
An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies

 , , ,
, , ,
Abstract
:1. Introduction
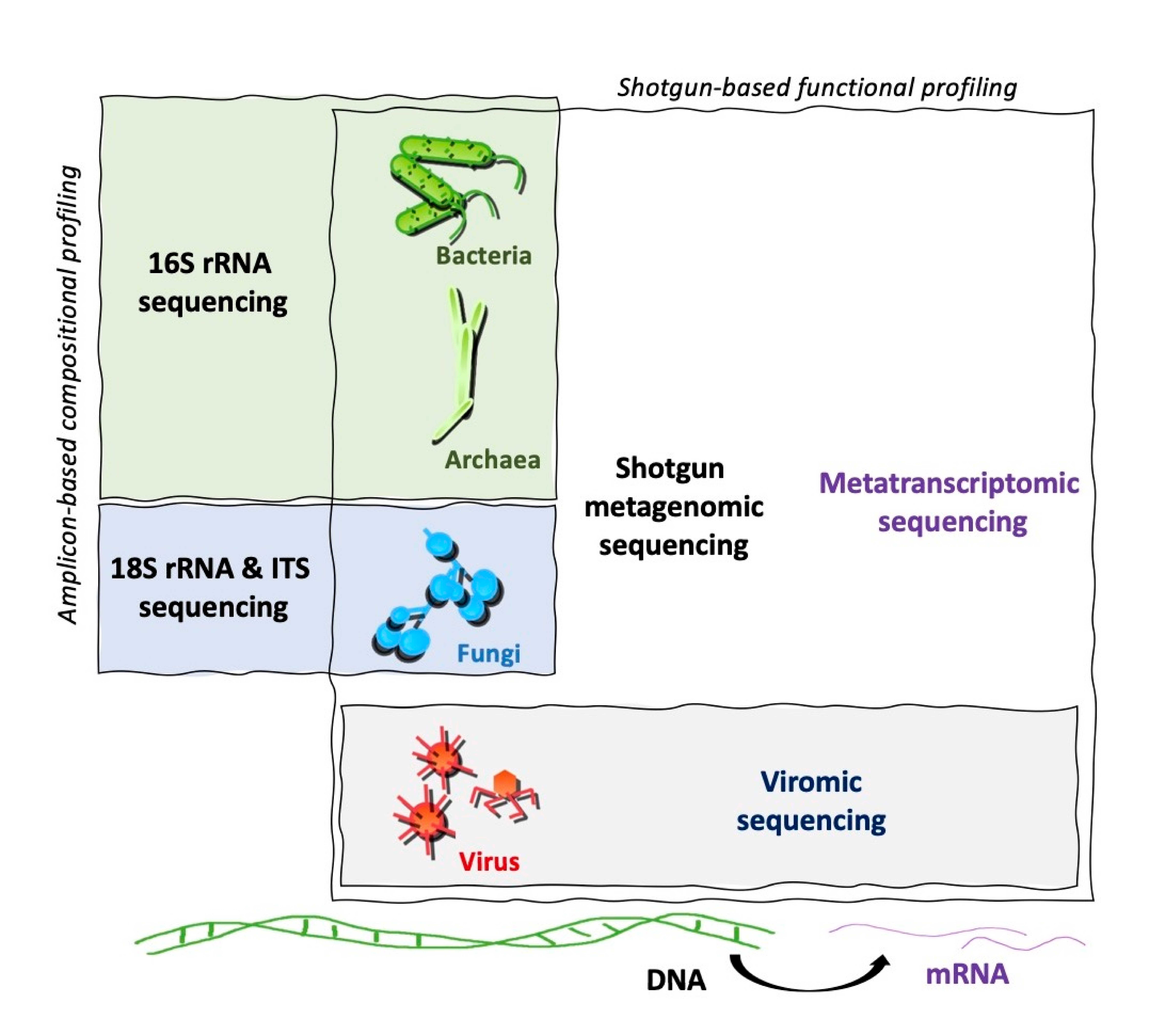
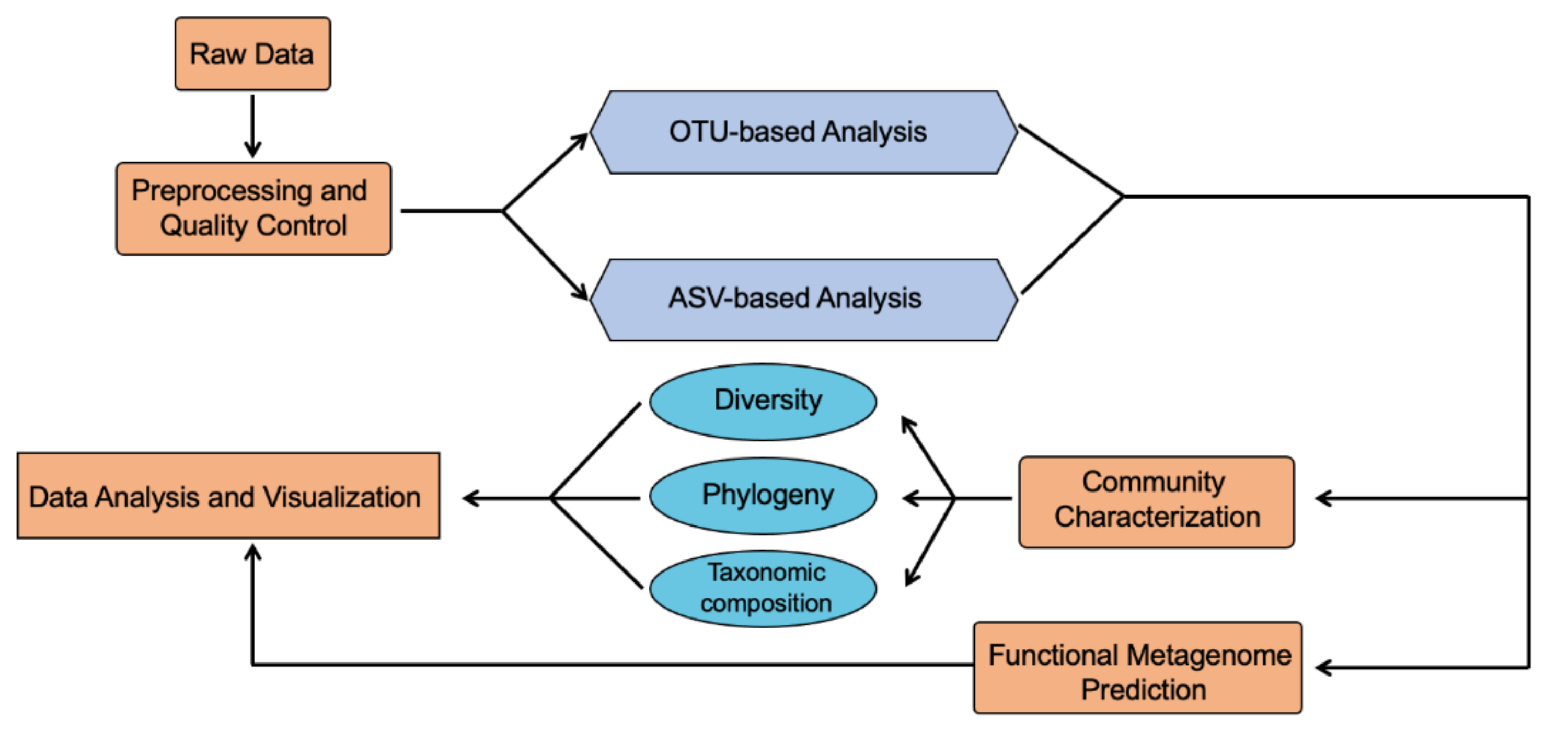
2. 16S rRNA Sequencing
- (1)
- Demultiplexing and quality filter, which assigns the multiplexed reads to each sample and filters sequences that cannot meet defined quality thresholds;
- (2)
- Chimera detection and filter, which applies ChimeraSlayer or USEARCH 6.1 to remove chimeric sequences;
- (3)
- OTU picking and taxonomy assignment, in which sequences will be clustered into OTUs based on their sequence similarity, and taxonomy will be assigned to each representative sequence of OTUs;
- (4)
- Community analysis, in which the community composition, phylogenetic tree, alpha- and beta-diversity can be computed or analyzed based on OTU tables.
3. 18S rRNA Amplicon Sequencing and Internal Transcribed Spacer (ITS) Sequencing
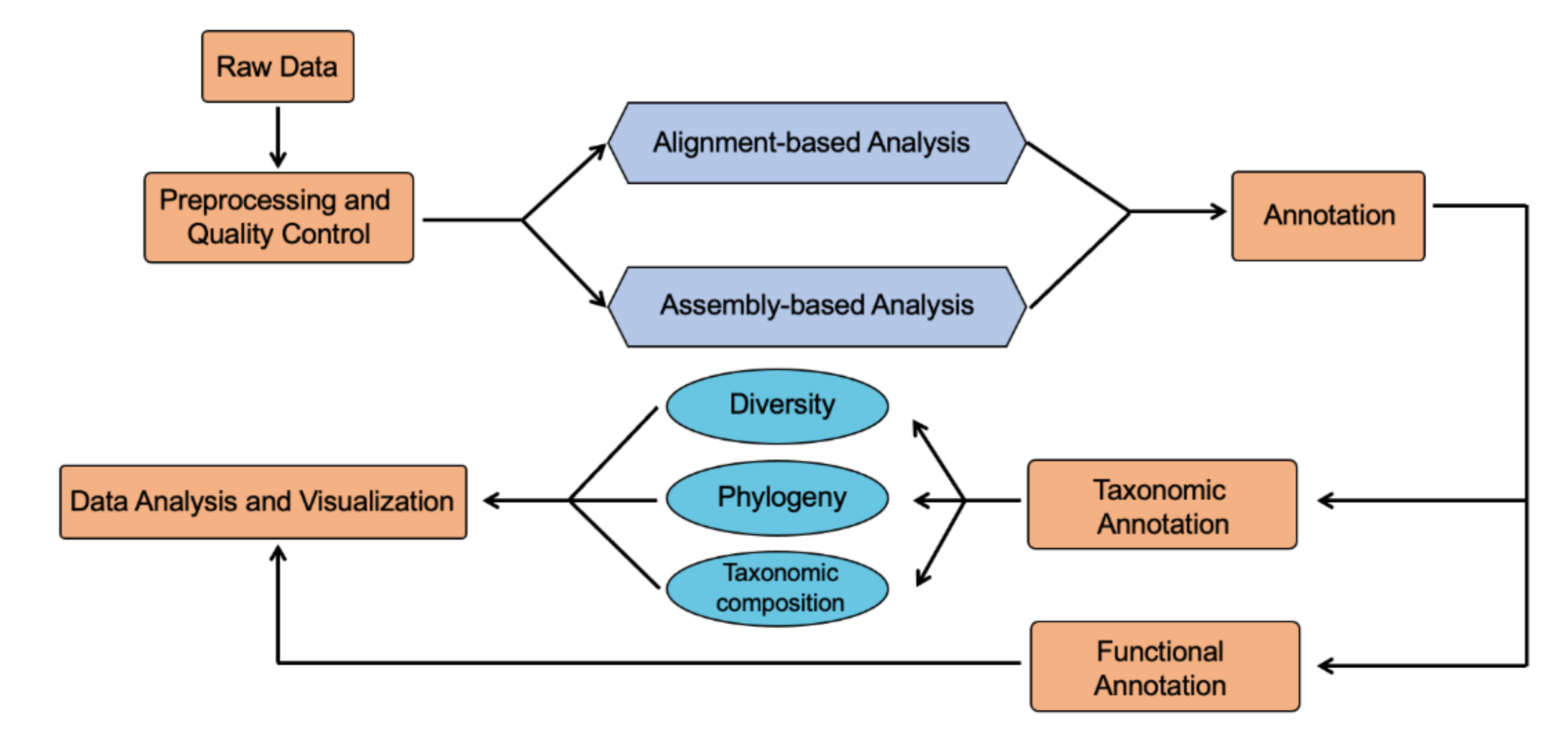
4. Shotgun Metagenomic and Metatranscriptomic Sequencing
5. Viromic Sequencing
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorkiewicz, G.; Moschen, A. Gut microbiome: A new player in gastrointestinal disease. Virchows Arch. 2018, 472, 159–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintz-Buschart, A.; Wilmes, P. Human Gut Microbiome: Function Matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Cresci, G.A.; Bawden, E. Gut Microbiome. Nutr. Clin. Prat. 2015, 30, 734–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The Long-Term Stability of the Human Gut Microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, S.; Duan, Y.; Liu, J.; Torralba, M.G.; Kuelbs, C.; Ventura-Cots, M.; Abraldes, J.G.; Bosques-Padilla, F.; Verna, E.C.; Robert, S.B., Jr.; et al. Intestinal Fungal Dysbiosis and Systemic Immune Response to Fungi in Patients With Alcoholic Hepatitis. Hepatology 2020, 71, 522–538. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Z.; Guo, R.; Chen, N.; Lu, H.; Huang, S.; Wang, J.; Li, L. Correlation between gastrointestinal fungi and varying degrees of chronic hepatitis B virus infection. Diagn. Microbiol. Infect. Dis. 2011, 70, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2014, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Allen, D.W.; Briese, T.; Couper, J.J.; Barry, S.C.; Colman, P.G.; Cotterill, A.M.; Davis, E.A.; Giles, L.C.; Harrison, L.C.; et al. Distinct Gut Virome Profile of Pregnant Women with Type 1 Diabetes in the ENDIA Study. Open Forum Infect. Dis. 2019, 6. [Google Scholar] [CrossRef]
- Malham, M.; Lilje, B.; Houen, G.; Winther, K.; Andersen, P.S.; Jakobsen, C. The microbiome reflects diagnosis and predicts disease severity in paediatric onset inflammatory bowel disease. Scand. J. Gastroenterol. 2019, 54, 969–975. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Härkönen, T.; Hämäläinen, A.-M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.; Zheng, Z.; Shao, T.; Liu, L.; Xie, Z.; Le Chatelier, E.; He, Z.; Zhong, W.; Fan, Y.; Zhang, L.; et al. Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 2017, 18, 142. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2018, 68, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Malan-Muller, S.; Valles-Colomer, M.; Raes, J.; Lowry, C.A.; Seedat, S.; Hemmings, S.M. The Gut Microbiome and Mental Health: Implications for Anxiety- and Trauma-Related Disorders. OMICS J. Integr. Biol. 2018, 22, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Genet. 2018, 16, 410–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Su, X.; Ning, K. Assessment of quality control approaches for metagenomic data analysis. Sci. Rep. 2014, 4, 6957. [Google Scholar] [CrossRef] [Green Version]
- Ewing, B.; Green, P. Base-Calling of Automated Sequencer Traces Using Phred. II. Error Probabilities. Genome Res 1998, 8, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Choi, H.-S.; Lee, B.; Chun, J.; Won, J.-H.; Yoon, S. hc-OTU: A Fast and Accurate Method for Clustering Operational Taxonomic Units Based on Homopolymer Compaction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 15, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cai, Y.; Liu, L.; Yu, F.; Farrell, M.L.; Mckendree, W.; Farmerie, W. ESPRIT: Estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009, 37, e76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Sun, Y. ESPRIT-Tree: Hierarchical clustering analysis of millions of 16S rRNA pyrosequences in quasilinear computational time. Nucleic Acids Res. 2011, 39, e95. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016, 081257. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Xu, Z.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, E.; Twin, J.; Bulach, D.M.; Garland, S.M.; Tabrizi, S.N. A Comparison of Three Bioinformatics Pipelines for the Analysis of Preterm Gut Microbiota using 16S rRNA Gene Sequencing Data. J. Proteom. Bioinform. 2015, 8. [Google Scholar] [CrossRef] [Green Version]
- López-García, A.; Pineda-Quiroga, C.; Atxaerandio, R.; Pérez, A.; Hernández, I.; García-Rodríguez, A.; González-Recio, O. Comparison of Mothur and QIIME for the Analysis of Rumen Microbiota Composition Based on 16S rRNA Amplicon Sequences. Front. Microbiol. 2018, 9, 3010. [Google Scholar] [CrossRef] [PubMed]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PIC-RUSt2: An improved and customizable approach for metagenome inference. bioRxiv 2020, 672295. [Google Scholar] [CrossRef] [Green Version]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data: Figure 1. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 2020, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 1–12. [Google Scholar] [CrossRef]
- Iwai, S.; Weinmaier, T.; Schmidt, B.L.; Albertson, D.G.; Poloso, N.J.; Dabbagh, K.; DeSantis, T.Z. Piphillin: Improved Prediction of Metagenomic Content by Direct Inference from Human Microbiomes. PLoS ONE 2016, 11, e0166104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, S.; Haque, M.M.; Mande, S.S. Vikodak—A Modular Framework for Inferring Functional Potential of Microbial Communities from 16S Metagenomic Datasets. PLoS ONE 2016, 11, e0148347. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, Z.D.; Mueller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef] [Green Version]
- Schwager, E.; Mallick, H.; Ventz, S.; Huttenhower, C. A Bayesian method for detecting pairwise associations in compositional data. PLoS Comput. Biol. 2017, 13, e1005852. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhya, I.; Hansen, R.; Meharg, C.; Thomson, J.M.; Russell, R.K.; Berry, S.H.; El-Omar, E.M.; Hold, G.L. The fungal microbiota of de-novo paediatric inflammatory bowel disease. Microbes Infect. 2015, 17, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Sovran, B.; Planchais, J.; Jegou, S.; Straube, M.; Lamas, B.; Natividad, J.M.; Agus, A.; Dupraz, L.; Glodt, J.; Da Costa, G.; et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 2018, 6, 152. [Google Scholar] [CrossRef] [PubMed]
- Chehoud, C.; Albenberg, L.G.; Judge, C.; Hoffmann, C.; Grunberg, S.; Bittinger, K.; Baldassano, R.N.; Lewis, J.D.; Bushman, F.D.; Wu, G.D. Fungal Signature in the Gut Microbiota of Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1948–1956. [Google Scholar] [CrossRef]
- Luan, C.; Xie, L.; Yang, X.; Miao, H.; Lv, N.; Zhang, R.; Xiao, X.; Hu, Y.; Liu, Y.; Wu, N.; et al. Dysbiosis of Fungal Microbiota in the Intestinal Mucosa of Patients with Colorectal Adenomas. Sci. Rep. 2015, 5, 7980. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabrò, A.; et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 2017, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Genet. 2019, 17, 95–109. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Leung, S.-Y.; To, K.K.W.; Chan, J.F.W.; Ngan, A.H.Y.; Cheng, V.C.C.; Lau, S.K.P.; Yuen, K.-Y. Internal Transcribed Spacer Region Sequence Heterogeneity inRhizopus microsporus: Implications for Molecular Diagnosis in Clinical Microbiology Laboratories. J. Clin. Microbiol. 2009, 48, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Ritland, C.E.; Ritland, K.; Straus, N.A. Variation in the ribosomal internal transcribed spacers (ITS1 and ITS2) among eight taxa of the Mimulus guttatus species complex. Mol. Biol. Evol. 1993, 10, 1273–1288. [Google Scholar] [CrossRef] [Green Version]
- Popovic, A.; Parkinson, J. Characterization of Eukaryotic Microbiome Using 18S Amplicon Sequencing. Adv. Struct. Saf. Stud. 2018, 1849, 29–48. [Google Scholar] [CrossRef]
- Tanabe, A.S.; Nagai, S.; Hida, K.; Yasuike, M.; Fujiwara, A.; Nakamura, Y.; Takano, Y.; Katakura, S. Comparative study of the validity of three regions of the 18S-rRNA gene for massively parallel sequencing-based monitoring of the planktonic eukaryote community. Mol. Ecol. Resour. 2015, 16, 402–414. [Google Scholar] [CrossRef]
- Xie, Q.; Lin, J.; Qin, Y.; Zhou, J.; Bu, W. Structural diversity of eukaryotic 18S rRNA and its impact on alignment and phylogenetic reconstruction. Protein Cell 2011, 2, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, A.; Todt, C.; Mikkelsen, N.T.; Lieb, B. Fast evolving 18S rRNA sequences from Solenogastres (Mollusca) resist standard PCR amplification and give new insights into mollusk substitution rate heterogeneity. BMC Evol. Biol. 2010, 10, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeger, F.; Wurzbacher, C.; Bourne, E.C.; Mazzoni, C.J.; Monaghan, M.T. Combining the 5.8S and ITS2 to Improve Classification of Fungi. Methods Ecol. Evol. 2019, 10, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, E. Structural RNA Homology Search and Alignment Using Covariance Models. Ph.D. Thesis, Washington University in Saint Louis, School of Medicine, St. Louis, MO, USA, 2009. [Google Scholar]
- Hildebrand, F.; Tadeo, R.; Voigt, A.Y.; Bork, P.; Raes, J. LotuS: An efficient and user-friendly OTU processing pipeline. Microbiome 2014, 2, 30. [Google Scholar] [CrossRef] [Green Version]
- Albanese, D.; Fontana, P.; De Filippo, C.; Cavalieri, D.; Donati, C. MICCA: A complete and accurate software for taxonomic profiling of metagenomic data. Sci. Rep. 2015, 5, 9743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafeiropoulos, H.; Viet, H.Q.; Vasileiadou, K.; Potirakis, A.; Arvanitidis, C.; Topalis, P.; Pavloudi, C.; Pafilis, E. PEMA: A flexible Pipeline for Environmental DNA Metabarcoding Analysis of the 16S/18S ribosomal RNA, ITS, and COI marker genes. GigaScience 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Ferro, M.; Antonio, E.A.; Souza, W.; Bacci, M. ITScan: A web-based analysis tool for Internal Transcribed Spacer (ITS) sequences. BMC Res. Notes 2014, 7, 857. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.J.G.M.; Sánchez-García, M.; Ebersberger, I.; De Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Rivers, A.R.; Weber, K.C.; Gardner, T.G.; Liu, S.; Armstrong, S.D. ITSxpress: Software to rapidly trim internally transcribed spacer sequences with quality scores for marker gene analysis. F1000Research 2018, 7, 1418. [Google Scholar] [CrossRef]
- Delgado-Serrano, L.; Restrepo, S.; Bustos, J.R.; Zambrano, M.M.; Anzola, J.M. Mycofier: A new machine learning-based classifier for fungal ITS sequences. BMC Res. Notes 2016, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, M.; Fosso, B.; Licciulli, F.; Balech, B.; Larini, I.; Grillo, G.; De Caro, G.; Liuni, S.; Pesole, G. ITSoneDB: A comprehensive collection of eukaryotic ribosomal RNA Internal Transcribed Spacer 1 (ITS1) sequences. Nucleic Acids Res. 2017, 46, D127–D132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Campo, J.; Kolisko, M.; Boscaro, V.; Santoferrara, L.F.; Nenarokov, S.; Massana, R.; Guillou, L.; Simpson, A.; Berney, C.; De Vargas, C.; et al. EukRef: Phylogenetic curation of ribosomal RNA to enhance understanding of eukaryotic diversity and distribution. PLoS Biol. 2018, 16, e2005849. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Vila, A.V.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Emami, A.; Zhou, R.; Lang, S.; Duan, Y.; Wang, Y.; Jiang, L.; Loomba, R.; Brenner, D.A.; Stärkel, P.; et al. Functional Microbial Responses to Alcohol Abstinence in Patients With Alcohol Use Disorder. Front. Physiol. 2020, 11, 370. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Duan, Y.; Lang, S.; Barupal, D.; Wu, T.; Valdiviez, L.; Roberts, B.; Choy, Y.Y.; Shen, T.; Byram, G.; et al. Functional Microbiomics Reveals Alterations of the Gut Microbiome and Host Co-Metabolism in Patients With Alcoholic Hepatitis. Hepatol. Commun. 2020, 4, 1168–1182. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 157, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; Fernández-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Shen, T.-C.D.; Chen, E.Z.; Bittinger, K.; Bailey, A.; Roggiani, M.; Sirota-Madi, A.; Friedman, E.S.; Chau, L.; Lin, A.; et al. A role for bacterial urease in gut dysbiosis and Crohn’s disease. Sci. Transl. Med. 2017, 9, eaah6888. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.D.; Chen, E.Z.; Baldassano, R.N.; Otley, A.R.; Griffiths, A.M.; Lee, D.; Bittinger, K.; Bailey, A.; Friedman, E.S.; Hoffmann, C.; et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 2015, 18, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 1–13. [Google Scholar] [CrossRef]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 132, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Feng, Y.; Wu, J.; Liu, F.; Zhang, Z.; Hao, Y.; Liang, S.; Li, B.; Li, J.; Lv, N.; et al. The Gut Microbiome Signatures Discriminate Healthy From Pulmonary Tuberculosis Patients. Front. Cell. Infect. Microbiol. 2019, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Sun, K. Ktrim: An extra-fast and accurate adapter- and quality-trimmer for sequencing data. Bioinformatics 2020, 36, 3561–3562. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Jünemann, S.; Kleinbölting, N.; Jaenicke, S.; Henke, C.; Hassa, J.; Nelkner, J.; Stolze, Y.; Albaum, S.P.; Schlüter, A.; Goesmann, A.; et al. Bioinformatics for NGS-based metagenomics and the application to biogas research. J. Biotechnol. 2017, 261, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. LBNL Report LBNL-7065E, Lawrence Berkeley National Laboratory. 2014. Available online: https://escholarship.org/uc/item/1h3515gn (accessed on 28 March 2021).
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Alvarez, R.V.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H. The UniProt Consortium UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. Meta-IDBA: A de Novo assembler for metagenomic data. Bioinformatics 2011, 27, i94–i101. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namiki, T.; Hachiya, T.; Tanaka, H.; Sakakibara, Y. MetaVelvet: An extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012, 40, e155. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Van Der Walt, A.J.; Van Goethem, M.W.; Ramond, J.-B.; Makhalanyane, T.P.; Reva, O.; Cowan, D.A. Assembling metagenomes, one community at a time. BMC Genom. 2017, 18, 1–13. [Google Scholar] [CrossRef]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Thomas, A.M.; Manghi, P.; Valles-Colomer, M.; et al. Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with Bi-oBakery 3. bioRxiv 2020. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Glass, E.M.; Wilkening, J.; Wilke, A.; Antonopoulos, D.; Meyer, F. Using the Metagenomics RAST Server (MG-RAST) for Analyzing Shotgun Metagenomes. Cold Spring Harb. Protoc. 2010, 2010, 5368. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.-M.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017, 45, D507–D516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goll, J.; Rusch, D.B.; Tanenbaum, D.M.; Thiagarajan, M.; Li, K.; Methé, B.A.; Yooseph, S. METAREP: JCVI metagenomics reports—An open source tool for high-performance comparative metagenomics. Bioinformatics 2010, 26, 2631–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Börnigen, D.; Morgan, X.C.; Franzosa, E.A.; Ren, B.; Xavier, R.J.; Garrett, W.S.; Huttenhower, C. Functional profiling of the gut microbiome in disease-associated inflammation. Genome Med. 2013, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-L.; Xu, S.-Y.; Ren, Z.-G.; Tao, L.; Jiang, J.-W.; Zheng, S.-S. Application of metagenomics in the human gut microbiome. World J. Gastroenterol. 2015, 21, 803–814. [Google Scholar] [CrossRef]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of Metatranscriptomics in Microbiome Research. Bioinform. Biol. Insights 2016, 10, BBI–S34610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakya, M.; Lo, C.-C.; Chain, P.S.G. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.Z.; Lanzen, A.; Bang-Andreasen, T.; Jacobsen, C.S. To assemble or not to resemble—A validated Comparative Metatranscriptomics Workflow (CoMW). GigaScience 2019, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.A.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungaro, F.; Massimino, L.; Furfaro, F.; Rimoldi, V.; Peyrin-Biroulet, L.; D’Alessio, S.; Danese, S. Metagenomic analysis of intestinal mucosa revealed a specific eukaryotic gut virome signature in early-diagnosed inflammatory bowel disease. Gut Microbes 2019, 10, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal Virome in Patients With Alcoholic Hepatitis. Hepatology 2020, 72, 2182–2196. [Google Scholar] [CrossRef]
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal Virome Signature Associated With Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159, 1839–1852. [Google Scholar] [CrossRef]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.-H.; Sugimura, N.; Lam, T.Y.-T.; et al. Alterations in Enteric Virome Are Associated with Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. mBio 2018, 9, e02248-18. [Google Scholar] [CrossRef] [Green Version]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Garmaeva, S.; Sinha, T.; Kurilshikov, A.; Fu, J.; Wijmenga, C.; Zhernakova, A. Studying the gut virome in the metagenomic era: Challenges and perspectives. BMC Biol. 2019, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bikel, S.; Valdez-Lara, A.; Cornejo-Granados, F.; Rico, K.; Canizales-Quinteros, S.; Soberón, X.; Del Pozo-Yauner, L.; Ochoa-Leyva, A. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: Towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 2015, 13, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Zárate, S.; Taboada, B.; Yocupicio-Monroy, M.; Arias, C.F. Human Virome. Arch. Med. Res. 2017, 48, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef]
- Yang, X.; Charlebois, P.; Gnerre, S.; Coole, M.G.; Lennon, N.J.; Levin, J.Z.; Qu, J.; Ryan, E.M.; Zody, M.C.; Henn, M.R. De novo assembly of highly diverse viral populations. BMC Genom. 2012, 13, 475. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Faubladier, M.; Mahul, A.; Paulhe, N.; Bernard, A.; Debroas, D.; Enault, F. Metavir: A web server dedicated to virome analysis. Bioinformatics 2011, 27, 3074–3075. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzi, H.A.; Hoover, J.; Inman, J.; Safford, T.; Murphy, S.; Kagan, L.; Williamson, S.J. TheViral MetaGenome Annotation Pipeline (VMGAP):an automated tool for the functional annotation of viral Metagenomic shotgun sequencing data. Stand. Genom. Sci. 2011, 4, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstens, A.; Roos, A.; Andreasson, A.; Magnuson, A.; Agréus, L.; Halfvarson, J.; Engstrand, L. Differential clustering of fecal and mucosa-associated microbiota in ‘healthy’ individuals. J. Dig. Dis. 2018, 19, 745–752. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Software | Short Description | Ref. |
|---|---|---|
| 16S rRNA, 18S rRNA and ITS sequencing data analysis | ||
| UCLUST/ UPARSE | UCLUST is an OTU-based clustering method. It employs USEARCH, and UPARSE is a subroutine of USEARCH which constructs OTUs de novo from next-generation reads. The general pipeline procedure of UPARSE is reads filtering, trimming, and then clustering and chimera filtering simultaneously. Pros: Able to perform de novo, closed-reference, and open-reference clustering. Cons: May filter out too many reads and result in inaccuracy of estimating the least abundant species. | [19,20] |
| CD-HIT | CD-HIT is one of the most used OTU-based clustering tool to decrease redundancy of sequence and improve the performance of other analysis. Pros: Uses novel parallelization strategy to achieve fast runtime; can handle extremely large databases. Cons: Diminished clustering accuracy. | [21] |
| Hc-OTU | Hc-OTU is an OTU-based clustering method for 16S rRNA sequence, employs homopolymer compaction and k-mer profiling. Pros: High accuracy. 7,000 times faster than MOTHUR and about six times faster than ESPRIT-TREE, while remaining the same accuracy level as MOTHUR. Supports user-specified k-mer distance threshold parameter value. Cons: Its worst-case computational complexity run time is O(n2), while UCLUST and CD-HIT are faster than hc-OTU with run time of O(n1.2). | [22] |
| ESPRIT | ESPRIT is an OTU-based hierarchical clustering method consisting of quality filtering, computing pairwise distance, hierarchical clustering and estimate with statistical interference. There are two version of ESPRIT, one for personal computer (small/medium size data) and one for computer clusters (large size data). Pros: Able to perform analysis on various size of data. Cons: Slow time O(n2) and space complexity. | [23] |
| ESPRIT-Tree | ESPRIT-Tree is an OTU-based online-learning-based hierarchical clustering method. ESPRIT-TREE improves on previous ESPRIT algorithm and uses a pseudometric-based partition tree. Pros: Improved runtime from ESPRIT: O(n1.17); relatively high accuracy. Cons: In terms of computational efficiency, UCLUST performs better than ESPRIT-Tree. | [24] |
| DADA2 | DADA2 is an ASV-based analysis pipeline for modeling and error-correcting Illumina sequence reads. Pros: High accuracy: able to resolve single nucleotide biological differences. Can perform species-level analysis. Runtime scales linearly as sample number increase, and reasonable memory requirements. Cons: Comparably slow denoising algorithm than UPARSE. | [26] |
| UNOISE2 | UNOISE 2 is an ASV-based tool for denoising (error-correcting) Illumina sequence reads. It is improved from UNOISE and clusters unique reads in the sequence. Pros: Higher accuracy and speed than DADA2. Cons: Does not use quality scores. | [27] |
| Deblur | Deblur is an ASV-based denoising tool, which uses error profiles to obtain putative error-free sequences. It operates independently on each sample. Pros: Able to obtain single-nucleotide resolution, faster than DADA2, better memory efficiency than DADA2 and UNOISE 2. Better sensitivity and specificity. Cons: Slower than UNOISE 2, limited by read length and sample sequences’ diversity. | [28] |
| QIIME/ QIIME2 | QIIME and QIIME2 are bioinformatics platforms for microbial community analysis and visualizations. QIIME 2 is engineered based on QIIME and replaced QIIME. QIIME2 use existing bioinformatics tools as subroutines, such as DADA2, deblur, etc. Pros: Have multiple interfaces, continues to grow and adapt to novel strategies. Cons: A large number of dependent programs need to be installed. | [29,30] |
| Mothur | Mothur is a software analyzing raw sequences and generating visualization tools to describe α and β diversity. It is a combination of multiple analytic tools for describing and comparing microbial communities. It provides examples for data acquired from different sequencing platforms. Pros: Able to perform both ASV-based and OTU-based analysis. Cons: Relatively slow runtime and space complexity. | [31] |
| PICRUSt/ PICRUSt2 | PICRUSt is a software for predicting functional composition based solely on marker gene sequence profiles. PICRUSt2 is the improved version of PICRUSt by having a larger reference database, enhanced prediction ability and more accurate de novo amplicon tree-building. PICRUSt2: Pros: Able to identify novel discoveries. Can process 18S and ITS rRNA sequence while the original version only supports 16s rRNA sequence analysis. Cons: Can only differentiate taxa the same level as the amplified marker gene sequence. Can be problematic if the interested microbial community’s majority phyla are not yet well-characterized. | [35,36] |
| Tax4Fun/ Tax4Fun2 | Tax4Fun is an R package for predicting functional profiles for 16S rRNA data on the basis of SILVA-labeled OUT abundances. Tax4Fun 2 is an improved version of Tax4Fun with more accurate and enhanced prediction power. Tax4Fun 2: Pros: Easy-to-use, platform-independent and highly memory-efficient. Tax4Fun2 has higher accuracies than PICRUSt and Tax4Fun. Cons: Availability of suitable reference genomes may limit Tax4Fun 2’s performance. Only supports prediction from 16S rRNA gene. | [37,39] |
| Piphillin | Piphillin is a web application that produces metagenome predictions based on the nearest-neighbor mappings of 16S rRNA sequences to genome. Pros: No local computational power requirements. High correlation with corresponding metagenomic data. Higher accuracy than PICRUSt2 Cons: Have high requirements on reference database. Only supports 16S rRNA gene prediction. | [40] |
| Vikodak/ iVikodak | Vikodak is a web service that provides functional prediction on 16S rRNA data. It contains 3 modules: Global Mapper, Inter Sample Feature Analyzer, and Local Mapper. With these 3 modules, it is able to perform functional prediction both globally and in detail and perform pair-wise comparative statistical analysis. iVikodak is an improved version of Vikodak. Pros: No local computational power requirements. No coding skill required. Allows for single pathway probing and gene quorum assumption. Cons: Only supports prediction from 16S rRNA gene. | [41] |
| SSU-ALIGN | SSU-ALIGN is designed primarily to align 16S and 18S small subunit ribosomal RNA, but can also be used for large subunit ribosomal RNA alignment. Pros: High sensitivity and specificity. Cons: Not capable of inferring phylogenetic trees. Computationally expensive. | [65] |
| LotuS2 | LotuS2 is a software pipeline for 16S/18S/ITS rRNA analysis. It is able to calculate denoised, chimera-checked OTUs and construct OTU phylogenetic tree. Pros: Fast and user friendly. Able to handle a wide variety of data sizes on a personal computer. Cons: Mapping speed limited by BLAST+. | [66] |
| MICCA | MICCS is a command-line software for the processing of 16S rRNA gene and ITS amplicon sequencing data, from raw sequences to OTU tables, taxonomic classification and phylogenetic tree inference. Pros: Can be used effectively on sample with a large portion of uncharacterized species. Low requirements for reference database. Memory efficient. Cons: Less estimated OTUs obtained as a comprise for high consistency. | [67] |
| PEMA | PEMA is a software pipeline for metabarcoding analysis based on third-party tools. Its function includes read pre-processing, OTU clustering, ASV inference, taxonomy assignment, and COI marker gene analysis. Pros: Allows partial re-execution. Fast execution time. Cons: Heavyweight computation. | [68] |
| ITScan | ITScan is an online pipeline for fungal diversity analysis and identification based on ITS sequences. Pros: Does not require coding skills. User friendly. Cons: Requires FASTA-formatted input file. | [69] |
| ITSx | ITSx is a software for detection and extraction of the ITS1 and ITS2 subregions from ITS sequences for fungi and other eukaryotes. It relies on HMMER for profile hidden Markov model analysis. Pros: Has a very high proportion of true-positive extractions and a low proportion of false-positive extractions. Cons: Requires FASTA-formatted input file. | [70] |
| ITSxpress | ITSxpress is a software for ITS1, ITS2 or the entire ITS region trimming. It implements HMMER and BBMerge. It is designed to support the calling of exact sequence variants rather than OTUs. Pros: Fast runtime. Processes FASTQ-formatted input file. | [71] |
| Mycofier | Mycofier is a machine-learning-based fungal ITS1 sequence classifier at the genus level. The final model was based on ITS1 sequences from 510 fungal genera using a Naïve Bayes algorithm. Pros: Doesn’t require pairwise sequence alignment. Cons: Only analyze fungal ITS1 sequences. BLAST approach provides higher classification accuracy. | [72] |
| Shotgun metagenomic and metatranscriptomic sequencing data analysis | ||
| Trimmomatic | Trimmomatic is a sequence trimmer for Illumina sequence data. It has multiple processing steps including detection and removal of adapter and other illumine-specific sequences, and quality filtering. Pros: Processes both paired end and single end data. Cons: Slower than Ktrim. | [89] |
| Ktrim | Ktrim provides both adapter- and quality-trimming of the sequencing data. Pros: Faster than Trimmomatic. Cons: Higher over-trimming rates than Trimmomatic. | [90] |
| Cutadapt | Cutadapt is a sequence trimmer which removes adapter sequences, primers and other types of unwanted sequence from high-throughput sequencing reads. Pros: Supports 454, Illumina and SOLiD (color space) data. Cons: Slow runtime. | [91] |
| MultiQC | MultiQC creates a summary report visualizing output from different tools across multiple samples, facilitating the identification of global trends and biases. Pros: Provides a global view instead of per-sample analysis. | [92] |
| Bowtie2 | Bowtie2 is a software for sequence alignment to reference genome. It supports gapped, local, and paired-end alignments. The software implements full-text minute index and SIMD dynamic programming. Pros: Memory efficient. High speed, sensitivity and accuracy. Cons: Alignment with short reads remains an active challenge (<50 bp). | [95] |
| DIAMOND | DIAMOND is a sequence aligner for protein and translated DNA searches. It aims to determine all significant alignments for a given input. DIAMOND uses double indexing and spaced seeds. Pros: Significantly higher speed with similar sensitivity to BLASTX. Cons: Heavy memory consuming. | [96] |
| BBMap | BBMap is a sequence aligner that can align DNA and RNA sequencing reads from multiple platforms, including Illumina, 454, Sanger, Ion Torrent, Pac Bio, and Nanopore. BBMap needs to index a reference before mapping to it. Pros: Fast and accurate, particularly for reads with long indels or highly mutated genomes. Has no upper limit to number of contigs or genome size. Cons: The indexing phase requires FASTA format only. | [97] |
| Meta-IDBA | Meta-IDBA is a de novo metagenomic assembler. It first constructs de Bruijn graph and then divides graph into connected components. Pros: Provides a multiple alignment of similar contigs from different subspecies in the same species. Cons: Unable to reconstruct the contigs of each single subspecies. | [105] |
| IDBA-UD | IDBA-UD is a de novo single-cell and metagenomic assembler, which can assemble sequences with highly uneven depth. It is based on de Bruijn graph approach. Pros: Implements local assembly. Cons: Sequence of species with high abundance is more likely to be misidentified as repeats. | [106] |
| MetaVelvet | MetaVelvet is a de novo short sequence metagenome assembler. It is extended upon the Velvet assembler (single-genome and de Bruijn-graph based) to overcome the limitations of single-genome assembler. Pros: Able to reconstruct scaffold sequences including low-abundance species. Cons: Has slightly higher percentages of chimeric scaffolds. | [107] |
| MegaHit | MegaHit is a de novo assembler for assembling metagenomics data. It implements succinct de Bruijn graphs. Pros: Fast and memory efficient. Available in both CPU-only and GPU-accelerated versions. Cons: Relatively biased towards the assembly of low abundant genome fragments. | [108] |
| MetaQUAST | MetaQUAST evaluates and compares the quality of metagenome assemblies. It is improved based on QUAST. Its metagenome specific features includes: unlimited number of reference genome, species content detection, chimeric detection, and visualizations. Pros: Can be fed with multiple assemblies. Cons: Reduced precision in order to get higher time/memory efficiency. | [110] |
| MEGAN | MEGAN is a BLAST-based automated pipeline for taxonomic and functional analysis of metagenomic and metatranscriptomic datasets. Pros: Allows laptop analysis of large metagenomic data sets. | [111] |
| MetaPhlAn/ MetaPhlAn2 | MetaPhlAn is an automated pipeline that profiles the microbial composition from shotgun metagenomic data at the species-level. The microbial community it can profile includes bacteria, archaea, eukaryotes and viruses. It accomplishes profiling with unique clade-specific marker genes. MetaPhlAn 2 is extended beyond the first version with enhanced metagenomic taxonomic profiling ability. Pros: Able to work with large-scale metagenome data. | [112] |
| HUMAnN2 | HUMAnN2 is an automated pipeline designed for functional analysis of metagenomic and metatranscriptomic data at the species-level. The general process of HUMAnN2 pipeline is identification of known species, alignment of reads to pangenomes, translated search on unclassified reads, and quantification of gene families and pathways. HUMAnN2 utilizes other pipelines such as MetaPhlAn2 to perform identification of known species. Pros: High accuracy, sensitivity, speed. Cons: A large proportion of sequencing reads remain unmapped and unintegrated. | [113] |
| MG-RAST | MG-RAST is a web-based fully automated system for metagenomic analysis. It provides phylogenic and functional analysis. Pros: Require only 75 bp or longer for gene prediction or similarity analysis that provides taxonomic binning and functional classification. Able to handle both assembled and unassembled data. Cons: MG-RAST has been optimized for use with the Firefox browser. There are some browser-to-browser issues with visualization of certain diagrams. | [114] |
| IMG/M | IGM/M is a web-based pipeline that provides comparative analysis for metagenome. It provides structural and functional annotation. Prefer assembled contigs. Pros: Integrates all datasets into a single protein level abstraction. In contrast to MG-RAST, IMG/M includes more computationally expensive tools such as hidden Markov model and BLASTX. Cons: Statistical analysis tool is only available as an on-demand computation to the registered IMG users of the Expert Review IMG site. | [115] |
| METAREP | METAREP is a suite of web-based tools to view and compare metagenomic annotated data including both functional and taxonomical assignments. Pros: Able to handle extremely large datasets. Able to perform comparison on up to 20+ datasets simultaneously. Cons: No inbuilt annotation workflow. Users need to upload existing annotations. | [116] |
| CuffDiff | Cufflinks is a suite of programs that assembles transcriptomes, estimates abundance, and performs gene expression differentiations. It implements a parsimony-based algorithm. Pros: High efficiency, sensitivity and precision. Cons: Not optimized for metatranscriptomics analysis. | [123] |
| Blast2GO | Blast2Go is a Blast-based software that provides automatic functional annotation on DNA/protein sequences. It has multiple annotation styles that can be used for various conditions. Pros: Combines multiple annotation strategies. Strong visualization tools. Con: Not optimized for large datasets with large number of genes. | [124] |
| Viromic sequencing data analysis | ||
| VICUNA | VICUNA is a de novo assembler targeting viral populations, which have high mutation rates. Its algorithm uses an overlap-layout-consensus based approach. The general process of VICUNA is trimming reads, constructing/clustering contigs, validating contigs, and then extending and merging contigs. Pros: Able to efficiently process ultra-deep sequence data. High accuracy and continuity. Cons: Limited accessibility due to its requirement of local computing power. | [135] |
| Metavir/ Metavir2 | Metavir is a web-based pipeline specifically for viral metagenome analysis. Metavir 2 is developed based on Metavir with additional features such as new tools for assembled virome sequence analysis and new dataset comparison strategies.Pros: User-friendly interface. Able to perform analysis on both raw reads and assembled virome sequencesCons: Focuses on the compositional analysis. Functional annotation is lacking. | [136,137] |
| VMGAP | VMGAP is an automated pipeline for functional annotation of viral shotgun metagenomic data. It first performs a database searches and then functional assignments. Pros: Uses specialized databases. Cons: Requires local installation of several open-source packages, programs and public databases. | [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, B.; Chi, L.; Zhu, Y.; Shi, X.; Tu, P.; Li, B.; Yin, J.; Gao, N.; Shen, W.; Schnabl, B. An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules 2021, 11, 530. https://doi.org/10.3390/biom11040530
Gao B, Chi L, Zhu Y, Shi X, Tu P, Li B, Yin J, Gao N, Shen W, Schnabl B. An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules. 2021; 11(4):530. https://doi.org/10.3390/biom11040530
Chicago/Turabian StyleGao, Bei, Liang Chi, Yixin Zhu, Xiaochun Shi, Pengcheng Tu, Bing Li, Jun Yin, Nan Gao, Weishou Shen, and Bernd Schnabl. 2021. "An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies" Biomolecules 11, no. 4: 530. https://doi.org/10.3390/biom11040530