.H.; Lee, Y.T.; et al. Cdk5 Directly Targets Nuclear p21CIP1 and Promotes Cancer Cell Growth. Cancer Res. 2016, 76, 6888–6900. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.S.; Hsu, F.N.; Chiang, M.C.; You, S.C.; Chen, M.C.; Lo, M.J.; Lin, H. The role of Cdk5 in retinoic acid-induced apoptosis of cervical cancer cell line. Chin. J. Physiol. 2009, 52, 23–30. [Google Scholar] [CrossRef]
- Lin, H.; Chen, M.C.; Ku, C.T. Cyclin-dependent kinase 5 regulates steroidogenic acute regulatory protein and androgen production in mouse Leydig cells. Endocrinology 2009, 150, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Juang, J.L.; Wang, P.S. Involvement of Cdk5/p25 in digoxin-triggered prostate cancer cell apoptosis. J. Biol. Chem. 2004, 279, 29302–29307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.C.; Gao, A.C.; Lai, C.H.; Hsieh, J.T.; Lin, H. Induction of neuroendocrine differentiation in castration resistant prostate cancer cells by adipocyte differentiation-related protein (ADRP) delivered by exosomes. Cancer Lett. 2017, 391, 74–82. [Google Scholar] [CrossRef]
- Oner, M.; Lin, E.; Chen, M.C.; Hsu, F.N.; Shazzad Hossain Prince, G.M.; Chiu, K.Y.; Teng, C.J.; Yang, T.Y.; Wang, H.Y.; Yue, C.H.; et al. Future Aspects of CDK5 in Prostate Cancer: From Pathogenesis to Therapeutic Implications. Int. J. Mol. Sci. 2019, 20, 3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, G.; Yang, T.Y.; Lin, H.; Chen, M.C. Mechanistic insight of cyclin-dependent kinase 5 in modulating lung cancer growth. Chin. J. Physiol. 2019, 62, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Chen, M.C.; Huang, C.Y.; Hsu, S.L.; Huang, W.J.; Lin, M.S.; Wu, J.C.; Lin, H. All-trans retinoic acid induces DU145 cell cycle arrest through Cdk5 activation. Cell. Physiol. Biochem. 2014, 33, 1620–1630. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Huang, C.Y.; Hsu, S.L.; Lin, E.; Ku, C.T.; Lin, H.; Chen, C.M. Retinoic Acid Induces Apoptosis of Prostate Cancer DU145 Cells through Cdk5 Overactivation. Evid. Based Complement. Altern. Med. 2012, 2012, 580736. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Lin, H.; Hsu, F.N.; Huang, P.H.; Lee, G.S.; Wang, P.S. Involvement of cAMP in nerve growth factor-triggered p35/Cdk5 activation and differentiation in PC12 cells. Am. J. Physiol. Cell Physiol. 2010, 299, C516–C527. [Google Scholar] [CrossRef]
- Lin, H.; Chen, M.C.; Chiu, C.Y.; Song, Y.M.; Lin, S.Y. Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. J. Biol. Chem. 2007, 282, 2776–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.M.; Chi, C.W.; Chang, H.M.; Wu, L.H.; Lee, T.K.; Lin, J.D.; Chen, S.T.; Lee, C.H. Effects of clodronate on cancer growth and Ca2+ signaling of human thyroid carcinoma cell lines. Anticancer Res. 2004, 24, 1617–1623. [Google Scholar]
- Ke, C.C.; Liu, R.S.; Yang, A.H.; Liu, C.S.; Chi, C.W.; Tseng, L.M.; Tsai, Y.F.; Ho, J.H.; Lee, C.H.; Lee, O.K. CD133-expressing thyroid cancer cells are undifferentiated, radioresistant and survive radioiodide therapy. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.C.; Li, J.H.; Chen, C.C.; Chen, C.W.; Lin, H.; Wang, P.S. Inhibitory Effects of Digoxin and Digitoxin on Cell Growth in Human Ovarian Cancer Cell Line SKOV-3. Integr. Cancer 2021, 20, 15347354211002662. [Google Scholar] [CrossRef]
- Chou, J.C.; Lieu, F.K.; Ho, D.M.; Shen, H.Y.; Lin, P.H.; Hu, S.; Wang, S.W.; Lin, H.; Wang, P.S. Regulation of extracellular and intracellular prolactin on cell proliferation and survival rate through GHR/JAK2/STAT3 pathway in NSCLC. Chemosphere 2021, 264, 128604. [Google Scholar] [CrossRef]
- Chang, C.H.; Chen, M.C.; Chiu, T.H.; Li, Y.H.; Yu, W.C.; Liao, W.L.; Oner, M.; Yu, C.R.; Wu, C.C.; Yang, T.Y.; et al. Arecoline Promotes Migration of A549 Lung Cancer Cells through Activating the EGFR/Src/FAK Pathway. Toxins 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.C.; Chen, K.C.; Chang, G.C.; Lin, H.; Wu, C.C.; Kao, W.H.; Teng, C.J.; Hsu, S.L.; Yang, T.Y. RAGE acts as an oncogenic role and promotes the metastasis of human lung cancer. Cell Death Dis. 2020, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Phay, J.E.; Shah, M.H. Targeting RET receptor tyrosine kinase activation in cancer. Clin. Cancer Res. 2010, 16, 5936–5941. [Google Scholar] [CrossRef] [Green Version]
- Knowles, P.P.; Murray-Rust, J.; Kjaer, S.; Scott, R.P.; Hanrahan, S.; Santoro, M.; Ibanez, C.F.; McDonald, N.Q. Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 2006, 281, 33577–33587. [Google Scholar] [CrossRef] [Green Version]
- Arighi, E.; Borrello, M.G.; Sariola, H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005, 16, 441–467. [Google Scholar] [CrossRef] [PubMed]
- Bronte, G.; Ulivi, P.; Verlicchi, A.; Cravero, P.; Delmonte, A.; Crino, L. Targeting RET-rearranged non-small-cell lung cancer: Future prospects. Lung Cancer 2019, 10, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Sherman, S.I.; Clary, D.O.; Elisei, R.; Schlumberger, M.J.; Cohen, E.E.; Schoffski, P.; Wirth, L.J.; Mangeshkar, M.; Aftab, D.T.; Brose, M.S. Correlative analyses of RET and RAS mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer 2016, 122, 3856–3864. [Google Scholar] [CrossRef] [PubMed]
- Simonds, W.F. Genetics of Hyperparathyroidism, Including Parathyroid Cancer. Endocrinol. Metab. Clin. N. Am. 2017, 46, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.J. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat. Rev. Cancer 2005, 5, 367–375. [Google Scholar] [CrossRef]
- Khatami, F.; Tavangar, S.M. Multiple Endocrine Neoplasia Syndromes from Genetic and Epigenetic Perspectives. Biomark Insights 2018, 13, 1177271918785129. [Google Scholar] [CrossRef]
- Elisei, R.; Tacito, A.; Ramone, T.; Ciampi, R.; Bottici, V.; Cappagli, V.; Viola, D.; Matrone, A.; Lorusso, L.; Valerio, L.; et al. Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations. Genes 2019, 10, 698. [Google Scholar] [CrossRef] [Green Version]
- Elisei, R.; Cosci, B.; Romei, C.; Bottici, V.; Renzini, G.; Molinaro, E.; Agate, L.; Vivaldi, A.; Faviana, P.; Basolo, F.; et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: A 10-year follow-up study. J. Clin. Endocrinol. Metab. 2008, 93, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef]
- Mologni, L.; Gambacorti-Passerini, C.; Goekjian, P.; Scapozza, L. RET kinase inhibitors: A review of recent patents (2012–2015). Expert Opin. Pat. 2017, 27, 91–99. [Google Scholar] [CrossRef]
- Loya-Lopez, S.; Sandoval, A.; Gonzalez-Ramirez, R.; Calderon-Rivera, A.; Avalos-Fuentes, A.; Rodriguez-Sanchez, M.; Caballero, R.; Tovar-Soto, D.; Felix, R.; Floran, B. Cdk5 phosphorylates CaV1.3 channels and regulates GABAA-mediated miniature inhibitory post-synaptic currents in striato-nigral terminals. Biochem. Biophys. Res. Commun. 2020, 524, 255–261. [Google Scholar] [CrossRef]
- Hsu, F.N.; Chen, M.C.; Chiang, M.C.; Lin, E.; Lee, Y.T.; Huang, P.H.; Lee, G.S.; Lin, H. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J. Biol. Chem. 2011, 286, 33141–33149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Lu, Z.; Mao, W.; Ahmed, A.A.; Yang, H.; Zhou, J.; Jennings, N.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Miranda, R.; et al. CDK5 Regulates Paclitaxel Sensitivity in Ovarian Cancer Cells by Modulating AKT Activation, p21Cip1- and p27Kip1-Mediated G1 Cell Cycle Arrest and Apoptosis. PLoS ONE 2015, 10, e0131833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozo, K.; Bibb, J.A. The Emerging Role of Cdk5 in Cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Lin, J.X.; Zhang, P.Y.; Sun, Y.Q.; Li, P.; Xie, J.W.; Wang, J.B.; Chen, Q.Y.; Cao, L.L.; Lin, Y.; et al. CDK5 suppresses the metastasis of gastric cancer cells by interacting with and regulating PP2A. Oncol. Rep. 2019, 41, 779–788. [Google Scholar] [CrossRef]
- Cao, L.; Zhou, J.; Zhang, J.; Wu, S.; Yang, X.; Zhao, X.; Li, H.; Luo, M.; Yu, Q.; Lin, G.; et al. Cyclin-dependent kinase 5 decreases in gastric cancer and its nuclear accumulation suppresses gastric tumorigenesis. Clin. Cancer Res. 2015, 21, 1419–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.; Meyer, D.; Guo, A.; Gundara, J.; Mettlach, G.; et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.H.; Kim, D.W.; Suh, J.M.; Kim, H.; Song, J.H.; Hwang, E.S.; Park, K.C.; Chung, H.K.; Kim, J.M.; Lee, T.H.; et al. Activation of signal transducer and activator of transcription 3 by oncogenic RET/PTC (rearranged in transformation/papillary thyroid carcinoma) tyrosine kinase: Roles in specific gene regulation and cellular transformation. Mol. Endocrinol. 2003, 17, 1155–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Chai, S.; Liang, Z.; Wang, Y.; Zhou, Y.; Xu, X.; Zhang, C.; Zhang, M.; Si, J.; Huang, F.; et al. Correction to: KIF5B-RET fusion kinase promotes cell growth by multilevel activation of STAT3 in lung cancer. Mol. Cancer 2019, 18, 164. [Google Scholar] [CrossRef]
- Schuringa, J.J.; Wojtachnio, K.; Hagens, W.; Vellenga, E.; Buys, C.H.; Hofstra, R.; Kruijer, W. MEN2A-RET-induced cellular transformation by activation of STAT3. Oncogene 2001, 20, 5350–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.L.; Guan, Y.J.; Wang, L.; Wei, W.; Kane, A.B.; Chin, Y.E. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol. Cell Biol. 2004, 24, 9390–9400. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.P.; Liang, Y.F.; Samaan, N.A. Intrinsic drug resistance in a human medullary thyroid carcinoma cell line: Association with overexpression of mdrl gene and low proliferation fraction. Anticancer Res. 1991, 11, 1065–1068. [Google Scholar]
- Kucerova, L.; Feketeova, L.; Kozovska, Z.; Poturnajova, M.; Matuskova, M.; Nencka, R.; Babal, P. In vivo 5FU-exposed human medullary thyroid carcinoma cells contain a chemoresistant CD133+ tumor-initiating cell subset. Thyroid 2014, 24, 520–532. [Google Scholar] [CrossRef]
- Giani, F.; Vella, V.; Tumino, D.; Malandrino, P.; Frasca, F. The Possible Role of Cancer Stem Cells in the Resistance to Kinase Inhibitors of Advanced Thyroid Cancer. Cancers 2020, 12, 2249. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, C.-H.; Oner, M.; Chiu, C.-Y.; Chen, M.-C.; Teng, C.-L.; Wang, H.-Y.; Hsieh, J.-T.; Lai, C.-H.; Lin, H. RET Regulates Human Medullary Thyroid Cancer Cell Proliferation through CDK5 and STAT3 Activation. Biomolecules 2021, 11, 860. https://doi.org/10.3390/biom11060860
Yue C-H, Oner M, Chiu C-Y, Chen M-C, Teng C-L, Wang H-Y, Hsieh J-T, Lai C-H, Lin H. RET Regulates Human Medullary Thyroid Cancer Cell Proliferation through CDK5 and STAT3 Activation. Biomolecules. 2021; 11(6):860. https://doi.org/10.3390/biom11060860
Chicago/Turabian StyleYue, Chia-Herng, Muhammet Oner, Chih-Yuan Chiu, Mei-Chih Chen, Chieh-Lin Teng, Hsin-Yi Wang, Jer-Tsong Hsieh, Chih-Ho Lai, and Ho Lin. 2021. "RET Regulates Human Medullary Thyroid Cancer Cell Proliferation through CDK5 and STAT3 Activation" Biomolecules 11, no. 6: 860. https://doi.org/10.3390/biom11060860