1. Introduction
As a deadly infectious disease, tuberculosis (TB) infected about 10 million people and caused an estimated 1.4 million deaths worldwide in 2019, and the responsible pathogen for TB is
Mycobacterium tuberculosis (
Mtb) [
1]. Globally, TB has developed resistance to traditional anti-TB drugs like isoniazid and rifampicin, an unfortunate complication to TB prevention and treatment [
2,
3]. Patients infected with multidrug-resistant TB (MDR-TB) require medicines at higher costs and a longer time for treatment, only receiving a treatment success rate of 57% [
1]. With the emergence and spread of MDR-TB strains, it is imminent to find clinical targets for developing new antimicrobials against
Mtb.
RimM, an important ribosome maturation factor protein existing in
Mtb, is a candidate target for anti-TB drugs. The RimM protein family is included in various bacterial species, but no ortholog is available in humans. Researches have been extensively conducted on explicit functions of RimM serving as one of bacterial biogenesis factors active in the ribosome assembly process [
4]. Knockout of the RimM gene caused a significant decrease in cell growth rate, accumulation of 16S rRNA precursors and ribosomal intermediates, and a reduction of polysome level [
5,
6,
7,
8]. Further researches showed that RimM does not bind to complete 70S ribosomes or mature 30S subunits but moderately binds to immature 30S intermediates [
4,
5,
9]. Suppressive mutation experiments on ribosomal proteins and 16S rRNA indicated that RimM might bind the 3′-domain of 16S rRNA [
5,
8]. Analyses on composition and structures of the immature 30S intermediates collected in the RimM knockout strain indicated that RimM plays a crucial role in assisting the late assembly of the head domain of the 30S subunit [
4,
10,
11]. Moreover, in vitro pull-down experiments confirmed the specificity with which RimM binds to S19, a ribosomal protein located at the 30S head domain [
8]. Kinetic experiments disclosed that RimM accelerates the binding of S19 to 16S rRNA by overcoming the potential well during the slow binding process [
12]. Together, these advances indicate that the RimM–S19 interaction plays a vital role in ribosome biosynthesis, for which RimM is a competent attacking target for antimicrobials against
Mtb.
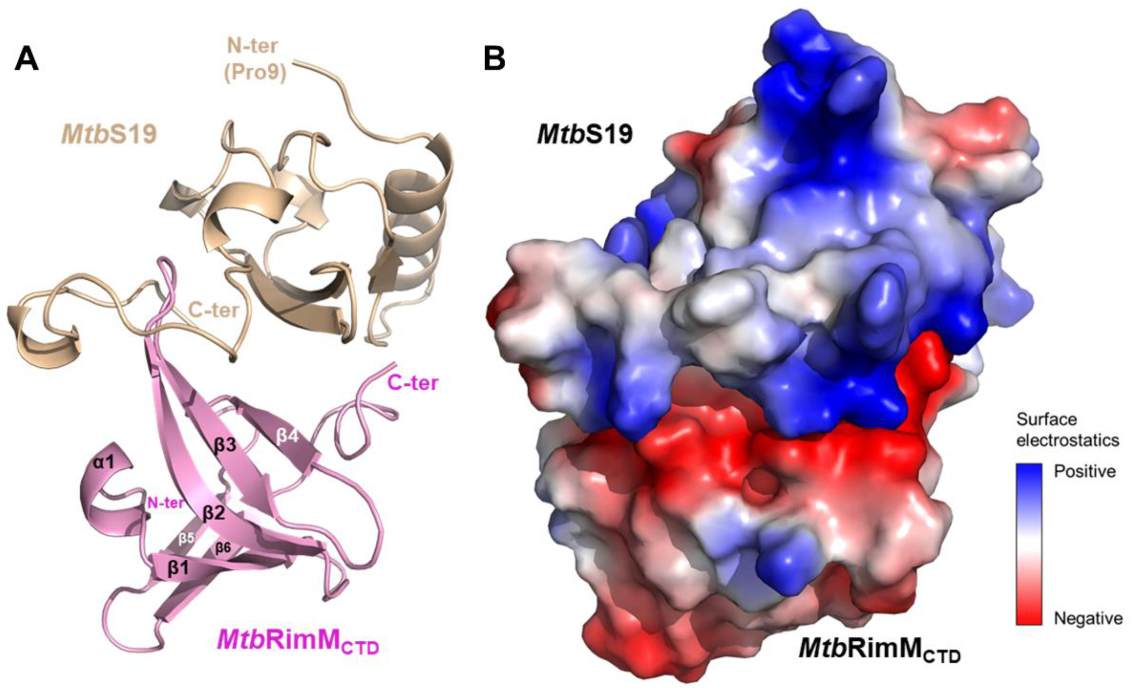
As is well known, the function of a protein is determined by its structure, and structural basis is required to mechanistically understand the protein function. So far, the three-dimensional (3D) structure of RimM from
Mtb (
MtbRimM) has not been determined. To our knowledge, the following 3D structures of RimM orthologs have been resolved and are accessible in the Protein Data Bank (PDB): (a) the crystal structures of RimM proteins in free form from
P. aeruginosa (3 mutations, PDB ID: 2F1L, termed
PaeRimM),
T. thermophilus HB8 (wild type, PDB ID: 2DYI, termed
TthRimM),
A. calcoaceticus (5 mutations, PDB ID: 2QGG, termed
AciRimM), and
H. influenzae (wild type, PDB ID: 3H9N, termed
HinRimM); (b) the crystal structure of the RimM–S19 complex from
T. thermophilus HB8 (both wild types, PDB ID: 3A1P, S19 from
T. thermophilus is termed
TthS19); (c) the solution structure of truncated RimM N-terminal domain from
T. thermophilus HB8 (wild type, PDB ID: 2DOG) [
13]. Inspection of these structures, in correspondence to earlier assertions based on multi-sequence alignments [
8,
14], shows that the structure of RimM is composed of an N-terminal domain (NTD), a C-terminal domain (CTD), and a short loop in between (
Figure 1A). Part of the RimM NTD shares sequence similarity with the RNA-binding KH domain, implying its potential interaction with RNA [
5,
15]. The RimM CTD was identified as a PRC-barrel domain [
14]. Originally, the PRC-barrels were discovered to be a mediator of quinone reduction within the photosynthetic reaction center complex from purple proteobacteria [
16]. However, PRC-barrels of the RimM family lack a decisive glutamate residue required for electron transfer in the redox reaction [
14]. Instead, they are likely to be involved in binding S19 during the maturation of the 30S ribosomal subunit [
8]. Previous nuclear magnetic resonance (NMR) titration assays showed that the CTD part of full-length RimM can significantly interact with S19 but the NTD part can not, indicating that the CTD part is primarily responsible for binding S19 [
13].
Suffering from very few available conformational restraints, the structural calculation of
TthRimM CTD failed to converge in the NMR structure ensemble, in sharp contrast with
TthRimM NTD [
13]. On the other hand, the aforementioned crystal structures of full-length RimM orthologs characterize a well-folded CTD, and the
TthRimM–S19 complex even describes a molecular model for the RimM–S19 interaction. However, sequence alignments show that the highest sequence identity between
MtbRimM CTD (termed
MtbRimM
CTD) and other CTDs from species with known structures is only 32.48% (
Figure 1B). Thus, the orthologous structures of RimM are insufficient for revealing the structural basis of
MtbRimM
CTD binding S19. Expectedly, such a structural basis would facilitate the design of RimM-based anti-TB drugs.
Here, we determined the solution structure of MtbRimMCTD that is primarily responsible for the interaction of MtbRimM with S19. We then analyzed dynamics features of MtbRimMCTD by NMR relaxation measurements and molecular dynamics (MD) simulation, and characterized biophysical properties of MtbRimMCTD binding S19 through NMR titration and surface plasmon resonance (SPR) assays. Based on the identified S19 binding sites on MtbRimMCTD, which were verified by mutagenesis experiments, we established a structural model of the MtbRimMCTD–S19 complex by molecular docking to illustrate the unique pattern of MtbRimMCTD binding S19. Our results shed light on the molecular mechanisms of the MtbRimM–S19 interaction and offer novel insights into drug development against tuberculosis.
2. Materials and Methods
2.1. Cloning, Expression, and Purification
The gene sequences of RimM and S19 from
M. tuberculosis strain H37Rv (
MtbRimM and
MtbS19) were obtained from the NCBI database (gene ID: 887188 for RimM and 888356 for S19). Recombinant plasmids harboring
MtbRimM gene (pET-22b, with a C-terminal His
6-tag) or
MtbS19 gene (pET-28a, with thrombin-cleavable N-terminal His
6-tag) were commercially synthesized (GenScript, Nanjing, China) Boundaries for the N-terminal domain (residues 4–93, NTD) and the C-terminal domain (residues 100–173, CTD) of
MtbRimM were determined by Pfam [
17]. To obtain truncations, individual gene fragments of the NTD of
MtbRimM (
MtbRimM
NTD, residues 1–93) and the CTD of
MtbRimM (
MtbRimM
CTD, residues 101–176) were extracted and cloned by PCR. The following primers were applied in the PCR: (a) 5′-TTAGGATCCATGGAGCTGGTTGTGG-3′ (restriction site BamH I) and (b) 5′-TAGCTCGAGTTAATCGTCCGCATCG-3′ (restriction site Xho I) for
MtbRimM
NTD, and (c) 5′-CGCCGGCATATGGATACCTACTATG-3′ (restriction site Nde I) and (d) 5′-TAGCTCGAGTTCCAGGTTCAGCAGA-3′ (restriction site Xho I) for
MtbRimM
CTD. PCR products for
MtbRimM
NTD were then ligated into the pET-28a-SUMO plasmid which contains an N-terminal His
6-tag followed by a SUMO fusion protein and a SUMO protease cutting site [
18], and those for
MtbRimM
CTD into the pET-22b plasmid which contains a C-terminal His
6-tag. Recombinant plasmids encoding
MtbRimM
CTD point mutants (pET-22b, with C-terminal His
6-tag) were commercially synthesized (Sangon Biotech, Shanghai, China). All plasmids used in this study were verified via DNA sequencing.
The plasmids bearing the genes of MtbRimM, MtbS19, MtbRimMNTD, MtbRimMCTD and point mutants were transformed into the E. Coli BL21(DE3) strain. Overexpression of these proteins was induced with 0.5 mM IPTG at OD600 = 0.6 and conducted at 25 °C for 10 h in either LB liquid media or M9 media. For uniform 15N- and/or 13C-labeling of the proteins, 0.1% (m/v) of 15NH4Cl and/or 0.3% (m/v) of 13C-glucose were added into M9 media.
The harvested cell pellet was resuspended in 50 mM Tris, pH 8.0, 950 mM NaCl, 2.0 mM imidazole, 0.1 mg/mL lysozyme, 1.0 mM phenylmethylsulfonyl fluoride (PMSF), and lysed on ice by sonication. The soluble fraction of the lysate was collected by centrifugation and loaded onto 5 mL Co-NTA resin, which was obtained by stripping the Ni-NTA resin (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) of coordinated Ni2+ with EDTA and re-cobaltizing it with CoCl2. In the affinity chromatography, recombinant protein was eluted with 50 mM Tris, pH 8.0, 250 mM NaCl, 300 mM imidazole. After that, the protein was buffer-exchanged into 20 mM potassium phosphate, pH 7.2, 100 mM KCl, 0.02% NaN3 (hereafter referred to as the universal buffer), and purified through size exclusion chromatography (SEC) using ÄKTA FPLC system with a Superdex 75 10/300 GL column (GE healthcare). Additionally, proteins with cleavable N-terminal His6-tags were treated with corresponding proteases until the tags were completely cleaved before repurified with Co-NTA and SEC. MtbRimMNTD was cleaved with 0.4 mg/mL SUMO-protease for 3 h at room temperature, and MtbS19 with 0.4 mg/mL thrombin overnight at 4 °C. Finally, the protein solution was concentrated to 600 μM for NMR experiments conducted in this study if not otherwise specified.
2.2. NMR Spectroscopy
NMR spectra were recorded at 298K on a Bruker Avance III 850 MHz spectrometer (Bruker BioSpin GmbH, Karlsruhe, Germany) equipped with a
1H-/
13C-/
15N- TCI cryogenic probe (Bruker AG, Fällanden, Switzerland). All protein samples for NMR spectroscopy were dissolved in the universal buffer. The protein samples included 10% D
2O (
v/
v) for recording general NMR spectra and 99% D
2O (
v/
v) for recording 3D
13C-edited NOESY-HSQC (nuclear Overhauser effect spectroscopy-heteronuclear singular quantum correlation) spectrum. The [
15N]-labeled protein sample was used for recording two-dimensional (2D)
1H-
15N HSQC and 3D
15N-edited NOESY-HSQC spectra, and the [
13C,
15N]-labeled protein sample was prepared for 2D
1H-
13C HSQC and other 3D NMR spectra. 2D
1H-
15N HSQC and
1H-
13C HSQC spectra were recorded on
MtbRimM,
MtbRimM
NTD and
MtbRimM
CTD. 3D HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO and HN(CA)CO spectra were recorded for performing backbone resonance assignments of
MtbRimM and
MtbRimM
CTD. 3D H(CCO)NH, CC(CO)NH, HBHA(CO)NH, H(C)CH-COSY, and (H)CCH-TOCSY spectra were recorded for conducting side-chain resonance assignments of
MtbRimM
CTD. 3D
13C-edited NOESY-HSQC and
15N-edited NOESY-HSQC spectra with a mixing time of 120 ms were recorded on
MtbRimM
CTD for obtaining NOE (nuclear Overhauser effect) restraints. All spectra were processed with NMRPipe [
19] and analyzed with NMRFAM-SPARKY [
20].
2.3. Structure Determination
Chemical shifts of
MtbRimM
CTD which had been previously deposited to BMRB (Accession ID: 36368) were used for NOE assignments (data under review) [
21]. Cross-peaks in
13C- and
15N-edited NOESY-HSQC spectra were either manually assigned or ambiguously assigned with the help of Aria 2.3 [
22]. NOE-derived distance restraints were generated from signal integrals in
13C- and
15N-edited NOESY-HSQC spectra. Backbone dihedral restraints (φ, ψ) were predicted from chemical shifts via the TALOS-N server [
23]. Altered MD parameters were adopted in Aria 2.3 setup (10,000, 4000, 80,000, and 64,000 for high temperature, refinement, cool1, and cool2 steps). Totally, 100 structures of
MtbRimM
CTD were calculated and refined with Aria 2.3, and 20 lowest-energy models were used as the ultimate structure ensemble. The C-terminal His
6-tag was not modeled in the final structures due to the lack of chemical shifts and NOE cross-peak assignments. PROCHECK [
24] was applied for structural quality evaluation, and Pymol [
25] for structure visualization.
2.4. NMR Relaxation Measurements
All recombinant proteins were dissolved in the universal buffer. A protein sample of [
15N]-labeled
MtbRimM
CTD at 600 μM was used to conduct NMR relaxation measurements of backbone amide groups including amide R
1 and R
2 relaxation rates and {
1H}-
15N heteronuclear steady-state NOEs (hNOEs). Two sets of pseudo-3D experiments incorporated with
1H-
15N HSQC spectra were recorded at 298K, 850 MHz for R
1 and R
2 measurements, and repeated 2D hNOE spectra were recorded at the same condition for hNOE measurements and error analysis. R
1 values were calculated with relaxation delays of 10, 50, 100 (×2), 200, 400, 600, 800 (×2), 1200, 1600, and 2000 ms, while R
2 values were determined with relaxation delays of 16.32, 32.64 (×2), 48.96, 65.28, 81.60, 97.92, 114.24, 130.56 (×2), 146.88, and 163.20 ms. The hNOEs were obtained in interleaved spectra with and without a 3-s
1H pre-saturation, the latter being replaced by a 3-s relaxation delay. Peak intensities were represented by peak heights for data analysis. NMRFAM-SPARKY [
20] was used to fit exponential decay curves to the experimental serial data for determining R
1 and R
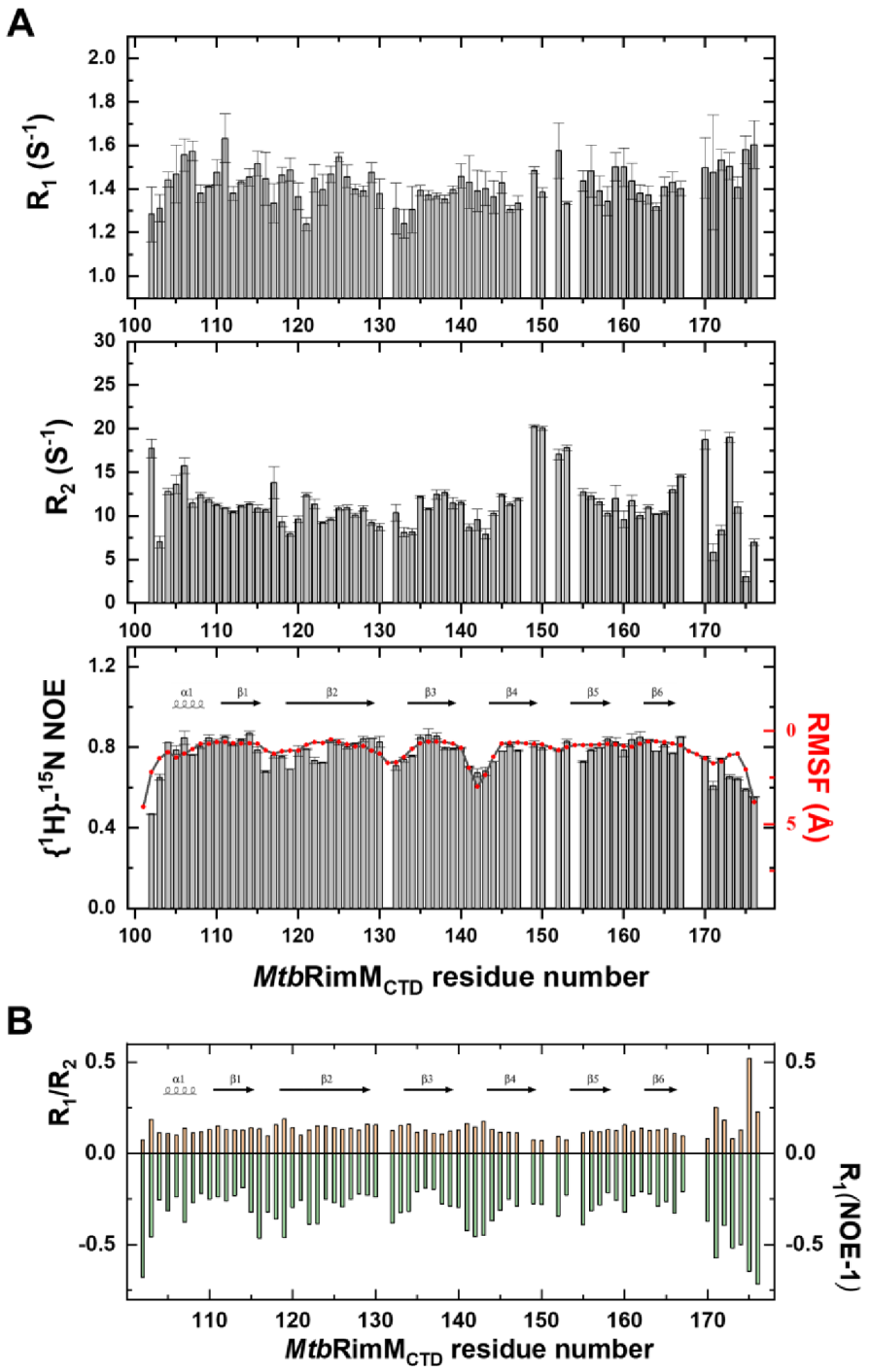
2 rates, where standard errors of rate constants were estimated by the spread in five repeated Gaussian distribution fits for random noise perturbing peak heights. Residues 131, 151, and 154 were subjugated to signal overlapping or broadening, thus unavailable for spin relaxation analysis. Therefore, a total of 71 residues were analyzed to access backbone dynamics, with prolines, the starting methionine, and the C-terminal His
6-tag excluded.
2.5. Model-Free Analysis
The FAST-Modelfree program (Version 1.3, Loria Lab, New Haven, CT, USA) [
26] was applied to extract dynamics parameters (S
2, τ
e, R
ex) based on relaxation data of
MtbRimM
CTD, which used estimated initial tensors of the protein as input. The Tensor 2 program was employed to estimate the initial tensors [
27]. Spin diffusion tensors were then iteratively optimized under an axially-symmetric model. The closest-to-average model in the
MtbRimM
CTD structure ensemble (hereafter referred to as the
MtbRimM
CTD representative structure) determined in this work was prepared as the structure input for both Tensor 2 and FAST-Modelfree automation. The values of grid-search steps and convergence limit in the FAST-Modelfree setup were 15 and 0.001 for each tensor, respectively. The S
2 cutoff was set to 0.4 for an all-encompassing characterization of residue spins.
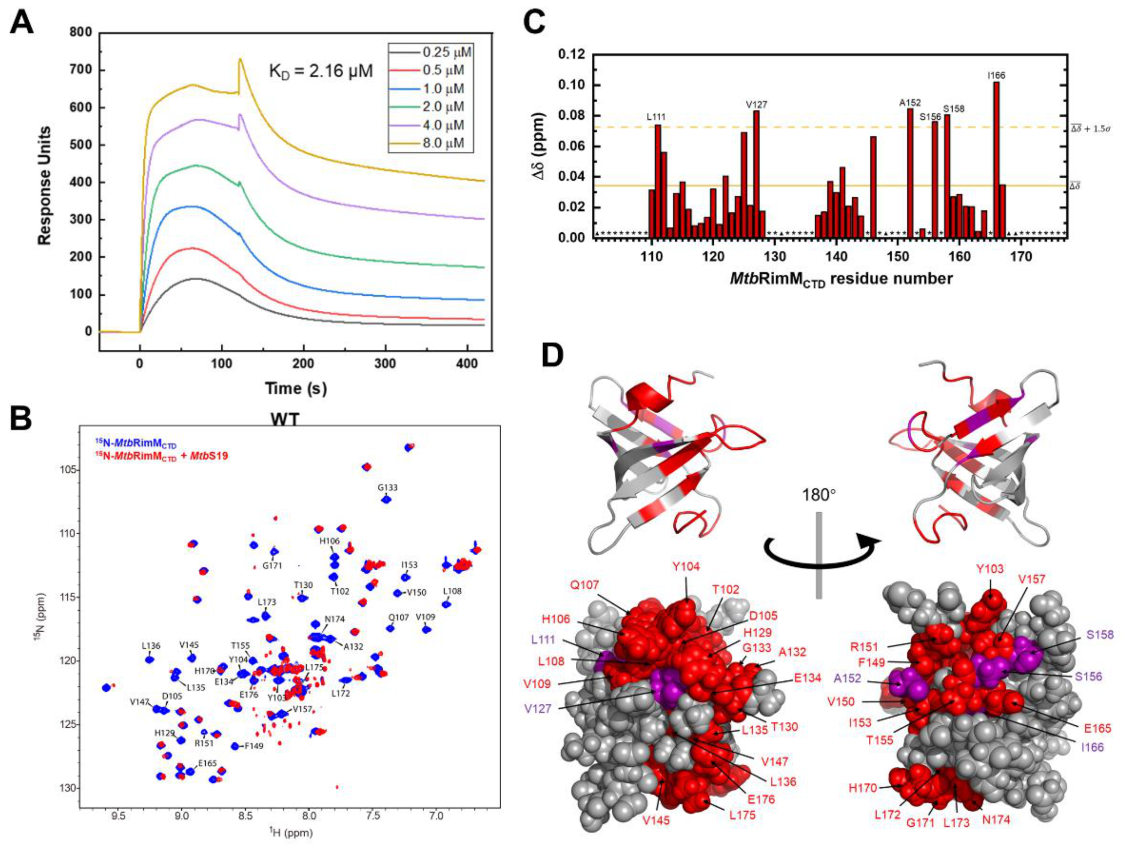
2.6. NMR Titration Assays
All recombinant proteins were dissolved in the universal buffer. Either 500 μM [
15N]-labeled
MtbRimM
NTD or 290 μM [
15N]-labeled
MtbRimM
CTD was titrated with unlabeled
MtbS19 to a respectively equimolar ratio. The titration of
MtbRimM
CTD into
MtbS19 yielded precipitation, a factor unfavorable for a titration assay with higher protein concentrations. For mutual equimolar titrations between
MtbRimM
NTD and
MtbRimM
CTD, both proteins at a concentration of 150 μM were used.
1H-
15N HSQC spectra were recorded at each titration point at 298K. The chemical shift perturbation (CSP) was determined with an empirical formula [
28] as
in which Δ
δH and Δ
δN represented chemical shift displacements for
1H and
15N nuclei observed upon titrations, respectively.
2.7. Sequence Alignments
Structure-based sequence alignments among the CTDs of RimM orthologs or among S19 orthologs were generated by Clustal X2 [
29]. Visualization of the alignments was performed with ESPript 3.0 [
30], where the secondary structure information was extracted by the STRIDE webserver using the determined PDB coordinates of
MtbRimM
CTD [
31].
2.8. MD Simulation
Molecular dynamics simulation was executed under the ff19SB force field [
32] integrated into the AmberTools20 suite [
33]. The representative structure of
MtbRimM
CTD was cleaned up with the
pdb4amber subroutine before the protein was solvated in an OPC water box [
34] extending 10.0 Å from the protein surface. Ten Na
+ ions were added to the system to neutralize the net charge of
MtbRimM
CTD. A two-stage energy minimization was performed to discard bad contacts: first, the water molecules alone; next, the entire system. After that, a three-stage system equilibration totaling 300 ps was conducted: the system was heated from 0 to 300 K under NVT ensemble for 100 ps and was run at 300 K under NVT and NPT ensembles successively for a respective 100 ps. Subsequently, a 120-ns MD simulation under NPT ensemble at 300 K was performed. Langevin dynamics was adopted for temperature regulation with a collision frequency of 1 ps
−1 and a time-based pseudo-random seed. Hydrogen-involving bonds were constrained by the SHAKE algorithm [
35] and omitted for force evaluation. The nonbonded cutoff was specified as 12 Å.
The
cpptraj subroutine [
36] incorporated within AmberTools20 was utilized for MD data extraction and analysis. Root mean square fluctuation (RMSF) per residue throughout the simulation was calculated with reference to the representative structure of
MtbRimM
CTD, to which the structures in all frames were RMS-fitted prior to calculation. Secondary structures were determined using the built-in DSSP engine [
37].
2.9. Molecular Docking
Due to possible backbone conformational changes upon
MtbRimM
CTD–S19 binding, molecular docking with backbone flexibility [
38] was carried out via RosettaDock 4.0 (Rosetta Commons) [
39]. As the two starting structures, the representative structure of
MtbRimM
CTD (termed dA) was chosen, and the structural model of
MtbS19 (termed dB) was built by homology modeling via the SWISS-MODEL server [
40], using the crystal structure of S19 in RimM-complexed form from
T. thermophilus HB8 (PDB: 3A1P) as the modeling template. The binding interface identified from chemical shift mapping described in this work was considered to engender a rough initial model (termed dC) containing both dA and dB. Totally, 100 conformational ensembles of each protein (termed dA′ and dB′, respectively) for backbone-flexible docking were generated using dA and dB under the unconstrained
relax protocol. dC was also
relaxed to spawn a clash-relieved model (termed dC′) after local refinement. Then, the
prepack protocol was run to optimize side-chain rotamers. Finally, with dA+dA′ and dB+dB′ as ensemble candidates for backbone switch and dC′ as the initial input structure, unconstrained backbone-flexible docking of
MtbRimM
CTD–S19 was performed and 30,000 docking models were calculated. The docking results were evaluated based on the RosettaDock interface energy score. In addition, the method developed by Kumar et al. can be applied to select a particular model out of a large number of docked models, which uses certain constraints for the docking and is then based on the lowest energy plot [
41]. Random perturbation subjugated to Gaussian distribution was applied to the input structure prior to every individual simulation, with standard deviations of 3 Å for translation and 8° for rotation.
2.10. SPR Affinity Assays
All recombinant proteins were dissolved in 20 mM potassium phosphate, pH 7.2, 100 mM KCl (also used as the system running buffer) for SPR affinity assays. All experiments were performed at 298 K on a Biacore T200 instrument (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). The sandwich approach was employed for the SPR assays: first, the anti-histidine antibody from the His Capture Kit (Cytiva Sweden AB, Uppsala, Sweden) was immobilized to the active surface of a CM5 sensor chip (Cytiva Sweden AB, Uppsala, Sweden) until saturation; second, excessive C-terminal His6-tagged MtbRimMCTD or its mutants flowed through and were captured by the anti-histidine antibody; last, a serial concentration of MtbS19 was injected into the system and captured by MtbRimMCTD. The control surface was treated in the same way, except that MtbS19 solution was substituted by blank running buffer. The following concentrations of MtbS19 were used for obtaining SPR assay curves: 0, 0.25, 0.50, 1.0, 2.0, 4.0, and 8.0 μM. Dissociation constants (KD) were determined by fitting the SPR assay curves to the steady-state model.
4. Discussion
TB is threatening the world healthcare with its MDR variants. Novel drug targets are urgently needed for effective prevention and treatment of MDR-TB. As is known, ribosomes are the organelle responsible for controlling protein biosynthesis. Critical protein components in bacterial ribosomes could be explored as potential drug targets [
47]. As one of the regulation factors vital for 30S ribosomal subunit assembly in vivo with S19-binding activities in vitro, RimM could be a potential target for the development of novel drugs against MDR-TB [
5,
8]. However, the 3D structure of
MtbRimM has not yet been determined. Considering that the CTD is primarily responsible for the interaction of
MtbRimM with S19, we herein clarified the structural basis of
MtbRimM
CTD binding S19 by applying several biophysical techniques. Based on the mapped S19 binding sites on
MtbRimM
CTD, we established the docking model of the
MtbRimM
CTD–S19 complex, and identified critical residues significantly contributing to the unique pattern of
MtbRimM
CTD binding S19.
4.1. MtbRimM CTD Is Structurally Independent of NTD and Primarily Responsible for Binding S19
Prior to structure determination, we firstly confirmed structural independence between the CTD and NTD of
MtbRimM. A previous study declared the structural independence of
TthRimM CTD on NTD by comparing the NMR structure ensemble and protein dynamics of a standalone NTD truncation with the NTD part of the full-length
ThtRimM [
13]. To examine the structural independence, we separately recorded
1H-
15N HSQC spectra on full-length
MtbRimM,
MtbRimM
NTD and
MtbRimM
CTD. Most peaks of both
MtbRimM
NTD and
MtbRimM
CTD overlapped well to those belonging to the full-length
MtbRimM, implying that the domain truncation almost did not affect the structure of either CTD or NTD (
Figure S8). Peaks of the N-terminal helix (residues 102–108) and inter-β-strand loops (F149, V150, L159, and D160) in
MtbRimM
CTD showed slight shifts relative to full-length
MtbRimM. Furthermore, the NMR titration of
MtbRimM
NTD into
MtbRimM
CTD and its reverse counterpart did not display observable peak changes (
Figure S9). We thereby proved the structural independence between the CTD and NTD of
MtbRimM. Additionally, the NMR titration of
MtbS19 into
MtbRimM
NTD did not show observable peak changes (
Figure S10), implying that S19 basically did not bind to
MtbRimM
NTD. Together, these results allow us to exploit the structural basis of
MtbRimM
CTD binding S19 without NTD interference.
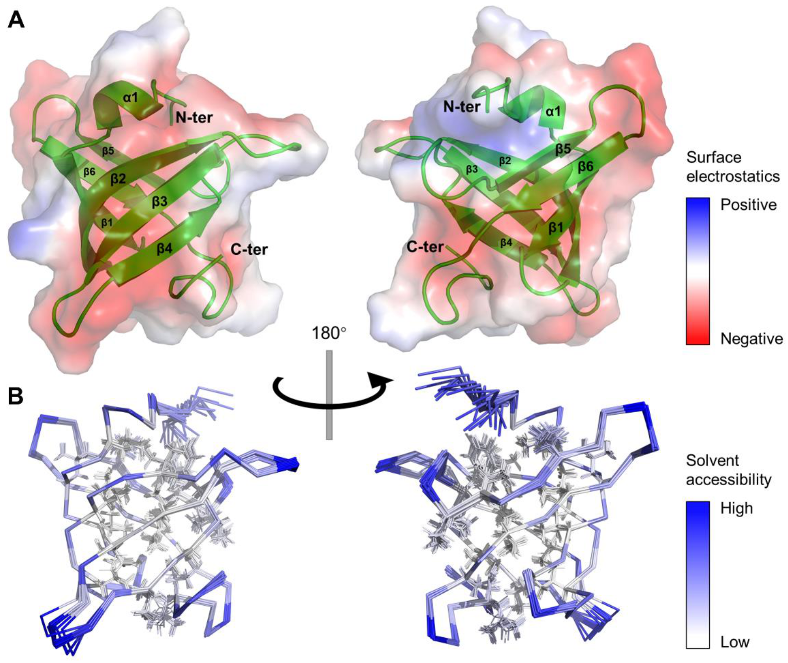
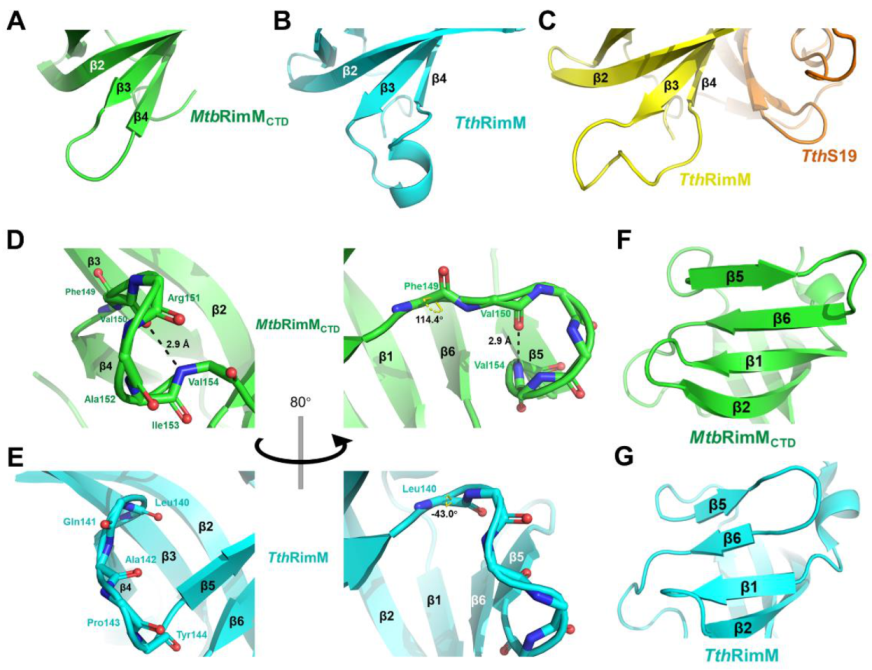
4.2. Unique Features of β3-β4 and β4-β5 Loops Characterize a Well-Folded MtbRimMCTD
The solution structure of
MtbRimM
CTD is comprised of a six-strand β-barrel and a short α-helix near the N-terminus, characterized by a hydrophobic core and a predominantly negatively charged surface. The tightly packed core consists of conservative non-polar residues mostly located on the six β-strands. The overall rigidity
of MtbRimM
CTD is reflected by the large all-residue mean S
2 value of 0.87 (the generalized order parameter), and the small RMSF of 1.08 Å in the MD simulation. Different from well-folded
MtbRimM
CTD,
TthRimM CTD is only partly folded in solution, as residues 125–143 showed few medium- or long-range NOEs [
13]. However, crystal structures of both free
TthRimM (PDB: 2DYI) and S19-complexed full-length
TthRimM (PDB: 3A1P) display a CTD of β-barrel fold resembling
MtbRimM
CTD, suggesting that the unfolded part of
TthRimM CTD in solution was not stabilized until trapped in a local energy minimum during crystallization. In fact, all five crystal structures of RimM orthologs available in PDB exhibit general structural similarity to
MtbRimM
CTD despite low sequence identity. Interestingly, the fragment in
MtbRimM
CTD corresponding to residues 125–143 in
TthRimM starts from the end of β3 and ends at the middle of β5, covering the two major conformational discrepancies between the two orthologs in the β3-β4 loop and β4-β5 loop. Moreover, the highly flexible β3-β4 loop of
TthRimM CTD showed R
1 values up to 4 s
−1 and low hNOE values around 0.5 [
13], in sharp contrast to the results obtained from the NMR relaxation measurement of
MtbRimM
CTD. Considering these results and the unique structural features of these two loops described above, the far shorter β3-β4 loop and the β4-β5 loop stabilized by a hydrogen bond (V154–V150) might facilitate the stable folding of
MtbRimM
CTD.
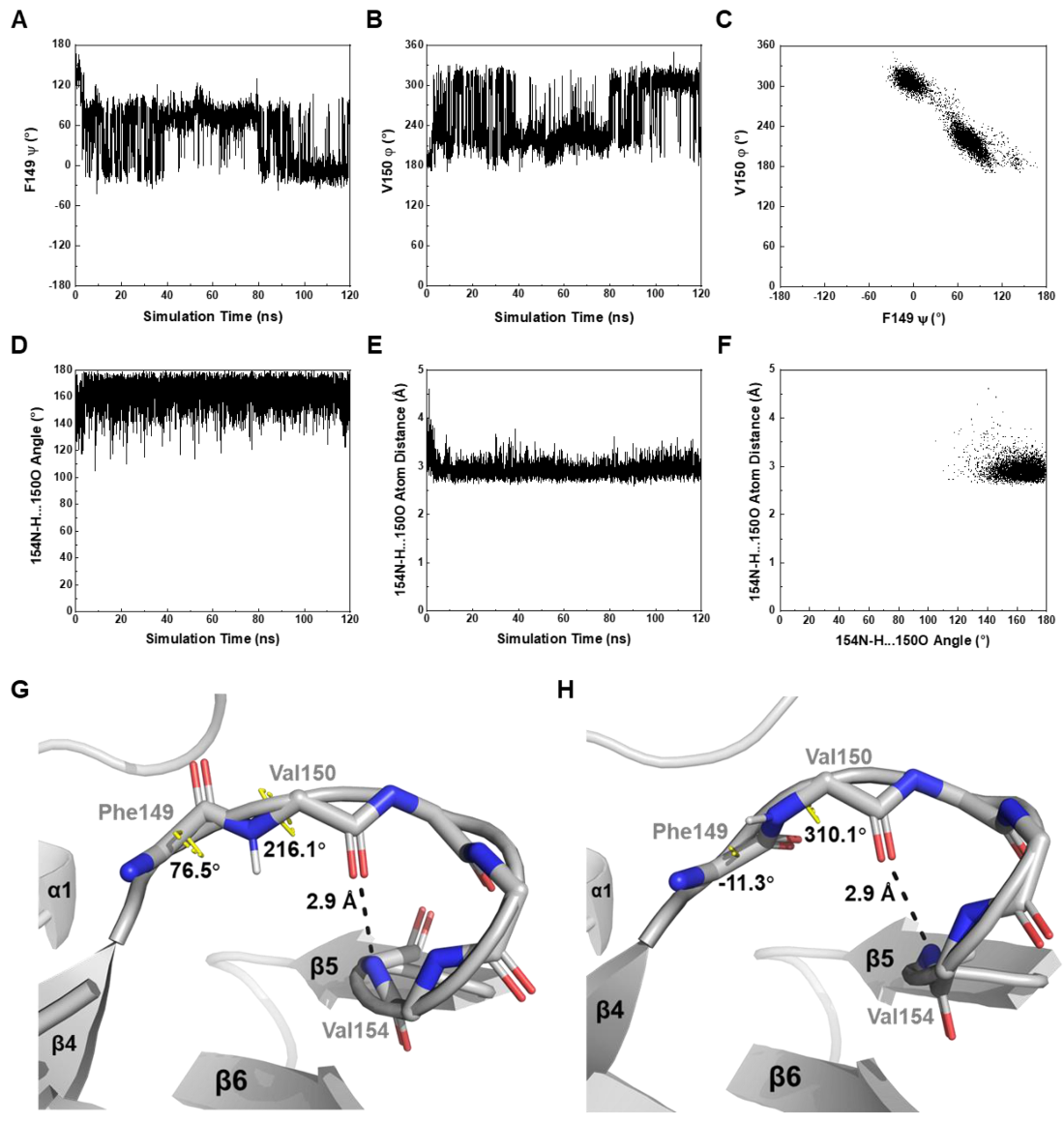
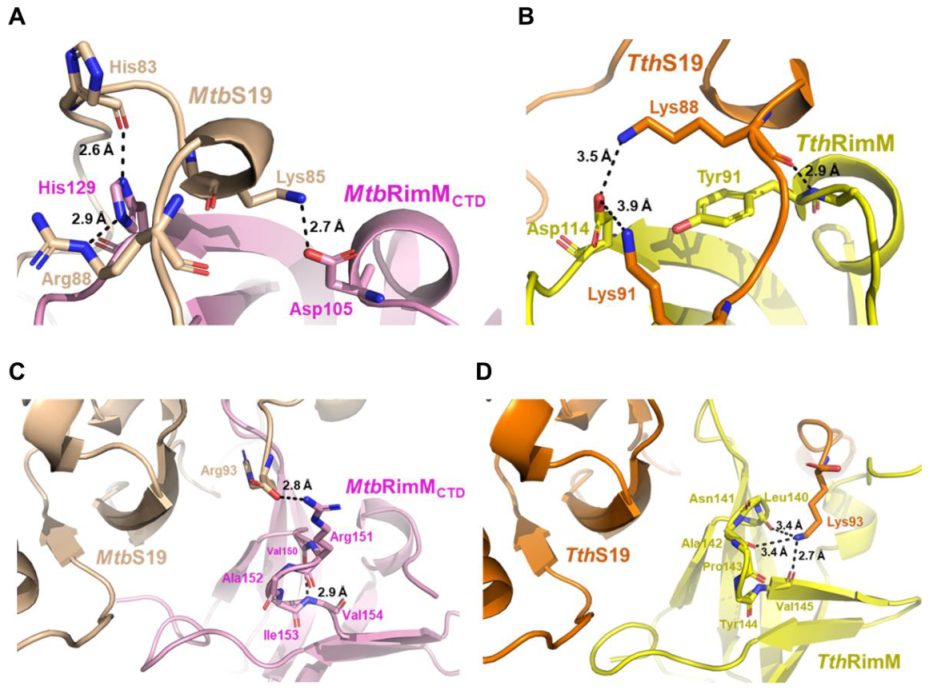
4.3. Both β4-β5 Loop and Nonconserved Key Residues Contribute to the Unique Pattern of MtbRimMCTD Binding S19
The docking model of
MtbRimM
CTD–S19 shows an interface containing surface charged residues, where the positively charged C-terminal tail of
MtbS19 binds to the negatively charged ring of
MtbRimM
CTD (
Figure 8B). The docking model displays a binding pattern different from that identified from the crystal structure of
TthRimM–S19 (PDB: 3A1P). The β4-β5 loop exhibits a smaller possibility to accommodate the C-terminus of
MtbS19 via multiple hydrogen bonds, forcing the terminal arginine to relocate to a different side of the 3A1P structure. Notably, conformational exchanges in this loop, which was intrinsically caused by the dihedral angle fluctuations of F149-V150, were revealed by NMR relaxation analysis and also confirmed by MD simulation (
Figure 5 and
Figure 6A–C). Comparatively, the experiment-based difference of the corresponding dihedral angle between
MtbRimM
CTD F149 ψ and
TthRimM L140 ψ was 157.4°, while the simulation-based fluctuation between the two dihedral angles individually related to two major orientations of
MtbRimM
CTD F149 ψ or V150 φ was nearly 90° (
Figure 6A,B). The orientation distinction, together with the stable hydrogen bond of V154-V150 (
Figure 3D), might provide a mechanistic understanding of the unique role of the helix-like β4-β5 loop in
MtbRimM
CTD binding S19, which is distinct from that in
TthRimM binding S19. Additionally, two nonconserved residues D105 and H129 in
MtbRimM
CTD could cause alteration of binding destination on the C-terminal tail of S19 (
Figure 8A,B). Although
MtbRimM
CTD H129 displays reverse charge relative to its counterparts in RimM orthologs (e.g., D114 in
TthRimM), its dual-binder function works well via the hydrogen bond rather than the salt bridge adopted by
TthRimM D114, as evidenced by the largest affinity decrease of the H129A mutant observed by SPR assays. Hence, the docking model reveals a unique pattern of
MtbRimM
CTD binding S19.
Notably, some residues in
MtbRimM
CTD that displayed peak broadening in the NMR titration experiment seemed not to directly contact
MtbS19 in the docking model of the
MtbRimM
CTD–S19 complex. One of the potential reasons is that not only
MtbRimM
CTD–S19 interaction directly cause significant line broadening in residues on the S19-binding surface of
MtbRimM
CTD, but also conformational changes induced by either S19 binding or non-specific interactions could cause observable line broadening in several residues around the S19-binding surface. Expectedly, single-site mutation experiments on a case-by-case basis can help distinguishing the non-specific interactions from the specific interactions. Furthermore, a previous work provides comprehensive methods for identifying nonspecific interactions in the protein-ligand complex, especially the ligand-induced modulation [
48].
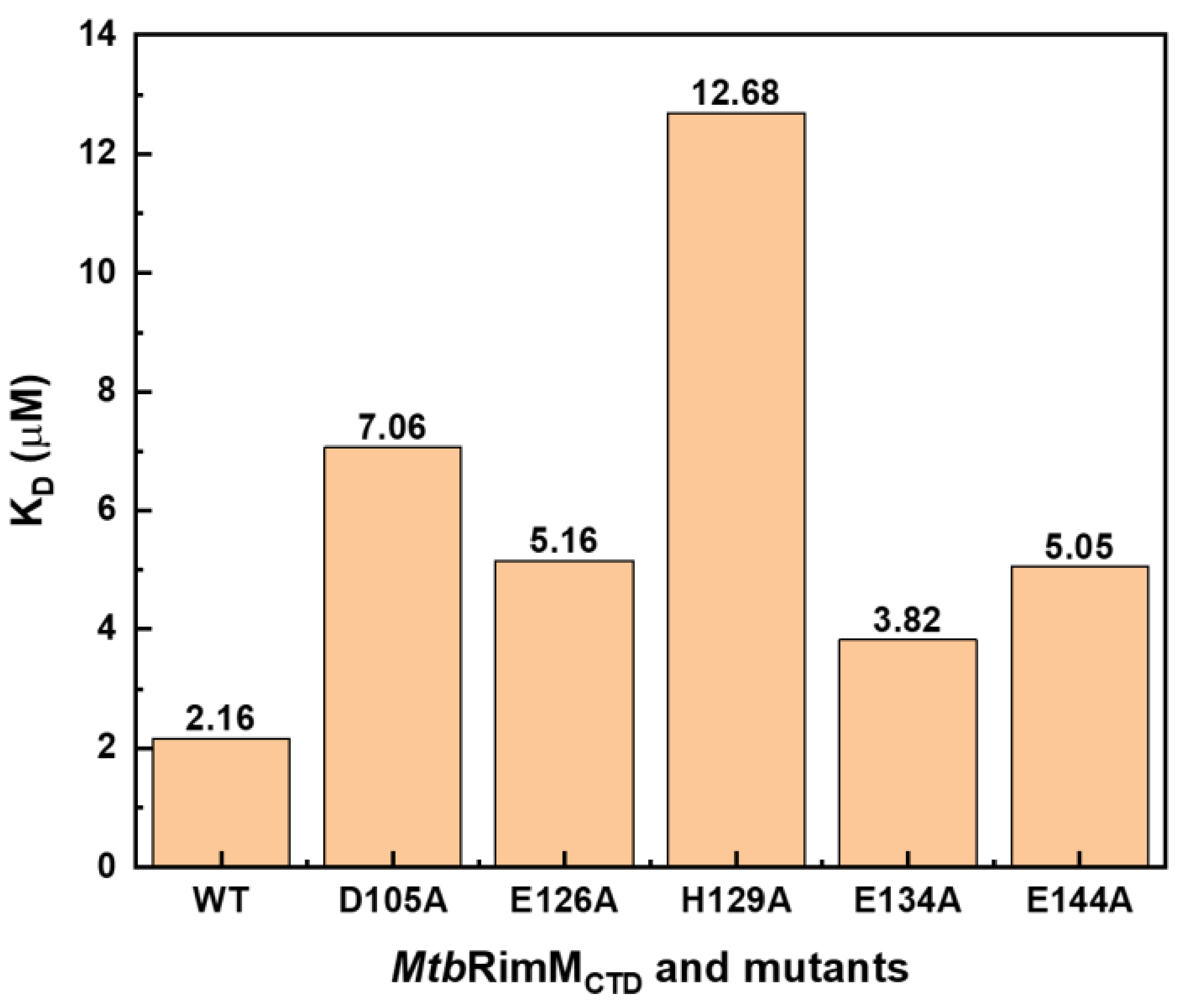
4.4. Partial Affinity Decrease of MtbRimMCTD–S19 by Single-Site Mutation Calls for Efficient Binding Inhibitors
While we have characterized the structural and interaction-related properties of
MtbRimM
CTD, some intriguing facts might hamper efforts of
MtbRimM-based drug design. As described above, the single-site mutations of five residues (D105A, E126A, H129A, E134A, and E144A), no matter they were conservative residues or not, observably reduced but did not neutralize the affinity of
MtbRimM
CTD binding S19. This result suggests that single-site mutation is insufficient for full inhibition of the
MtbRimM
CTD–S19 interaction. The logic behind this observation might lie in fault tolerance of RimM introduced during species evolution. Interestingly, even though RimM CTDs exhibit low conservation except for the residues forming the hydrophobic core, S19 orthologs among several bacterial species are highly conservative (
Figure S11). In comparison, the human 40S ribosomal protein S15 shares a lower sequence identity with
MtbS19 despite both proteins belong to the S19 protein family. Thus, relative low side effects on human hosts could be expected for potential drugs designed to specifically target
Mycobacterium tuberculosis and significantly breaking the
MtbRimM–S19 interaction. Additionally, the design of new anti-TB drugs eliminating the
MtbRimM
CTD–S19 interaction should focus on aiming key residues on the S19-binding pockets in
MtbRimM
CTD, or alternatively, on targeting the more conservative
MtbS19.
Furthermore, we predicted the preliminary druggability of the
MtbRimM
CTD–S19 complex by using the PockDrug webserver [
49] based on the docking model of
MtbRimM
CTD–S19. The prediction identified two potential pockets, showing moderate druggability probabilities for both pockets (
Figure S12,
Table S1). Notably, pocket 1 involves
MtbRimM
CTD E126 and H129 and
MtbS19 H83, and pocket 2 involves
MtbRimM
CTD R151 and
MtbS19 R93. These residues highly cover with the key residues on the interaction interface between
MtbRimM
CTD and S19, implicated in the pharmacal significance of
MtbRimM as a potential anti-TB drug target.
Expectedly, lead compounds capable of profoundly interfering with the hydrogen bonds and/or electrostatic interactions might inhibit the
MtbRimM–S19 interaction and break this complex. Moreover, the convex hull volumes of both predicted drug pockets are in the order of 100 Å
3 (
Table S1), indicative of the accommodation ability of small ligands. Taken together, these considerations imply that hydrophilic, charged, and slim molecules with capacities of blocking both drug pockets could be explored to be potential drugs targeting the
MtbRimM–S19 interaction.
As the heteronuclear 3D NMR spectra recorded on full-length MtbRimM exhibited severe peak overlap or peak broaden even disappearance, it is a difficult task to complete resonance assignments and structural determination for the full-length protein in solution. Nevertheless, we have demonstrated the structural independence between the NTD and CTD of MtbRimM. The current study on the standalone MtbRimMCTD will facilitate the future study on solution structure, dynamics and intermolecular interaction of full-length MtbRimM. Our results provide new insights into the molecular mechanisms of the RimM function regarding the assembly of S19 into the ribosome. Moreover, the structural basis of MtbRimMCTD binding S19 revealed in this study may be beneficial to the development of novel drugs against MDR-TB.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}