
Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases


Abstract
:1. Introduction
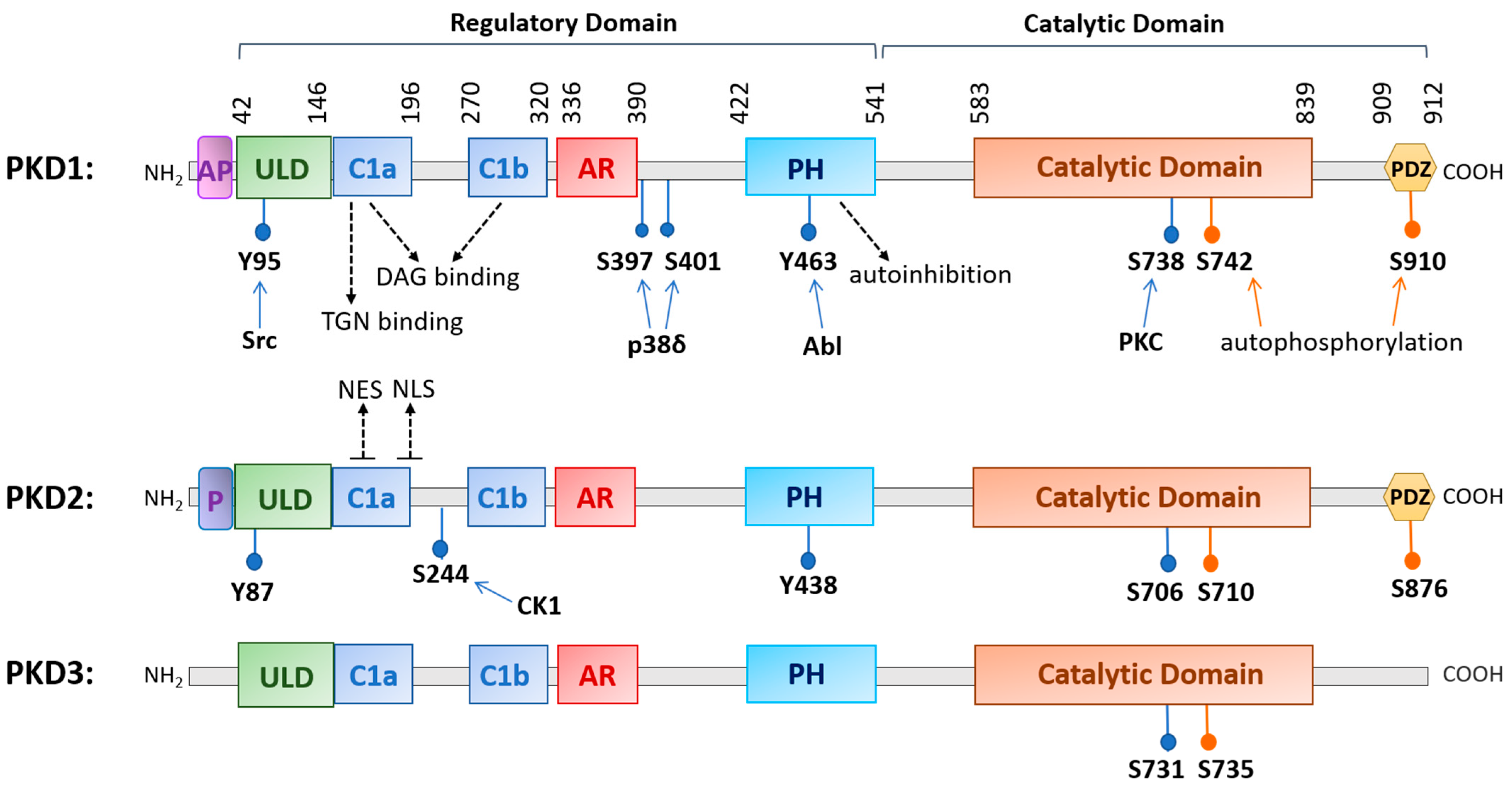
2. PKD Structure and Regulation
2.1. Structure, Isoforms, and Expression/Tissue Distribution
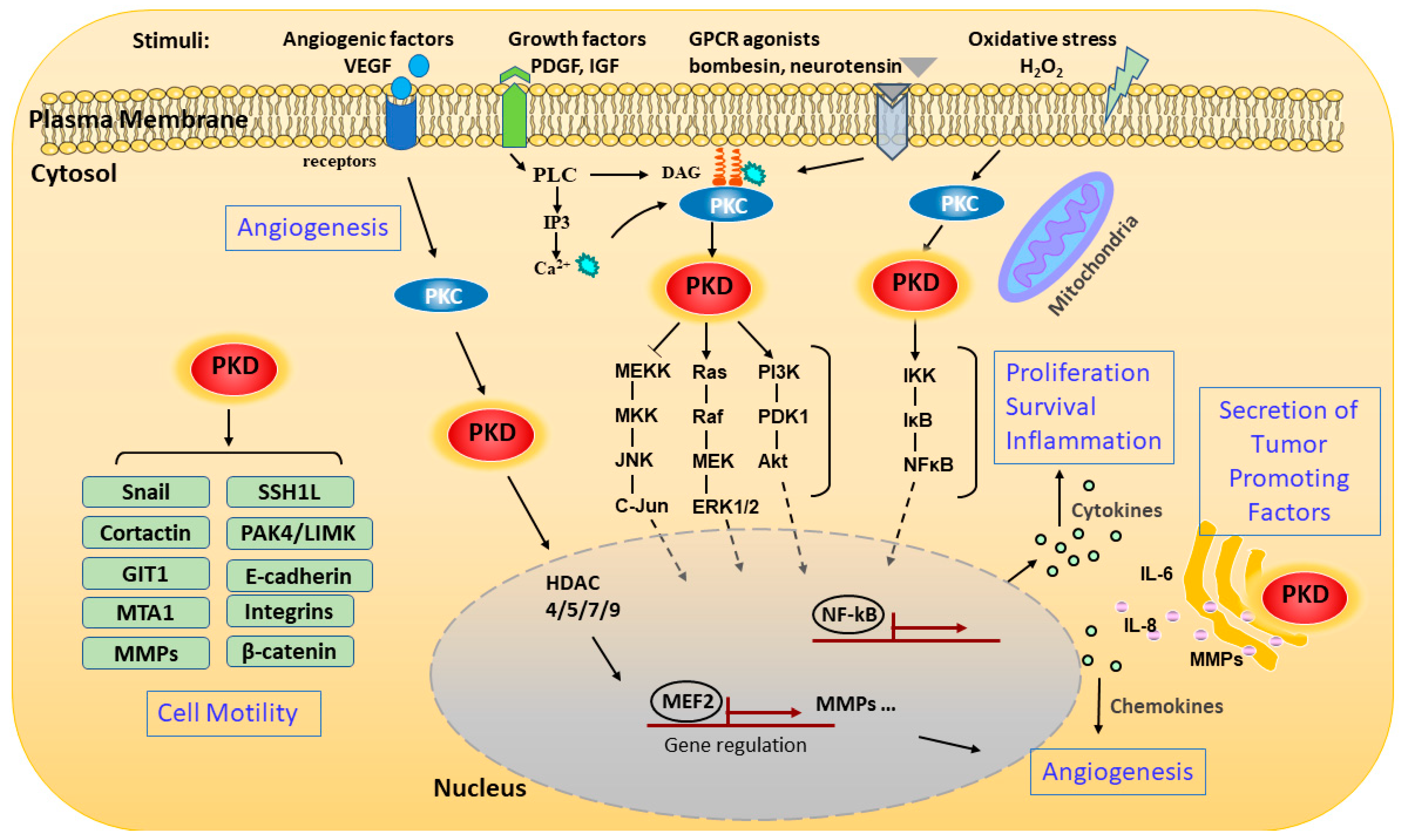
2.2. Mechanisms of Regulation
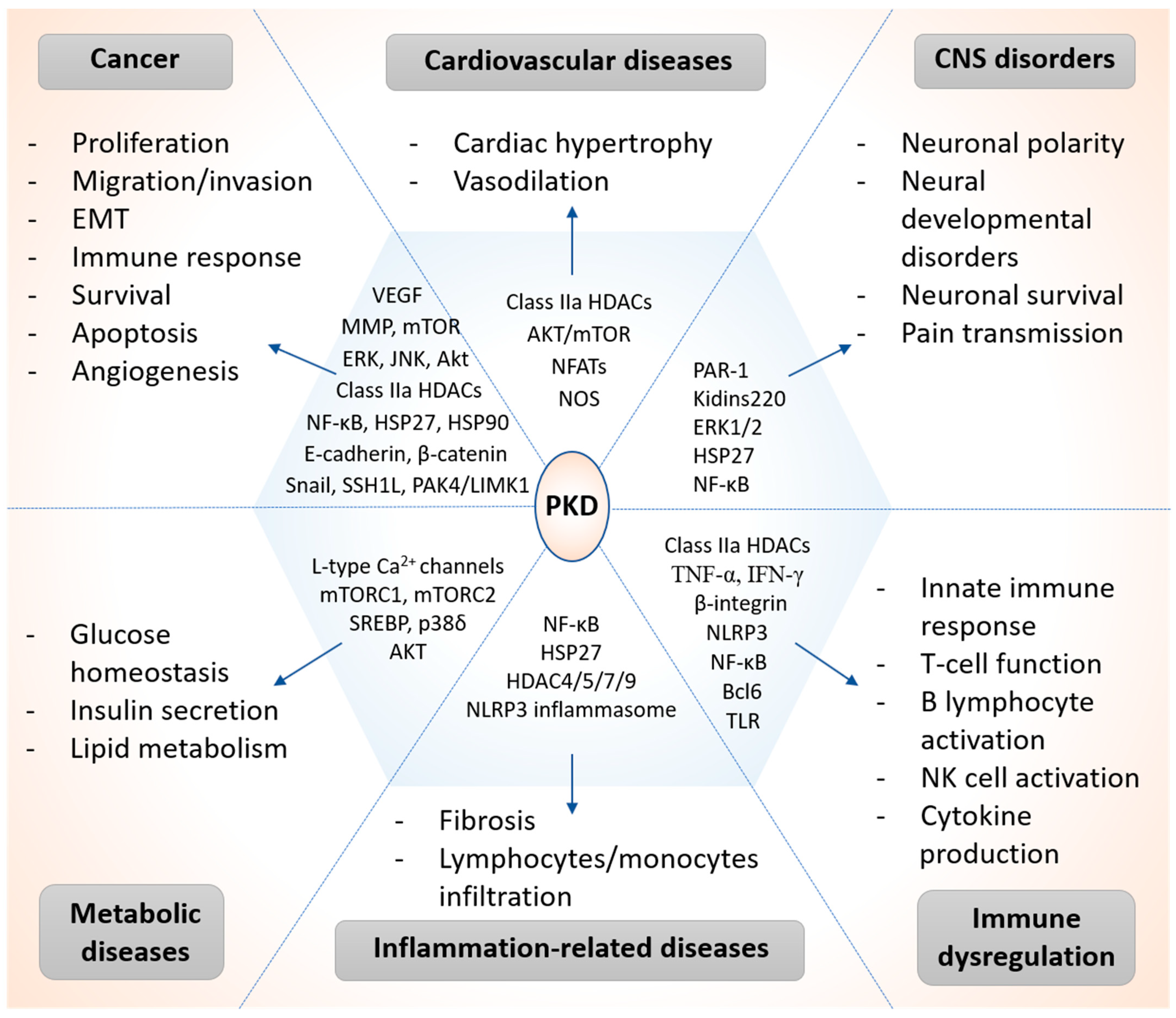
3. PKD in Pathological Processes and Human Diseases
3.1. Cancer
3.1.1. Cell Growth and Proliferation
3.1.2. Cell Survival and Apoptosis
3.1.3. Cell Adhesion, EMT, Migration, and Invasion
3.1.4. Angiogenesis
3.1.5. Immune Responses in Cancer
3.2. Cardiovascular Diseases
3.3. CNS Disorders
3.4. Metabolic Diseases
3.5. Inflammation-Related Diseases
3.6. Immune Dysregulation
4. Targeted Inhibition of PKD in Diseases
5. Perspectives and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PKD | protein kinase D |
| DAG | diacylglycerol |
| CNS | central nervous system |
| CAMK | Ca2+/calmodulin-dependent protein kinase |
| GPCR | G protein-coupled receptor |
| PH | pleckstrin homology |
| ULD | ubiquitin-like domain |
| PLC | phospholipase C |
| PKC | protein kinase C |
| TGN | trans-Golgi network |
| ROS | reactive oxygen species |
| ERK | extracellular-regulated protein kinase |
| NF-κB | nuclear factor-κB |
| mTOR | mammalian target of rapamycin |
| HDAC | histone deacetylase |
| MMP | matrix metalloproteinase |
| EMT | epithelial-to-mesenchymal transition |
| SSH1L | slingshot protein phosphatase 1L |
| LIMK1 | LIM domain kinase 1 |
| MTA1 | metastasis-associated protein 1 |
| ECM | extracellular matrix |
| VEGF | vascular endothelial growth factor |
| MEF2 | myocyte enhancer factor 2 |
References
- Johannes, F.J.; Prestle, J.; Eis, S.; Oberhagemann, P.; Pfizenmaier, K. PKCu is a novel, atypical member of the protein kinase C family. J. Biol. Chem. 1994, 269, 6140–6148. [Google Scholar] [CrossRef]
- Valverde, A.M.; Sinnett-Smith, J.; Van Lint, J.; Rozengurt, E. Molecular cloning and characterization of protein kinase D: A target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. USA 1994, 91, 8572–8576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, A.; Seki, N.; Hattori, A.; Kozuma, S.; Saito, T. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim. Biophys. Acta 1999, 1450, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Sturany, S.; Van Lint, J.; Muller, F.; Wilda, M.; Hameister, H.; Hocker, M.; Brey, A.; Gern, U.; Vandenheede, J.; Gress, T.; et al. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J. Biol. Chem. 2001, 276, 3310–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, R.; Truebestein, L.; Schmidt, H.A.; Leonard, T.A. It Takes Two to Tango: Activation of Protein Kinase D by Dimerization. Bioessays 2020, 42, e1900222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azoitei, N.; Cobbaut, M.; Becher, A.; Van Lint, J.; Seufferlein, T. Protein kinase D2: A versatile player in cancer biology. Oncogene 2018, 37, 1263–1278. [Google Scholar] [CrossRef]
- Roy, A.; Ye, J.; Deng, F.; Wang, Q.J. Protein kinase D signaling in cancer: A friend or foe? Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 283–294. [Google Scholar] [CrossRef]
- Chen, J.; Giridhar, K.V.; Zhang, L.; Xu, S.; Wang, Q.J. A protein kinase C/protein kinase D pathway protects LNCaP prostate cancer cells from phorbol ester-induced apoptosis by promoting ERK1/2 and NF-{kappa}B activities. Carcinogenesis 2011, 32, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaValle, C.R.; George, K.M.; Sharlow, E.R.; Lazo, J.S.; Wipf, P.; Wang, Q.J. Protein kinase D as a potential new target for cancer therapy. Biochim. Biophys. Acta 2010, 1806, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Waldron, R.T.; Rozengurt, E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J. Biol. Chem. 2003, 278, 154–163. [Google Scholar]
- Sundram, V.; Chauhan, S.C.; Jaggi, M. Emerging roles of protein kinase D1 in cancer. Mol. Cancer Res. 2011, 9, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Elsner, D.J.; Siess, K.M.; Gossenreiter, T.; Hartl, M.; Leonard, T.A. A ubiquitin-like domain controls protein kinase D dimerization and activation by trans-autophosphorylation. J. Biol. Chem. 2019, 294, 14422–14441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, B.M.; Bossuyt, J. Emergency Spatiotemporal Shift: The Response of Protein Kinase D to Stress Signals in the Cardiovascular System. Front. Pharmacol. 2017, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, N.; Borges, S.; Storz, P. Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion. J. Clin. Med. 2016, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Rozengurt, E.; Rey, O.; Waldron, R.T. Protein kinase D signaling. J. Biol. Chem. 2005, 280, 13205–13208. [Google Scholar] [CrossRef] [Green Version]
- Simsek Papur, O.; Sun, A.; Glatz, J.F.C.; Luiken, J.; Nabben, M. Acute and Chronic Effects of Protein Kinase-D Signaling on Cardiac Energy Metabolism. Front. Cardiovasc Med. 2018, 5, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozengurt, E. Protein kinase D signaling: Multiple biological functions in health and disease. Physiology (Bethesda) 2011, 26, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Prestle, J.; Pfizenmaier, K.; Brenner, J.; Johannes, F.J. Protein kinase C mu is located at the Golgi compartment. J. Cell Biol. 1996, 134, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Liljedahl, M.; Maeda, Y.; Colanzi, A.; Ayala, I.; Van Lint, J.; Malhotra, V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell 2001, 104, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Bossard, C.; Bresson, D.; Polishchuk, R.S.; Malhotra, V. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J. Cell Biol. 2007, 179, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Jamora, C.; Yamanouye, N.; Van Lint, J.; Laudenslager, J.; Vandenheede, J.R.; Faulkner, D.J.; Malhotra, V. Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 1999, 98, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Baron, C.L.; Malhotra, V. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 2002, 295, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Beznoussenko, G.V.; Van Lint, J.; Mironov, A.A.; Malhotra, V. Recruitment of protein kinase D to the trans-Golgi network via the first cysteine-rich domain. Embo J. 2001, 20, 5982–5990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Hausser, A.; Storz, P.; Martens, S.; Link, G.; Toker, A.; Pfizenmaier, K. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIbeta at the Golgi complex. Nat. Cell Biol. 2005, 7, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Rey, O.; Yuan, J.; Young, S.H.; Rozengurt, E. Protein kinase C nu/protein kinase D3 nuclear localization, catalytic activation, and intracellular redistribution in response to G protein-coupled receptor agonists. J. Biol. Chem. 2003, 278, 23773–23785. [Google Scholar] [CrossRef] [Green Version]
- Rey, O.; Sinnett-Smith, J.; Zhukova, E.; Rozengurt, E. Regulated nucleocytoplasmic transport of protein kinase D in response to G protein-coupled receptor activation. J. Biol. Chem. 2001, 276, 49228–49235. [Google Scholar] [CrossRef] [Green Version]
- Sin, Y.Y.; Martin, T.P.; Wills, L.; Currie, S.; Baillie, G.S. Small heat shock protein 20 (Hsp20) facilitates nuclear import of protein kinase D 1 (PKD1) during cardiac hypertrophy. Cell Commun. Signal. 2015, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Auer, A.; von Blume, J.; Sturany, S.; von Wichert, G.; Van Lint, J.; Vandenheede, J.; Adler, G.; Seufferlein, T. Role of the regulatory domain of protein kinase D2 in phorbol ester binding, catalytic activity, and nucleocytoplasmic shuttling. Mol. Biol Cell 2005, 16, 4375–4385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Blume, J.; Knippschild, U.; Dequiedt, F.; Giamas, G.; Beck, A.; Auer, A.; Van Lint, J.; Adler, G.; Seufferlein, T. Phosphorylation at Ser244 by CK1 determines nuclear localization and substrate targeting of PKD2. Embo J. 2007, 26, 4619–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, O.; Papazyan, R.; Waldron, R.T.; Young, S.H.; Lippincott-Schwartz, J.; Jacamo, R.; Rozengurt, E. The nuclear import of protein kinase D3 requires its catalytic activity. J. Biol. Chem. 2006, 281, 5149–5157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P.; Doppler, H.; Johannes, F.J.; Toker, A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J. Biol. Chem. 2003, 278, 17969–17976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doppler, H.; Storz, P. A novel tyrosine phosphorylation site in protein kinase D contributes to oxidative stress-mediated activation. J. Biol. Chem. 2007, 282, 31873–31881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P.; Toker, A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J. 2003, 22, 109–120. [Google Scholar] [CrossRef]
- Cowell, C.F.; Doppler, H.; Yan, I.K.; Hausser, A.; Umezawa, Y.; Storz, P. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J. Cell Sci 2009, 122, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell Biol. 2005, 25, 8520–8530. [Google Scholar] [CrossRef] [Green Version]
- Cobbaut, M.; Van Lint, J. Function and Regulation of Protein Kinase D in Oxidative Stress: A Tale of Isoforms. Oxid. Med. Cell Longev. 2018, 2018, 2138502. [Google Scholar] [CrossRef] [Green Version]
- Doppler, H.; Storz, P. Mitochondrial and Oxidative Stress-Mediated Activation of Protein Kinase D1 and Its Importance in Pancreatic Cancer. Front. Oncol. 2017, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Youssef, I.; Ricort, J.M. Deciphering the Role of Protein Kinase D1 (PKD1) in Cellular Proliferation. Mol. Cancer Res. 2019, 17, 1961–1974. [Google Scholar] [CrossRef]
- Yuan, J.; Pandol, S.J. PKD signaling and pancreatitis. J. Gastroenterol. 2016, 51, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Rashel, M.; Alston, N.; Ghazizadeh, S. Protein kinase D1 has a key role in wound healing and skin carcinogenesis. J. Investig. Dermatol 2014, 134, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Durand, N.; Borges, S.; Storz, P. Functional and therapeutic significance of protein kinase D enzymes in invasive breast cancer. Cell. Mol. Life Sci 2015, 72, 4369–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, S.; Tanasanvimon, S.; Sinnett-Smith, J.; Rozengurt, E. Role of protein kinase D signaling in pancreatic cancer. Biochem. Pharmacol. 2010, 80, 1946–1954. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, N.; Frohling, S.; Scholl, C.; Seufferlein, T. PRKD2: A two-pronged kinase crucial for the tumor-supporting activity of HSP90. Mol. Cell Oncol. 2015, 2, e981444. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, N.; Diepold, K.; Brunner, C.; Rouhi, A.; Genze, F.; Becher, A.; Kestler, H.; van Lint, J.; Chiosis, G.; Koren, J., 3rd; et al. HSP90 supports tumor growth and angiogenesis through PRKD2 protein stabilization. Cancer Res. 2014, 74, 7125–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B. Protein Kinase D1 Signaling in Angiogenic Gene Expression and VEGF-Mediated Angiogenesis. Front. Cell Dev. Biol. 2016, 4, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Qian, J.; Zeng, F.; Li, S.; Guo, W.; Chen, L.; Li, G.; Zhang, Z.; Wang, Q.J.; Deng, F. Protein kinase Ds promote tumor angiogenesis through mast cell recruitment and expression of angiogenic factors in prostate cancer microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 114. [Google Scholar] [CrossRef]
- Scheiter, M.; Bulitta, B.; van Ham, M.; Klawonn, F.; Konig, S.; Jansch, L. Protein Kinase Inhibitors CK59 and CID755673 Alter Primary Human NK Cell Effector Functions. Front. Immunol. 2013, 4, 66. [Google Scholar] [CrossRef] [Green Version]
- Aicart-Ramos, C.; He, S.D.; Land, M.; Rubin, C.S. A Novel Conserved Domain Mediates Dimerization of Protein Kinase D (PKD) Isoforms: DIMERIZATION IS ESSENTIAL FOR PKD-DEPENDENT REGULATION OF SECRETION AND INNATE IMMUNITY. J. Biol. Chem. 2016, 291, 23516–23531. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Chen, J.; Luo, M.; Wang, L.; Chen, H.; Kang, Y.; Wang, J.; Zhou, X.; Feng, Y.; Zhang, P. Protein kinase D3 regulates the expression of the immunosuppressive protein, PDL1, through STAT1/STAT3 signaling. Int. J. Oncol. 2020, 56, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Kisfalvi, K.; Hurd, C.; Guha, S.; Rozengurt, E. Induced overexpression of protein kinase D1 stimulates mitogenic signaling in human pancreatic carcinoma PANC-1 cells. J. Cell Physiol. 2010, 223, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Doppler, H.; Braun, U.B.; Panayiotou, R.; Scotti Buzhardt, M.; Radisky, D.C.; Crawford, H.C.; Fields, A.P.; Murray, N.R.; Wang, Q.J.; et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 2015, 6, 6200. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, Z.; Liu, Y.; Xu, S.; Tandon, M.; Appelboom, B.; LaValle, C.R.; Chiosea, S.I.; Wang, L.; Sen, M.; et al. Analysis of oncogenic activities of protein kinase D1 in head and neck squamous cell carcinoma. BMC Cancer 2018, 18, 1107. [Google Scholar] [CrossRef]
- McEneaney, V.; Dooley, R.; Harvey, B.J.; Thomas, W. Protein kinase D stabilizes aldosterone-induced ERK1/2 MAP kinase activation in M1 renal cortical collecting duct cells to promote cell proliferation. J. Steroid Biochem. Mol. Biol. 2010, 118, 18–28. [Google Scholar] [CrossRef]
- Tandon, M.; Johnson, J.; Li, Z.; Xu, S.; Wipf, P.; Wang, Q.J. New pyrazolopyrimidine inhibitors of protein kinase d as potent anticancer agents for prostate cancer cells. PLoS ONE 2013, 8, e75601. [Google Scholar] [CrossRef]
- Tandon, M.; Salamoun, J.M.; Carder, E.J.; Farber, E.; Xu, S.; Deng, F.; Tang, H.; Wipf, P.; Wang, Q.J. SD-208, a novel protein kinase D inhibitor, blocks prostate cancer cell proliferation and tumor growth in vivo by inducing G2/M cell cycle arrest. PLoS ONE 2015, 10, e0119346. [Google Scholar] [CrossRef] [Green Version]
- Karam, M.; Legay, C.; Auclair, C.; Ricort, J.M. Protein kinase D1 stimulates proliferation and enhances tumorigenesis of MCF-7 human breast cancer cells through a MEK/ERK-dependent signaling pathway. Exp. Cell Res. 2012, 318, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Karam, M.; Bieche, I.; Legay, C.; Vacher, S.; Auclair, C.; Ricort, J.M. Protein kinase D1 regulates ERalpha-positive breast cancer cell growth response to 17beta-estradiol and contributes to poor prognosis in patients. J. Cell Mol. Med. 2014, 18, 2536–2552. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.H.; Du, C.; Zhang, C.; Straubhaar, J.; Languino, L.R.; Balaji, K.C. Protein kinase D1 inhibits cell proliferation through matrix metalloproteinase-2 and matrix metalloproteinase-9 secretion in prostate cancer. Cancer Res. 2010, 70, 2095–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickkholgh, B.; Sittadjody, S.; Ordonez, K.; Rothberg, M.B.; Balaji, K.C. Protein kinase D1 induces G1-phase cell-cycle arrest independent of Checkpoint kinases by phosphorylating Cell Division Cycle Phosphatase 25. Prostate 2019, 79, 1053–1058. [Google Scholar] [CrossRef]
- Sundram, V.; Ganju, A.; Hughes, J.E.; Khan, S.; Chauhan, S.C.; Jaggi, M. Protein kinase D1 attenuates tumorigenesis in colon cancer by modulating beta-catenin/T cell factor activity. Oncotarget 2014, 5, 6867–6884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Wang, L.; Zhang, J.; Pang, Z.; Liu, Q.; Du, J. PKD1 is downregulated in non-small cell lung cancer and mediates the feedback inhibition of mTORC1-S6K1 axis in response to phorbol ester. Int. J. Biochem. Cell Biol. 2015, 60, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Veroli, M.V.; Prasad, S.; Wang, Q.J. Protein Kinase D2 Modulates Cell Cycle By Stabilizing Aurora A Kinase at Centrosomes. Mol. Cancer Res. 2018, 16, 1785–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, N.; Chu, E.; Wipf, P.; Schmitz, J.C. Protein kinase d as a potential chemotherapeutic target for colorectal cancer. Mol. Cancer Ther 2014, 13, 1130–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Xue, P.; Yang, M.; Shi, H.; Lu, D.; Wang, Z.; Shi, Q.; Hu, J.; Xie, S.; Zhan, W.; et al. Protein kinase D2 promotes the proliferation of glioma cells by regulating Golgi phosphoprotein 3. Cancer Lett 2014, 355, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Huck, B.; Duss, S.; Hausser, A.; Olayioye, M.A. Elevated protein kinase D3 (PKD3) expression supports proliferation of triple-negative breast cancer cells and contributes to mTORC1-S6K1 pathway activation. J. Biol. Chem. 2014, 289, 3138–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Song, H.; Yu, S.; Huang, K.H.; Ma, X.; Zhou, Y.; Yu, S.; Zhang, J.; Chen, L. Protein Kinase D3 promotes the cell proliferation by activating the ERK1/c-MYC axis in breast cancer. J. Cell Mol. Med. 2020, 24, 2135–2144. [Google Scholar] [CrossRef]
- Hao, Q.; McKenzie, R.; Gan, H.; Tang, H. Protein kinases D2 and D3 are novel growth regulators in HCC1806 triple-negative breast cancer cells. Anticancer Res. 2013, 33, 393–399. [Google Scholar]
- Kumari, S.; Khan, S.; Sekhri, R.; Mandil, H.; Behrman, S.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. Protein kinase D1 regulates metabolic switch in pancreatic cancer via modulation of mTORC1. Br. J. Cancer 2020, 122, 121–131. [Google Scholar] [CrossRef]
- Trauzold, A.; Schmiedel, S.; Sipos, B.; Wermann, H.; Westphal, S.; Roder, C.; Klapper, W.; Arlt, A.; Lehnert, L.; Ungefroren, H.; et al. PKCmu prevents CD95-mediated apoptosis and enhances proliferation in pancreatic tumour cells. Oncogene 2003, 22, 8939–8947. [Google Scholar] [CrossRef] [Green Version]
- Wei, N.; Chu, E.; Wu, S.Y.; Wipf, P.; Schmitz, J.C. The cytotoxic effects of regorafenib in combination with protein kinase D inhibition in human colorectal cancer cells. Oncotarget 2015, 6, 4745–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihailovic, T.; Marx, M.; Auer, A.; Van Lint, J.; Schmid, M.; Weber, C.; Seufferlein, T. Protein kinase D2 mediates activation of nuclear factor kappaB by Bcr-Abl in Bcr-Abl+ human myeloid leukemia cells. Cancer Res. 2004, 64, 8939–8944. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Deng, F.; Singh, S.V.; Wang, Q.J. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res. 2008, 68, 3844–3853. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Zhang, C.; Hassan, S.; Biswas, M.H.; Balaji, K.C. Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res. 2010, 70, 7810–7819. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Shen, M.; Zha, Y.L.; Li, W.; Wei, Y.; Blanco, M.A.; Ren, G.; Zhou, T.; Storz, P.; Wang, H.Y.; et al. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell 2014, 26, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Jaggi, M.; Rao, P.S.; Smith, D.J.; Wheelock, M.J.; Johnson, K.R.; Hemstreet, G.P.; Balaji, K.C. E-cadherin phosphorylation by protein kinase D1/protein kinase C{mu} is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res. 2005, 65, 483–492. [Google Scholar] [PubMed]
- Du, C.; Jaggi, M.; Zhang, C.; Balaji, K.C. Protein kinase D1-mediated phosphorylation and subcellular localization of beta-catenin. Cancer Res. 2009, 69, 1117–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganju, A.; Chauhan, S.C.; Hafeez, B.B.; Doxtater, K.; Tripathi, M.K.; Zafar, N.; Yallapu, M.M.; Kumar, R.; Jaggi, M. Protein kinase D1 regulates subcellular localisation and metastatic function of metastasis-associated protein 1. Br. J. Cancer 2018, 118, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Christoforides, C.; Rainero, E.; Brown, K.K.; Norman, J.C.; Toker, A. PKD controls alphavbeta3 integrin recycling and tumor cell invasive migration through its substrate Rabaptin-5. Dev. Cell 2012, 23, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Eiseler, T.; Doppler, H.; Yan, I.K.; Goodison, S.; Storz, P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009, 11, R13. [Google Scholar] [CrossRef] [Green Version]
- Peterburs, P.; Heering, J.; Link, G.; Pfizenmaier, K.; Olayioye, M.A.; Hausser, A. Protein kinase D regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer Res. 2009, 69, 5634–5638. [Google Scholar] [CrossRef] [Green Version]
- Merzoug-Larabi, M.; Spasojevic, C.; Eymard, M.; Hugonin, C.; Auclair, C.; Karam, M. Protein kinase C inhibitor Go6976 but not Go6983 induces the reversion of E- to N-cadherin switch and metastatic phenotype in melanoma: Identification of the role of protein kinase D1. BMC Cancer 2017, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Zeng, F.; Xu, W.; Wang, C.; Ke, Z.; Wang, Q.J.; Deng, F. PKD2 and PKD3 promote prostate cancer cell invasion by modulating NF-kappaB- and HDAC1-mediated expression and activation of uPA. J. Cell Sci. 2012, 125, 4800–4811. [Google Scholar] [CrossRef] [Green Version]
- Wille, C.; Kohler, C.; Armacki, M.; Jamali, A.; Gossele, U.; Pfizenmaier, K.; Seufferlein, T.; Eiseler, T. Protein kinase D2 induces invasion of pancreatic cancer cells by regulating matrix metalloproteinases. Mol. Biol. Cell 2014, 25, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Bernhart, E.; Damm, S.; Wintersperger, A.; DeVaney, T.; Zimmer, A.; Raynham, T.; Ireson, C.; Sattler, W. Protein kinase D2 regulates migration and invasion of U87MG glioblastoma cells in vitro. Exp. Cell Res. 2013, 319, 2037–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Cheng, Y.; Guo, Y.; Chen, J.; Chen, F.; Luo, R.; Li, A. Protein kinase D2 contributes to TNF-alpha-induced epithelial mesenchymal transition and invasion via the PI3K/GSK-3beta/beta-catenin pathway in hepatocellular carcinoma. Oncotarget 2016, 7, 5327–5341. [Google Scholar] [CrossRef] [Green Version]
- LaValle, C.R.; Zhang, L.; Xu, S.; Eiseman, J.L.; Wang, Q.J. Inducible silencing of protein kinase D3 inhibits secretion of tumor-promoting factors in prostate cancer. Mol. Cancer Ther. 2012, 11, 1389–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doppler, H.; Bastea, L.I.; Borges, S.; Spratley, S.J.; Pearce, S.E.; Storz, P. Protein kinase d isoforms differentially modulate cofilin-driven directed cell migration. PLoS ONE 2014, 9, e98090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huck, B.; Kemkemer, R.; Franz-Wachtel, M.; Macek, B.; Hausser, A.; Olayioye, M.A. GIT1 phosphorylation on serine 46 by PKD3 regulates paxillin trafficking and cellular protrusive activity. J. Biol. Chem. 2012, 287, 34604–34613. [Google Scholar] [CrossRef] [Green Version]
- Ochi, N.; Tanasanvimon, S.; Matsuo, Y.; Tong, Z.; Sung, B.; Aggarwal, B.B.; Sinnett-Smith, J.; Rozengurt, E.; Guha, S. Protein kinase D1 promotes anchorage-independent growth, invasion, and angiogenesis by human pancreatic cancer cells. J. Cell Physiol. 2011, 226, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yuan, Y.; Opansky, C.; Chen, Y.; Aguilera-Barrantes, I.; Wu, S.; Yuan, R.; Cao, Q.; Cheng, Y.C.; Sahoo, D.; et al. Diet-induced obesity links to ER positive breast cancer progression via LPA/PKD-1-CD36 signaling-mediated microvascular remodeling. Oncotarget 2017, 8, 22550–22562. [Google Scholar] [CrossRef]
- Azoitei, N.; Pusapati, G.V.; Kleger, A.; Moller, P.; Kufer, R.; Genze, F.; Wagner, M.; van Lint, J.; Carmeliet, P.; Adler, G.; et al. Protein kinase D2 is a crucial regulator of tumour cell-endothelial cell communication in gastrointestinal tumours. Gut 2010, 59, 1316–1330. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Feng, Y.; Lu, L.; Wang, H.; Dai, L.; Li, Y.; Zhang, P. Interferon-gamma-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology 2012, 217, 385–393. [Google Scholar] [CrossRef]
- Zheng, H.; Qian, J.; Baker, D.P.; Fuchs, S.Y. Tyrosine phosphorylation of protein kinase D2 mediates ligand-inducible elimination of the Type 1 interferon receptor. J. Biol. Chem. 2011, 286, 35733–35741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhukova, E.; Sinnett-Smith, J.; Rozengurt, E. Protein kinase D potentiates DNA synthesis and cell proliferation induced by bombesin, vasopressin, or phorbol esters in Swiss 3T3 cells. J. Biol. Chem. 2001, 276, 40298–40305. [Google Scholar] [CrossRef] [Green Version]
- Rennecke, J.; Rehberger, P.A.; Furstenberger, G.; Johannes, F.J.; Stohr, M.; Marks, F.; Richter, K.H. Protein-kinase-Cmu expression correlates with enhanced keratinocyte proliferation in normal and neoplastic mouse epidermis and in cell culture. Int. J. Cancer 1999, 80, 98–103. [Google Scholar] [CrossRef]
- Yuan, J.; Slice, L.; Walsh, J.H.; Rozengurt, E. Activation of protein kinase D by signaling through the alpha subunit of the heterotrimeric G protein G(q). J. Biol. Chem. 2000, 275, 2157–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Slice, L.W.; Gu, J.; Rozengurt, E. Cooperation of Gq, Gi, and G12/13 in protein kinase D activation and phosphorylation induced by lysophosphatidic acid. J. Biol. Chem. 2003, 278, 4882–4891. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Slice, L.W.; Rozengurt, E. Activation of protein kinase D by signaling through Rho and the alpha subunit of the heterotrimeric G protein G13. J. Biol. Chem. 2001, 276, 38619–38627. [Google Scholar] [CrossRef] [Green Version]
- Sinnett-Smith, J.; Jacamo, R.; Kui, R.; Wang, Y.M.; Young, S.H.; Rey, O.; Waldron, R.T.; Rozengurt, E. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J. Biol. Chem. 2009, 284, 13434–13445. [Google Scholar] [CrossRef] [Green Version]
- Sinnett-Smith, J.; Zhukova, E.; Hsieh, N.; Jiang, X.; Rozengurt, E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J. Biol. Chem. 2004, 279, 16883–16893. [Google Scholar] [CrossRef] [Green Version]
- Sinnett-Smith, J.; Zhukova, E.; Rey, O.; Rozengurt, E. Protein kinase D2 potentiates MEK/ERK/RSK signaling, c-Fos accumulation and DNA synthesis induced by bombesin in Swiss 3T3 cells. J. Cell Physiol. 2007, 211, 781–790. [Google Scholar] [CrossRef]
- Sinnett-Smith, J.; Ni, Y.; Wang, J.; Ming, M.; Young, S.H.; Rozengurt, E. Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: Role in mitogenic signaling. Am. J. Physiol. Cell Physiol. 2014, 306, C961–C971. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Han, L.; Sinnett-Smith, J.; Han, L.L.; Stevens, J.V.; Rozengurt, N.; Young, S.H.; Rozengurt, E. Positive cross talk between protein kinase D and beta-catenin in intestinal epithelial cells: Impact on beta-catenin nuclear localization and phosphorylation at Ser552. Am. J. Physiol. Cell Physiol. 2016, 310, C542–C557. [Google Scholar] [CrossRef] [Green Version]
- Papazyan, R.; Doche, M.; Waldron, R.T.; Rozengurt, E.; Moyer, M.P.; Rey, O. Protein kinase D isozymes activation and localization during mitosis. Exp. Cell Res. 2008, 314, 3057–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienzle, C.; Eisler, S.A.; Villeneuve, J.; Brummer, T.; Olayioye, M.A.; Hausser, A. PKD controls mitotic Golgi complex fragmentation through a Raf-MEK1 pathway. Mol. Biol Cell 2013, 24, 222–233. [Google Scholar] [CrossRef]
- Zhang, T.; Braun, U.; Leitges, M. PKD3 deficiency causes alterations in microtubule dynamics during the cell cycle. Cell Cycle 2016, 15, 1844–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Leon, E.; Amable, G.; Jacamo, R.; Picco, M.E.; Anaya, L.; Rozengurt, E.; Rey, O. Protein kinase D1 inhibition interferes with mitosis progression. J. Cell Physiol. 2019, 234, 20510–20519. [Google Scholar] [CrossRef]
- Li, Q.Q.; Hsu, I.; Sanford, T.; Railkar, R.; Balaji, N.; Sourbier, C.; Vocke, C.; Balaji, K.C.; Agarwal, P.K. Protein kinase D inhibitor CRT0066101 suppresses bladder cancer growth in vitro and xenografts via blockade of the cell cycle at G2/M. Cell. Mol. Life Sci 2018, 75, 939–963. [Google Scholar] [CrossRef]
- Pang, Z.; Wang, Y.; Ding, N.; Chen, X.; Yang, Y.; Wang, G.; Liu, Q.; Du, J. High PKD2 predicts poor prognosis in lung adenocarcinoma via promoting Epithelial-mesenchymal Transition. Sci Rep. 2019, 9, 1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azoitei, N.; Kleger, A.; Schoo, N.; Thal, D.R.; Brunner, C.; Pusapati, G.V.; Filatova, A.; Genze, F.; Moller, P.; Acker, T.; et al. Protein kinase D2 is a novel regulator of glioblastoma growth and tumor formation. Neuro-Oncology 2011, 13, 710–724. [Google Scholar] [CrossRef]
- Nickkholgh, B.; Sittadjody, S.; Rothberg, M.B.; Fang, X.; Li, K.; Chou, J.W.; Hawkins, G.A.; Balaji, K.C. Beta-catenin represses protein kinase D1 gene expression by non-canonical pathway through MYC/MAX transcription complex in prostate cancer. Oncotarget 2017, 8, 78811–78824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Gyabaah, K.; Nickkholgh, B.; Cline, J.M.; Balaji, K.C. Novel In Vivo model for combinatorial fluorescence labeling in mouse prostate. Prostate 2015, 75, 988–1000. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lu, L.; Feng, Y.; Wang, H.; Dai, L.; Li, Y.; Zhang, P. PKD2 mediates multi-drug resistance in breast cancer cells through modulation of P-glycoprotein expression. Cancer Lett. 2011, 300, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shen, Q.; Labow, M.; Gaither, L.A. Protein kinase D3 sensitizes RAF inhibitor RAF265 in melanoma cells by preventing reactivation of MAPK signaling. Cancer Res. 2011, 71, 4280–4291. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invesig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Bastea, L.I.; Doppler, H.; Balogun, B.; Storz, P. Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PLoS ONE 2012, 7, e30459. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Stossel, T.P.; Fenteany, G.; Hartwig, J.H. Cell surface actin remodeling. J. Cell Sci. 2006, 119, 3261–3264. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.W.; Olson, M.F. LIM kinases: Function, regulation and association with human disease. J. Mol. Med. (Berl.) 2007, 85, 555–568. [Google Scholar] [CrossRef]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Eiseler, T.; Doppler, H.; Yan, I.K.; Kitatani, K.; Mizuno, K.; Storz, P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 2009, 11, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Spratley, S.J.; Bastea, L.I.; Doppler, H.; Mizuno, K.; Storz, P. Protein kinase D regulates cofilin activity through p21-activated kinase 4. J. Biol. Chem. 2011, 286, 34254–34261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, S.; Eiseler, T.; Scholz, R.P.; Beck, A.; Link, G.; Hausser, A. A novel protein kinase D phosphorylation site in the tumor suppressor Rab interactor 1 is critical for coordination of cell migration. Mol. Biol. Cell 2011, 22, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Bastea, L.I.; Long, J.; Doppler, H.; Ling, K.; Storz, P. Protein Kinase D1 regulates focal adhesion dynamics and cell adhesion through Phosphatidylinositol-4-phosphate 5-kinase type-l gamma. Sci. Rep. 2016, 6, 35963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doppler, H.R.; Bastea, L.I.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z.; Storz, P. Protein kinase D1-mediated phosphorylations regulate vasodilator-stimulated phosphoprotein (VASP) localization and cell migration. J. Biol. Chem. 2013, 288, 24382–24393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P. Protein kinase D1: Gatekeeper of the epithelial phenotype and key regulator of cancer metastasis? Br. J. Cancer 2018, 118, 459–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onodera, Y.; Nam, J.M.; Hashimoto, A.; Norman, J.C.; Shirato, H.; Hashimoto, S.; Sabe, H. Rab5c promotes AMAP1-PRKD2 complex formation to enhance beta1 integrin recycling in EGF-induced cancer invasion. J. Cell Biol. 2012, 197, 983–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Molder, L.; Sonnenberg, A. PKD2 and RSK1 Regulate Integrin beta4 Phosphorylation at Threonine 1736. PLoS ONE 2015, 10, e0143357. [Google Scholar] [CrossRef] [PubMed]
- Borges, S.; Doppler, H.; Perez, E.A.; Andorfer, C.A.; Sun, Z.; Anastasiadis, P.Z.; Thompson, E.; Geiger, X.J.; Storz, P. Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 2013, 15, R66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpsoy, A.; Gunduz, U. Protein kinase D2 silencing reduced motility of doxorubicin-resistant MCF7 cells. Tumour Biol. 2015, 36, 4417–4426. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, X.; Hamblin, M.H.; Yin, K.J. Long Non-Coding RNA Malat1 Regulates Angiogenesis in Hindlimb Ischemia. Int. J. Mol. Sci 2018, 19, 1723. [Google Scholar] [CrossRef] [Green Version]
- Wulff, C.; Weigand, M.; Kreienberg, R.; Fraser, H.M. Angiogenesis during primate placentation in health and disease. Reproduction 2003, 126, 569–577. [Google Scholar] [CrossRef]
- Sun, P.; Zhang, K.; Hassan, S.H.; Zhang, X.; Tang, X.; Pu, H.; Stetler, R.A.; Chen, J.; Yin, K.J. Endothelium-Targeted Deletion of microRNA-15a/16-1 Promotes Poststroke Angiogenesis and Improves Long-Term Neurological Recovery. Circ. Res. 2020, 126, 1040–1057. [Google Scholar] [CrossRef]
- Martiny-Baron, G.; Marme, D. VEGF-mediated tumour angiogenesis: A new target for cancer therapy. Curr. Opin. Biotechnol. 1995, 6, 675–680. [Google Scholar] [CrossRef]
- Zhang, X.; Nie, D.; Chakrabarty, S. Growth factors in tumor microenvironment. Front. Biosci. (Landmark Ed.) 2010, 15, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Jia, Q.; Song, H.Y.; Warren, R.S.; Donner, D.B. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J. Biol. Chem. 1995, 270, 6729–6733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hoeppner, L.H.; Angom, R.S.; Wang, E.; Dutta, S.; Doeppler, H.R.; Wang, F.; Shen, T.; Scarisbrick, I.A.; Guha, S.; et al. Protein kinase D up-regulates transcription of VEGF receptor-2 in endothelial cells by suppressing nuclear localization of the transcription factor AP2beta. J. Biol. Chem. 2019, 294, 15759–15767. [Google Scholar] [CrossRef]
- Zhao, D.; Desai, S.; Zeng, H. VEGF stimulates PKD-mediated CREB-dependent orphan nuclear receptor Nurr1 expression: Role in VEGF-induced angiogenesis. Int. J. Cancer 2011, 128, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.A.; Navarro, M.N.; Sinclair, L.V.; Emslie, E.; Feijoo-Carnero, C.; Cantrell, D.A. Unique functions for protein kinase D1 and protein kinase D2 in mammalian cells. Biochem. J. 2010, 432, 153–163. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, W.; Wang, H.; Peng, H.; Liu, X.; Guo, W.; Su, G.; Zhao, Z. PKD knockdown inhibits pressure overload-induced cardiac hypertrophy by promoting autophagy via AKT/mTOR pathway. Int. J. Biol. Sci 2017, 13, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Vega, R.B.; Harrison, B.C.; Meadows, E.; Roberts, C.R.; Papst, P.J.; Olson, E.N.; McKinsey, T.A. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell Biol. 2004, 24, 8374–8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Qi, X.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 3059–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, B.C.; Kim, M.S.; van Rooij, E.; Plato, C.F.; Papst, P.J.; Vega, R.B.; McAnally, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N.; et al. Regulation of cardiac stress signaling by protein kinase d1. Mol. Cell Biol. 2006, 26, 3875–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aicart-Ramos, C.; Sanchez-Ruiloba, L.; Gomez-Parrizas, M.; Zaragoza, C.; Iglesias, T.; Rodriguez-Crespo, I. Protein kinase D activity controls endothelial nitric oxide synthesis. J. Cell Sci. 2014, 127, 3360–3372. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Li, J.; Cai, X.; Sun, H.; Jiao, J.; Bai, T.; Zhou, X.W.; Chen, X.; Gill, D.L.; Tang, X.D. Protein kinase D3 is a pivotal activator of pathological cardiac hypertrophy by selectively increasing the expression of hypertrophic transcription factors. J. Biol. Chem. 2011, 286, 40782–40791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Tang, B.L. Protein trafficking mechanisms associated with neurite outgrowth and polarized sorting in neurons. J. Neurochem. 2001, 79, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Wang, Y. Protein kinase D: A new player among the signaling proteins that regulate functions in the nervous system. Neurosci. Bull. 2014, 30, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.M.; Huang, Y.H.; Zhu, Y.B.; Wang, Y. Both the establishment and maintenance of neuronal polarity require the activity of protein kinase D in the Golgi apparatus. J. Neurosci. 2008, 28, 8832–8843. [Google Scholar] [CrossRef]
- Higuero, A.M.; Sanchez-Ruiloba, L.; Doglio, L.E.; Portillo, F.; Abad-Rodriguez, J.; Dotti, C.G.; Iglesias, T. Kidins220/ARMS modulates the activity of microtubule-regulating proteins and controls neuronal polarity and development. J. Biol. Chem. 2010, 285, 1343–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, J.L.; Lewandowski, K.T.; Meek, S.E.; Storz, P.; Toker, A.; Piwnica-Worms, H. Phosphorylation of the Par-1 polarity kinase by protein kinase D regulates 14-3-3 binding and membrane association. Proc. Natl. Acad. Sci. USA 2008, 105, 18378–18383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, K.; Baba, M.; Nagayasu, K.; Yamamoto, K.; Kondo, M.; Kitagawa, K.; Takemoto, T.; Seiriki, K.; Kasai, A.; Ago, Y.; et al. Autism-associated protein kinase D2 regulates embryonic cortical neuron development. Biochem. Biophys. Res. Commun 2019, 519, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Allou, L.; Lambert, L.; Amsallem, D.; Bieth, E.; Edery, P.; Destree, A.; Rivier, F.; Amor, D.; Thompson, E.; Nicholl, J.; et al. 14q12 and severe Rett-like phenotypes: New clinical insights and physical mapping of FOXG1-regulatory elements. Eur. J. Hum. Genet. 2012, 20, 1216–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vineeth, V.S.; Dutta, U.R.; Tallapaka, K.; Das Bhowmik, A.; Dalal, A. Whole exome sequencing identifies a novel 5Mb deletion at 14q12 region in a patient with global developmental delay, microcephaly and seizures. Gene 2018, 673, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Ellaway, C.J.; Ho, G.; Bettella, E.; Knapman, A.; Collins, F.; Hackett, A.; McKenzie, F.; Darmanian, A.; Peters, G.B.; Fagan, K.; et al. 14q12 microdeletions excluding FOXG1 give rise to a congenital variant Rett syndrome-like phenotype. Eur. J. Hum. Genet. 2013, 21, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Montilla, A.; Mata, G.P.; Matute, C.; Domercq, M. Contribution of P2X4 Receptors to CNS Function and Pathophysiology. Int. J. Mol. Sci. 2020, 21, 5562. [Google Scholar] [CrossRef]
- Pose-Utrilla, J.; Garcia-Guerra, L.; Del Puerto, A.; Martin, A.; Jurado-Arjona, J.; De Leon-Reyes, N.S.; Gamir-Morralla, A.; Sebastian-Serrano, A.; Garcia-Gallo, M.; Kremer, L.; et al. Excitotoxic inactivation of constitutive oxidative stress detoxification pathway in neurons can be rescued by PKD1. Nat. Commun. 2017, 8, 2275. [Google Scholar] [CrossRef] [Green Version]
- Stetler, R.A.; Gao, Y.; Zhang, L.; Weng, Z.; Zhang, F.; Hu, X.; Wang, S.; Vosler, P.; Cao, G.; Sun, D.; et al. Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury. J. Neurosci. 2012, 32, 2667–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetler, R.A.; Cao, G.; Gao, Y.; Zhang, F.; Wang, S.; Weng, Z.; Vosler, P.; Zhang, L.; Signore, A.; Graham, S.H.; et al. Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J. Neurosci. 2008, 28, 13038–13055. [Google Scholar] [CrossRef]
- Kolczynska, K.; Loza-Valdes, A.; Hawro, I.; Sumara, G. Diacylglycerol-evoked activation of PKC and PKD isoforms in regulation of glucose and lipid metabolism: A review. Lipids Health Dis. 2020, 19, 113. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, V.; Ghislain, J.; Vivot, K.; Tamarina, N.; Philipson, L.H.; Fielitz, J.; Poitout, V. Deletion of Protein Kinase D1 in Pancreatic beta-Cells Impairs Insulin Secretion in High-Fat Diet-Fed Mice. Diabetes 2018, 67, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Wang, C.; Chen, J.Y.; Lu, F.; Wang, J.; Hou, N.; Hu, X.; Zeng, F.; Ma, D.; Sun, X.; et al. Deficiency of PRKD2 triggers hyperinsulinemia and metabolic disorders. Nat. Commun. 2018, 9, 2015. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.A.; Li, J.; Silva, S.R.; Jackson, L.N.; Zhou, Y.; Watanabe, H.; Ives, K.L.; Hellmich, M.R.; Evers, B.M. PKD3 Is the Predominant Protein Kinase D Isoform in Mouse Exocrine Pancreas and Promotes Hormone-induced Amylase Secretion. J. Biol. Chem. 2009, 284, 2459–2471. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.E.; Loffler, M.C.; Loza Valdes, A.E.; Schmitz, W.; El-Merahbi, R.; Viera, J.T.; Erk, M.; Zhang, T.; Braun, U.; Heikenwalder, M.; et al. The kinase PKD3 provides negative feedback on cholesterol and triglyceride synthesis by suppressing insulin signaling. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Kim, Y.I.; Park, J.E.; Brand, D.D.; Fitzpatrick, E.A.; Yi, A.K. Protein kinase D1 is essential for the proinflammatory response induced by hypersensitivity pneumonitis-causing thermophilic actinomycetes Saccharopolyspora rectivirgula. J. Immunol. 2010, 184, 3145–3156. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, K.; Gon, Y.; Shimokawa, T.; Nunomura, S.; Endo, D.; Miyata, N.; Hashimoto, S.; Van Lint, J.; Ra, C. High affinity receptor for IgE stimulation activates protein kinase D augmenting activator protein-1 activity for cytokine producing in mast cells. Int. Immunopharmacol. 2010, 10, 277–283. [Google Scholar] [CrossRef]
- Salamon, P.; Shefler, I.; Hershko, A.Y.; Mekori, Y.A. The Involvement of Protein Kinase D in T Cell-Induced Mast Cell Activation. Int. Arch. Allergy Immunol. 2016, 171, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Chung, C.; Slice, L.; Sinnett-Smith, J.; Rozengurt, E. Protein kinase D mediates synergistic expression of COX-2 induced by TNF-{alpha} and bradykinin in human colonic myofibroblasts. Am. J. Physiol Cell Physiol. 2009, 297, C1576–C1587. [Google Scholar] [CrossRef] [Green Version]
- Chiu, T.T.; Leung, W.Y.; Moyer, M.P.; Strieter, R.M.; Rozengurt, E. Protein kinase D2 mediates lysophosphatidic acid-induced interleukin 8 production in nontransformed human colonic epithelial cells through NF-kappaB. Am. J. Physiol. Cell Physiol. 2007, 292, C767–C777. [Google Scholar] [CrossRef]
- Hao, Q.; Wang, L.; Tang, H. Vascular endothelial growth factor induces protein kinase D-dependent production of proinflammatory cytokines in endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 296, C821–C827. [Google Scholar] [CrossRef] [PubMed]
- Diebold, Y.; Chen, L.L.; Tepavcevic, V.; Ferdman, D.; Hodges, R.R.; Dartt, D.A. Lymphocytic infiltration and goblet cell marker alteration in the conjunctiva of the MRL/MpJ-Fas(lpr) mouse model of Sjogren’s syndrome. Exp. Eye Res. 2007, 84, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Hao, F.; Xu, X.; Chisolm, G.M.; Cui, M.Z. Lysophosphatidylcholine activates a novel PKD2-mediated signaling pathway that controls monocyte migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Saini, S.; Liu, T.; Yoo, J. Bradykinin stimulates protein kinase D-mediated colonic myofibroblast migration via cyclooxygenase-2 and heat shock protein 27. J. Surg. Res. 2017, 209, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gamez, M.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef]
- Yuan, J.; Tan, T.; Geng, M.; Tan, G.; Chheda, C.; Pandol, S.J. Novel Small Molecule Inhibitors of Protein Kinase D Suppress NF-kappaB Activation and Attenuate the Severity of Rat Cerulein Pancreatitis. Front. Physiol. 2017, 8, 1014. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Liu, Y.; Tan, T.; Guha, S.; Gukovsky, I.; Gukovskaya, A.; Pandol, S.J. Protein kinase d regulates cell death pathways in experimental pancreatitis. Front. Physiol. 2012, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Yang, Y.; Zhang, H.; Han, Y.; Li, Y.; Zhang, Y.; Yin, D.; He, Q.; Zhao, Z.; Blumberg, P.M.; et al. Interaction between protein kinase D1 and transient receptor potential V1 in primary sensory neurons is involved in heat hypersensitivity. Pain 2008, 137, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, K.; Park, J.E.; Yoon, T.W.; Halder, P.; Kim, Y.I.; Metcalfe, V.; Talati, A.J.; English, B.K.; Yi, A.K. Group B Streptococci Induce Proinflammatory Responses via a Protein Kinase D1-Dependent Pathway. J. Immunol. 2017, 198, 4448–4457. [Google Scholar] [CrossRef]
- Rezaee, F.; DeSando, S.A.; Ivanov, A.I.; Chapman, T.J.; Knowlden, S.A.; Beck, L.A.; Georas, S.N. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J. Virol. 2013, 87, 11088–11095. [Google Scholar] [CrossRef] [Green Version]
- Amadesi, S.; Grant, A.D.; Cottrell, G.S.; Vaksman, N.; Poole, D.P.; Rozengurt, E.; Bunnett, N.W. Protein kinase D isoforms are expressed in rat and mouse primary sensory neurons and are activated by agonists of protease-activated receptor 2. J. Comp. Neurol. 2009, 516, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Gan, H.; McKenzie, R.; Hao, Q.; Idell, S.; Tang, H. Protein kinase D is increased and activated in lung epithelial cells and macrophages in idiopathic pulmonary fibrosis. PLoS ONE 2014, 9, e101983. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H.; Yin, M.; Pei, X.; Hausser, A.; Ishikawa, E.; Yamasaki, S.; Jin, Z.G. Deletion of Protein Kinase D3 Promotes Liver Fibrosis in Mice. Hepatology 2020, 72, 1717–1734. [Google Scholar] [CrossRef]
- Geng, J.; Zhao, Z.; Kang, W.; Wang, W.; Zhang, Y.; Zhiming, G.E. Atorvastatin reverses cardiac remodeling possibly through regulation of protein kinase D/myocyte enhancer factor 2D activation in spontaneously hypertensive rats. Pharmacol. Res. 2010, 61, 40–47. [Google Scholar] [CrossRef]
- Shao, W.; Li, D.; Peng, J.; Chen, S.; Zhou, C.; Cheng, Z.; Yu, Y.; Li, H.; Li, C.; You, Y.; et al. Inhibitory effect of ethyl acetate extract of Aristolochia yunnanensis on cardiac fibrosis through extracellular signal-regulated kinases 1/2 and transforming growth factor beta/small mother against decapentaplegic signaling pathways. Transl. Res. 2014, 163, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Davidson-Moncada, J.K.; Lopez-Lluch, G.; Segal, A.W.; Dekker, L.V. Involvement of protein kinase D in Fc gamma-receptor activation of the NADPH oxidase in neutrophils. Biochem. J. 2002, 363, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.R.; Legere, H.J., 3rd; Katz, H.R. Activation of protein kinase D1 in mast cells in response to innate, adaptive, and growth factor signals. J. Immunol. 2007, 179, 7876–7882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Kim, Y.I.; Yi, A.K. Protein kinase D1 is essential for MyD88-dependent TLR signaling pathway. J. Immunol. 2009, 182, 6316–6327. [Google Scholar] [CrossRef] [PubMed]
- Parra, M.; Kasler, H.; McKinsey, T.A.; Olson, E.N.; Verdin, E. Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J. Biol. Chem. 2005, 280, 13762–13770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, S.A.; Liu, P.; Spitaler, M.; Olson, E.N.; McKinsey, T.A.; Cantrell, D.A.; Scharenberg, A.M. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol. Cell Biol. 2006, 26, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, R.B.; Dickey, D.M.; Chung, H.; Quale, A.C.; Nagarajan, L.R.; Billadeau, D.D.; Shimizu, Y. Protein kinase D1 and the beta 1 integrin cytoplasmic domain control beta 1 integrin function via regulation of Rap1 activation. Immunity 2005, 23, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haxhinasto, S.A.; Bishop, G.A. A novel interaction between protein kinase D and TNF receptor-associated factor molecules regulates B cell receptor-CD40 synergy. J. Immunol. 2003, 171, 4655–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, A.; Chen, Y.Z.; Tsukamoto, H.; Jotsuka, T.; Masuda, M.; Nishimura, Y. Unique T cell proliferation associated with PKCmu activation and impaired ZAP-70 phosphorylation in recognition of overexpressed HLA/partially agonistic peptide complexes. Eur. J. Immunol. 2003, 33, 1497–1507. [Google Scholar] [CrossRef]
- Spitaler, M.; Emslie, E.; Wood, C.D.; Cantrell, D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity 2006, 24, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, E.; Kosako, H.; Yasuda, T.; Ohmuraya, M.; Araki, K.; Kurosaki, T.; Saito, T.; Yamasaki, S. Protein kinase D regulates positive selection of CD4(+) thymocytes through phosphorylation of SHP-1. Nat. Commun. 2016, 7, 12756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, T.; SoRelle, J.A.; Choi, J.H.; Yue, T.; Wang, K.W.; McAlpine, W.; Wang, J.; Liu, A.; Tabeta, K.; Turer, E.E.; et al. Mutual inhibition between Prkd2 and Bcl6 controls T follicular helper cell differentiation. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.; Gatenby, P.A.; Wilson, A.; Malik, S.; Fulcher, D.A.; Tangye, S.G.; Manku, H.; Vyse, T.J.; Roncador, G.; Huttley, G.A.; et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum 2010, 62, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Cook, M.C.; Angelucci, C.; Athanasopoulos, V.; Rui, L.; Hill, K.M.; Yu, D.; Domaschenz, H.; Whittle, B.; Lambe, T.; et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005, 435, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Tanaka, S.; Motomura, Y.; Harada, Y.; Ohno, S.; Ohno, S.; Yanagi, Y.; Inoue, H.; Kubo, M. The 3’ enhancer CNS2 is a critical regulator of interleukin-4-mediated humoral immunity in follicular helper T cells. Immunity 2012, 36, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Sharlow, E.R.; Giridhar, K.V.; LaValle, C.R.; Chen, J.; Leimgruber, S.; Barrett, R.; Bravo-Altamirano, K.; Wipf, P.; Lazo, J.S.; Wang, Q.J. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J. Biol. Chem. 2008, 283, 33516–33526. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Altamirano, K.; George, K.M.; Frantz, M.C.; Lavalle, C.R.; Tandon, M.; Leimgruber, S.; Sharlow, E.R.; Lazo, J.S.; Wang, Q.J.; Wipf, P. Synthesis and Structure-Activity Relationships of Benzothienothiazepinone Inhibitors of Protein Kinase D. ACS Med. Chem. Lett. 2011, 2, 154–159. [Google Scholar] [CrossRef] [PubMed]
- George, K.M.; Frantz, M.C.; Bravo-Altamirano, K.; Lavalle, C.R.; Tandon, M.; Leimgruber, S.; Sharlow, E.R.; Lazo, J.S.; Wang, Q.J.; Wipf, P. Design, Synthesis, and Biological Evaluation of PKD Inhibitors. Pharmaceutics 2011, 3, 186–228. [Google Scholar] [CrossRef] [Green Version]
- Lavalle, C.R.; Bravo-Altamirano, K.; Giridhar, K.V.; Chen, J.; Sharlow, E.; Lazo, J.S.; Wipf, P.; Wang, Q.J. Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility. BMC Chem. Biol. 2010, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Sharlow, E.R.; Mustata Wilson, G.; Close, D.; Leimgruber, S.; Tandon, M.; Reed, R.B.; Shun, T.Y.; Wang, Q.J.; Wipf, P.; Lazo, J.S. Discovery of diverse small molecule chemotypes with cell-based PKD1 inhibitory activity. PLoS ONE 2011, 6, e25134. [Google Scholar] [CrossRef] [Green Version]
- Monovich, L.; Vega, R.B.; Meredith, E.; Miranda, K.; Rao, C.; Capparelli, M.; Lemon, D.D.; Phan, D.; Koch, K.A.; Chapo, J.A.; et al. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2010, 584, 631–637. [Google Scholar] [CrossRef]
- Meredith, E.L.; Beattie, K.; Burgis, R.; Capparelli, M.; Chapo, J.; Dipietro, L.; Gamber, G.; Enyedy, I.; Hood, D.B.; Hosagrahara, V.; et al. Identification of potent and selective amidobipyridyl inhibitors of protein kinase D. J. Med. Chem. 2010, 53, 5422–5438. [Google Scholar] [CrossRef]
- Meredith, E.L.; Ardayfio, O.; Beattie, K.; Dobler, M.R.; Enyedy, I.; Gaul, C.; Hosagrahara, V.; Jewell, C.; Koch, K.; Lee, W.; et al. Identification of orally available naphthyridine protein kinase D inhibitors. J. Med. Chem. 2010, 53, 5400–5421. [Google Scholar] [CrossRef] [PubMed]
- Gamber, G.G.; Meredith, E.; Zhu, Q.; Yan, W.; Rao, C.; Capparelli, M.; Burgis, R.; Enyedy, I.; Zhang, J.H.; Soldermann, N.; et al. 3,5-diarylazoles as novel and selective inhibitors of protein kinase D. Bioorg. Med. Chem. Lett. 2011, 21, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ochi, N.; Tong, Z.; Deorukhkar, A.; Sung, B.; Kelland, L.; Jamieson, S.; Sutherland, R.; Raynham, T.; et al. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1136–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, I.M.; Bagherzadeh, A.; Charles, M.; Raynham, T.; Ireson, C.; Boakes, A.; Kelland, L.; Zachary, I.C. Characterization of the biological effects of a novel protein kinase D inhibitor in endothelial cells. Biochem. J. 2010, 429, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Borges, S.; Perez, E.A.; Thompson, E.A.; Radisky, D.C.; Geiger, X.J.; Storz, P. Effective Targeting of Estrogen Receptor-Negative Breast Cancers with the Protein Kinase D Inhibitor CRT0066101. Mol. Cancer Ther. 2015, 14, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Y.; Yu, S.; Zhou, Y.; Ma, X.; Su, Q.; An, L.; Wang, F.; Shi, A.; Zhang, J.; et al. The Role and Mechanism of CRT0066101 as an Effective Drug for Treatment of Triple-Negative Breast Cancer. Cell Physiol. Biochem. 2019, 52, 382–396. [Google Scholar] [CrossRef]
- Pasquier, A.; Vivot, K.; Erbs, E.; Spiegelhalter, C.; Zhang, Z.; Aubert, V.; Liu, Z.; Senkara, M.; Maillard, E.; Pinget, M.; et al. Lysosomal degradation of newly formed insulin granules contributes to beta cell failure in diabetes. Nat. Commun. 2019, 10, 3312. [Google Scholar] [CrossRef]
- Venardos, K.; De Jong, K.A.; Elkamie, M.; Connor, T.; McGee, S.L. The PKD inhibitor CID755673 enhances cardiac function in diabetic db/db mice. PLoS ONE 2015, 10, e0120934. [Google Scholar] [CrossRef] [PubMed]
- Renton, M.C.; McGee, S.L.; Howlett, K.F. The role of protein kinase D (PKD) in intracellular nutrient sensing and regulation of adaptive responses to the obese environment. Obes. Rev. 2020. [Google Scholar] [CrossRef]
- Chen, J.; Cui, B.; Fan, Y.; Li, X.; Li, Q.; Du, Y.; Feng, Y.; Zhang, P. Protein kinase D1 regulates hypoxic metabolism through HIF-1alpha and glycolytic enzymes incancer cells. Oncol. Rep. 2018, 40, 1073–1082. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pathological Events | PKDs | Proposed Function | Cancer Types | Target Gene/Mechanisms | Ref |
|---|---|---|---|---|---|
| Proliferation | PKD1 | Positive | - Pancreatic | - Stimulates accumulation of c-Fos, DNA synthesis via strengthening ERK while suppressing JNK/c-Jun signaling | [51] |
| - Drives the formation of acinar-to-ductal metaplasia and further progression to pancreatic intraepithelial neoplasia | [52] | ||||
| - Prolongs ERK1/2 activation | [53,54] | ||||
| - Cell cycle regulation | [55,56] | ||||
| - Head and neck squamous cell; Kidney | - MEK/ERK-dependent signaling pathway | [57] | |||
| - Prostate | - Increases ERα expression and cell sensitivity to 17β-estradiol | [58] | |||
| - Breast | - Contributes to hyperplastic and inflammatory responses to topical phorbol ester | [41] | |||
| Negative | - Prostate | - Increases MMP-2, MMP-9 secretion | [59] | ||
| - Induces G1-phase arrest by phosphorylating cell-division cycle phosphatase 25 | [60] | ||||
| - Colon | - Induces nuclear exclusion of β-catenin | [61] | |||
| - Lung | - Negative regulator of mTORC1-S6K1 signalling | [62] | |||
| PKD2 | Positive | - Prostate | - Activated during G2-M, co-localizes with/regulate Aurora A kinase at the centrosome | [63] | |
| - Colon | - Stimulates NF-κB activity via AKT and ERK signalling | [64] | |||
| - Glioblastoma | - Regulates Golgi phosphoprotein 3 | [65] | |||
| PKD3 | Positive | - Breast | - Regulates mTORC1-S6 kinase 1 signalling | [66] | |
| - Activates ERK1/c-Myc axis | [67] | ||||
| - Phosphorylates HSP27 and HDAC4/5/7 | [68] | ||||
| Survival | PKD1 | Positive | - Pancreatic | - Activates glucose transporter 1 and mTORC1 | [69] |
| - Induces anti-apoptotic proteins survivin and c-FLIPL | [70] | ||||
| - Prostate | - Activates ERK1/2 and NF-κB signalling | [8] | |||
| PKD2 | Positive | - Prostate; Colon; Leukemia | - Stimulates NF-κB activity | [8,71,72] | |
| - Colon; Breast | - Reverses HSP90 inhibition-induced apoptotic effects | [45] | |||
| PKD3 | Positive | - Prostate | - Akt and ERK1/2 | [73] | |
| EMT Migration Invasion | PKD1 | Negative | - Prostate; Breast | - Inactivates transcription factor Snail | [74,75] |
| - Prostate | - Phosphorylates junctional proteins (E-cadherin and β-catenin) | [76,77] | |||
| - Polyubiquitination and proteasomal degradation of MTA1 | [78] | ||||
| - Breast | - Promotes ɑvβ3 integrin recycling via phosphorylating Rabaptin-5 | [79] | |||
| - Represses the expression of MMPs | [80] | ||||
| - Melanoma | - Phosphorylates SSH1L, block cofilin dephosphorylation | [81] | |||
| - Regulates E-cadherin expression and β-catenin localization | [82] | ||||
| PKD2 | Positive | - Prostate | - Phosphorylates IKKβ, nuclear translocation and activation of NFκB | [83] | |
| - Pancreatic- Glioblastoma | - Stimulates expression and secretion of MMP-7 and MMP-9 | [84] | |||
| - Liver | - Regulates MMP-1 and integrin expression | [85] | |||
| - Prostate | - Promotes PI3K/Akt/GSK-3β signalling | [86] | |||
| PKD3 | Positive | - Breast | - Activates NFκB and deactivate HDAC1 | [83] | |
| - Secretion of MMP-9 and tumor-promoting cytokines | [87] | ||||
| - Activates PAK4/LIMK signaling | [88] | ||||
| - Regulates cytoskeletal remodeling by phosphorylating GIT1 | [89] | ||||
| Angiogenesis | PKD1 | Positive | - Pancreatic | - Induces the secretion of VEGF and CXC chemokines | [90] |
| - Breast | - LPA/PKD-1-CD36 signaling | [91] | |||
| PKD2 | Positive | - Gastrointestinal | - Regulates tumor-endothelial cell communication | [92] | |
| - Colon; Breast | - Stabilizes Hsp90; NF-κB/VEGF-A | [44,45] | |||
| PKD3 | Positive | - Prostate | - Regulates mast cell recruitment | [47] | |
| Immune response | PKD2 | Positive | - Oral squamous | - Regulates PD-L1 surface expression | [93] |
| - Fibrosarcoma | - Phosphorylates and degrades IFNAR1 | [94] | |||
| PKD3 | Positive | - Oral squamous cell carcinoma | - Regulates IFN-γ induced PD-L1 expression | [50] |
| Disease | PKDs | Functions | Diseases/Pathologies | Targets | Ref |
|---|---|---|---|---|---|
| Cardiovascular disease | PKD1 | - PKD1 activation leads to cardiac hypertrophy. | - Cardiac hypertrophy | - HDAC4, 5, 7, 9; MEF2 | [146] |
| - AKT/mTOR regulated autophagy | [144] | ||||
| - Regulates VEGF-mediated angiogenesis. | - Vasodilation | - Nitric oxide synthase | [148] | ||
| PKD3 | - Mediates glucose uptake during cardiac hypertrophy. | - Cardiac hypertrophy | - NFATc4, Nkx2.5, GATA4, MEF2 | [149] | |
| CNS disorders | PKD1 | - Maintains polarity of hippocampal neurons | - Neuronal polarization and development | - Kidins220, Par-1 | [153,154,155] |
| - Neuronal survival | - Neurodegeneration | - NF-κB | [161] | ||
| - Ischemic stroke | - Hsp27 | [162] | |||
| - Mediates neurogenic inflammation and pain transmission | - Hyperalgesia | - TRPV | [184] | ||
| PKD2 | - Maintains neuronal polarity | - Neuronal polarization and development | - Kidins220 | [154] | |
| - Contributes to autism spectrum disorder | - ASD, RTT | - ERK1/2 | [156] | ||
| - Mediates neurogenic inflammation and pain transmission | - Hyperalgesia | - TRPV | [184] | ||
| PKD3 | - Expressed in primary sensory neurons that mediate neurogenic inflammation and pain transmission | - Hyperalgesia | - TRPV | [184] | |
| Metabolic disease | PKD1 | - Regulates insulin secretion and pancreatic β cell survival; Insulin exocytosis at TGN | - Type 2 diabetes, obesity | - Inhibitory phosphorylation by p38δ | [24] |
| PKD2 | - PKD2 inhibition leads to insulin resistance | - Hyperinsulinemia | - L-type Ca2+ channels | [166] | |
| PKD3 | - Suppresses insulin signalling in liver and promotes insulin resistance | - Type 2 diabetes | - Akt/mTORC1 and mTORC2 | [168] | |
| Inflammatory disease | PKD1 | - Contributes to bacteria-induced proinflammatory immune responses and neutrophil influx | - Hypersensitivity pneumonitis | - MAPK, NF-κB | [169] |
| - Inflammatory cell infiltration | - Pancreatitis | - NF-κB, IL-6, MCP-1 | [179] | ||
| - Contributes to fibrosis | - Fibrosis | - HDACs, MEF2 | [146] | ||
| PKD3 | - Liver fibrosis, hepatic macrophage polarization | - Liver fibrosis | - TGFβ | [187] | |
| Immune dysregulation | PKD1 | - Mast cell activation | - Allergic reaction | - MCP-1 | [191] |
| - Activated by TLR ligands, and is MyD88-dependent | - Proinflammatory immune responses | - TRAF6, TAK1, MAPKs | [192] | ||
| - Transcriptional activates Nur77 during thymocyte activation | - T-cell receptor activation | - HDAC7 | [195] | ||
| PKD2 | - Excessive cell autonomous T follicular helper cell development | - Germinal center development | - Bcl6 | [200] | |
| - Nature killer cell activation | - Innate immune response | - IFN-γ, TNF-α | [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Connelly, J.; Chao, Y.; Wang, Q.J. Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases. Biomolecules 2021, 11, 483. https://doi.org/10.3390/biom11030483
Zhang X, Connelly J, Chao Y, Wang QJ. Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases. Biomolecules. 2021; 11(3):483. https://doi.org/10.3390/biom11030483
Chicago/Turabian StyleZhang, Xuejing, Jaclyn Connelly, Yapeng Chao, and Qiming Jane Wang. 2021. "Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases" Biomolecules 11, no. 3: 483. https://doi.org/10.3390/biom11030483