
Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations

Abstract
:1. Introduction
1.1. PDE4 Structural Biology
1.2. An Overview of Selective PDE4 Inhibitors and Their Side Effects
1.3. PDE4D as a Therapeutic Target for the Treatment of Alzheimer’s Disease
1.4. Natural Polyphenolic Compounds as Potential PDE4D Inhibitors
2. Computational Methods
2.1. Molecular Docking
2.2. Preparation of Initial Complexes
2.3. Molecular Dynamics Simulations
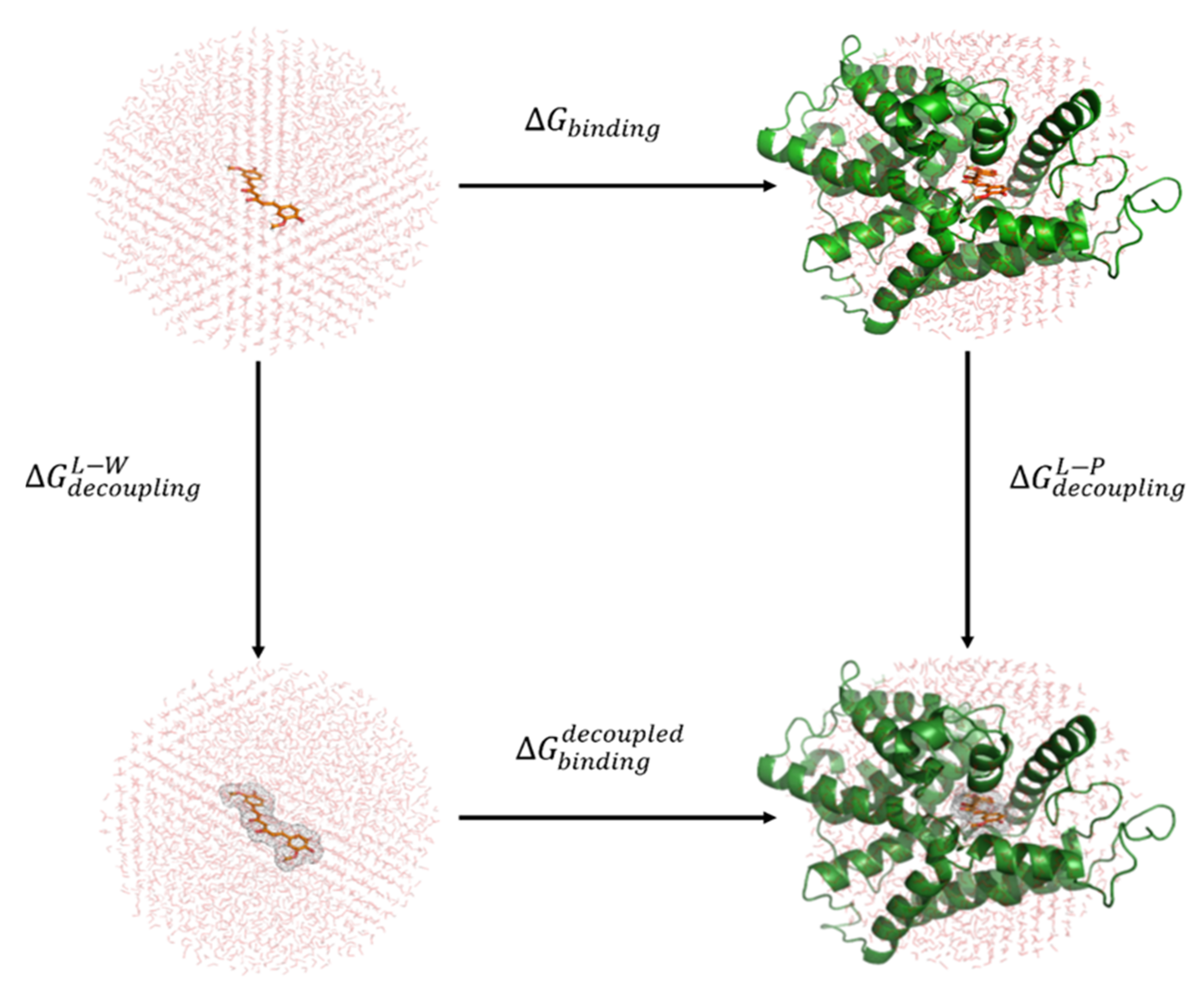
2.4. Binding Free Energy Calculations with Linear Response Approximation (LRA) Method
3. Results and Discussion
3.1. Molecular Docking
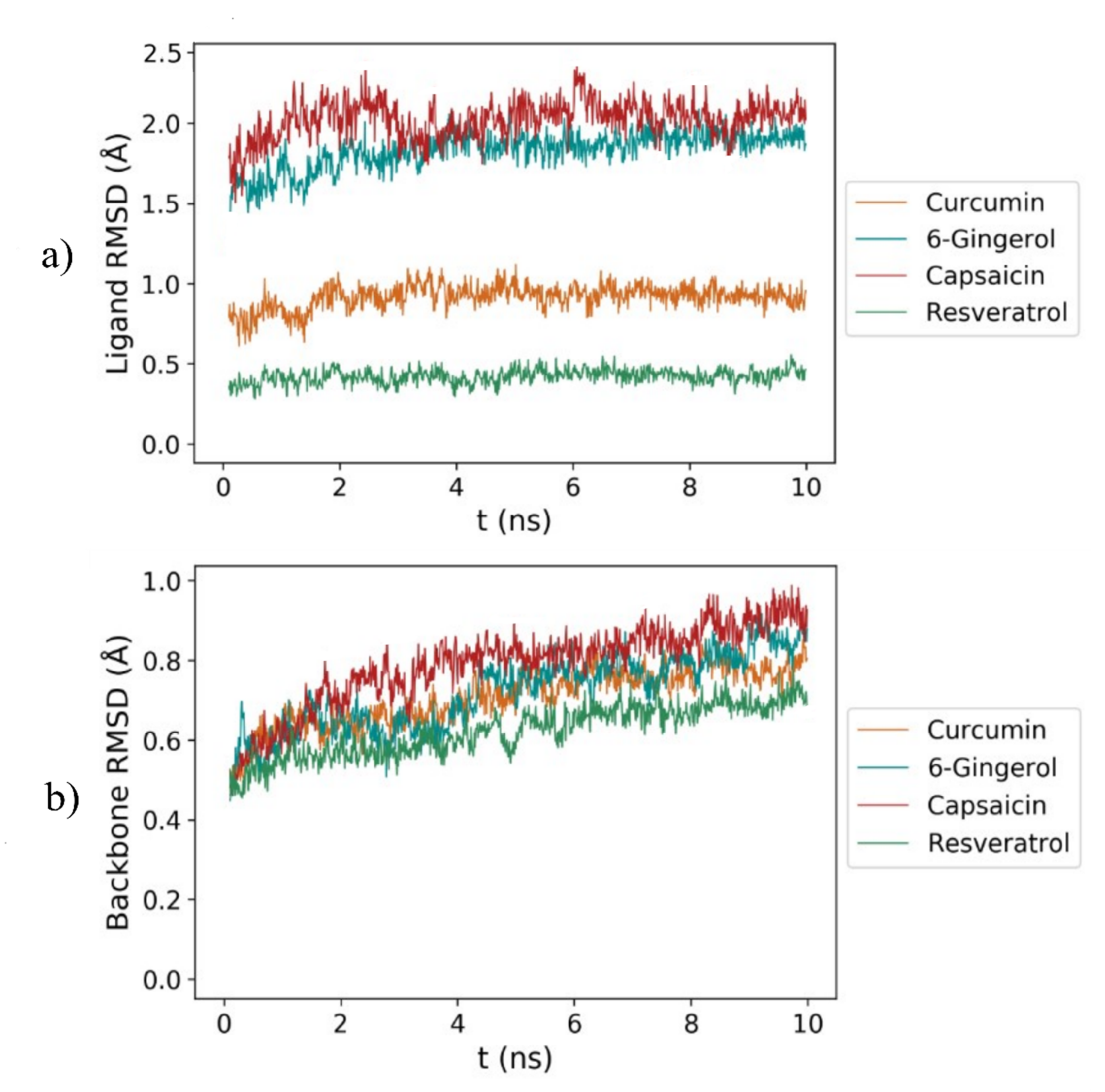
3.2. RMSD Analysis
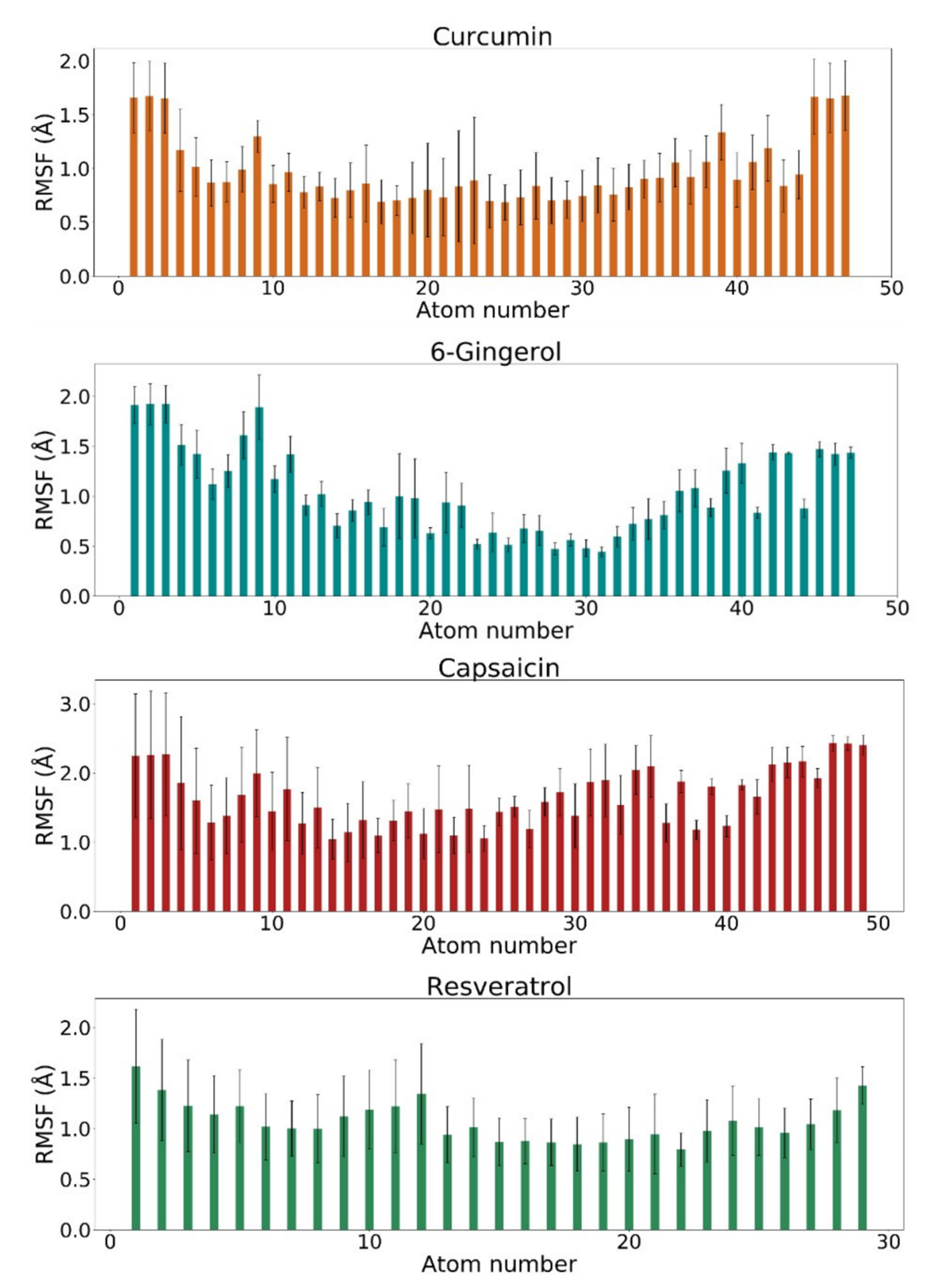
3.3. RMSF Analysis
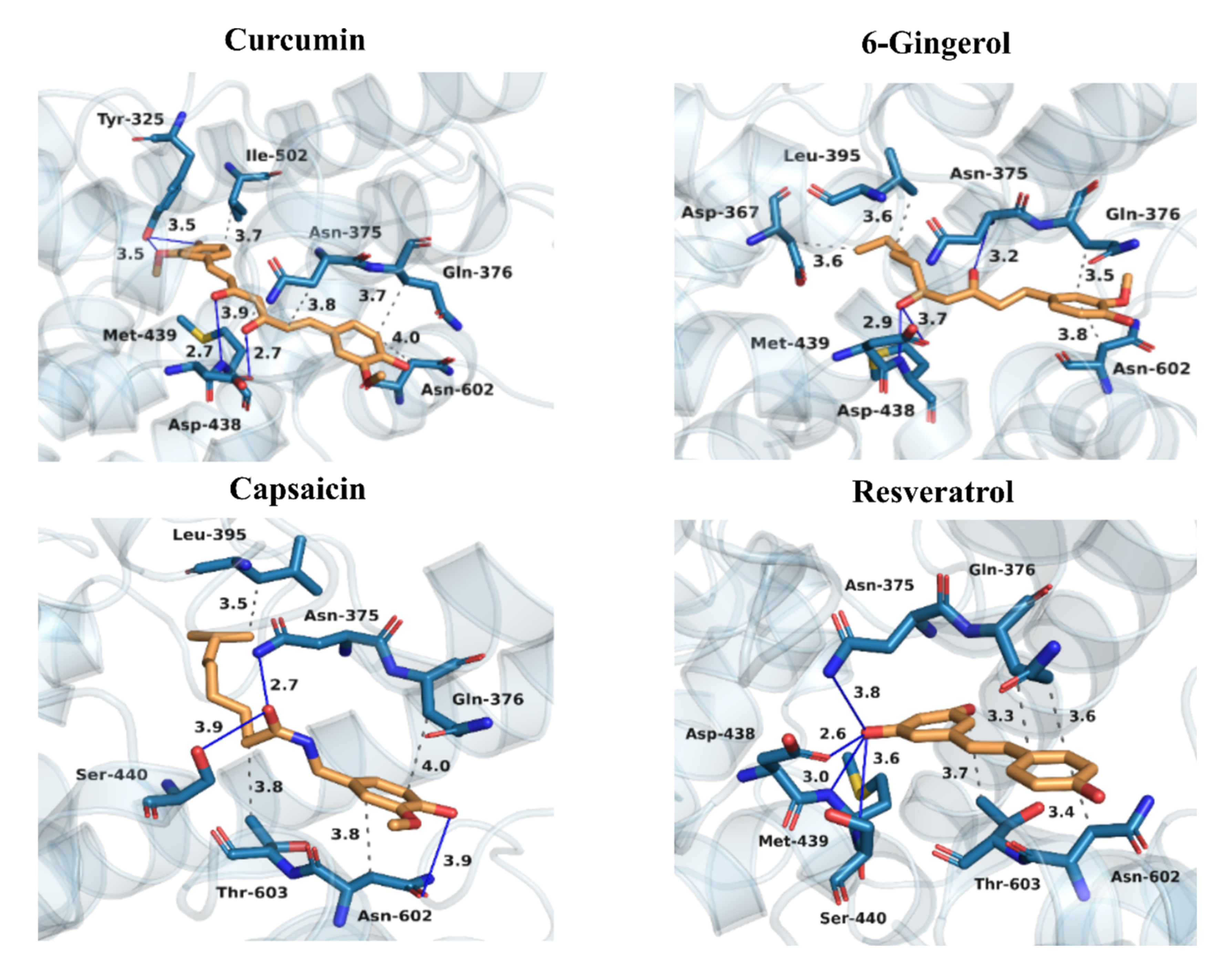
3.4. Binding Patterns of Curcumin, 6-Gingerol, Capsaicin, and Resveratrol to PDE4D
3.5. Results of Binding Free Energy Calculations with the LRA Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β peptide |
| Bcl-2 | B-cell lymphoma 2 |
| BDNF | Brain-derived neurotrophic factor |
| cAMP | Cyclic adenosine monophosphate |
| CNS | Central nervous system |
| COPD | Chronic obstructive pulmonary disease |
| CR3 | Control region 3 |
| CREB | cAMP response element binding protein |
| ele | electrostatic interactions |
| Epac1/2 | Exchange factor directly activated by cAMP 1 and 2 |
| ESP | Electrostatic potential |
| FDA | Food and Drug Administration |
| FEP | Free energy perturbation |
| GAFF | General AMBER force field |
| HF | Hartree-Fock |
| IBD | Inflammatory bowel diseases |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| LIE | Linear interaction energy |
| LRA | Linear response approximation |
| LRF | Local reaction field |
| MD | Molecular dynamics |
| PDB | Protein data bank |
| PDE4 | Phosphodiesterase 4 |
| PKA | Protein kinase A |
| RA | Rheumatic arthritis |
| RESP | Restricted electrostatic charge fitting procedure |
| PLIP | Protein ligand interaction profiler |
| RMR6 | Radial-mean-reduced scoring function at a cutoff radius of 6 Å from each atom of the ligand |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| ROS | Reactive oxygen species |
| SCAAS | Surface constraint all-atom solvent |
| UCR1 | Upstream conserved region 1 |
| UCR2 | Upstream conserved region 2 |
| vdW | van der Waals interactions |
References
- Chiricozzi, A.; Caposiena, D.; Garofalo, V.; Cannizzaro, M.V.; Chimenti, S.; Saraceno, R. A new therapeutic for the treatment of moderate-to-severe plaque psoriasis: Apremilast. Expert Rev. Clin. Immunol. 2016, 12, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, T.; Qi, B.; He, J.; Ke, H.; Shi, J. Advances in the development of phosphodiesterase-4 inhibitors. J. Med. Chem. 2020, 19, 10594–10617. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I.; Mavropoulos, A.; Bogdanos, D.P. Phosphodiesterase 4 inhibitors in immune-mediated diseases: Mode of action, clinical applications, current and future perspectives. Curr. Med. Chem. 2017, 24, 3054–3067. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.M.; Talarek, S.; Listos, J.; Nabavi, S.F.; Devi, K.P.; de Oliveira, M.R.; Tewari, D.; Argüelles, S.; Mehrzadi, S.; Hosseinzadeh, A. Phosphodiesterase inhibitors say NO to Alzheimer’s disease. Food Chem. Toxicol. 2019, 134, 110822. [Google Scholar] [CrossRef]
- Richter, W.; Conti, M. Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs). J. Biol. Chem. 2002, 277, 40212–40221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef]
- Cedervall, P.; Aulabaugh, A.; Geoghegan, K.F.; McLellan, T.J.; Pandit, J. Engineered stabilization and structural analysis of the autoinhibited conformation of PDE4. Proc. Natl. Acad. Sci. USA 2015, 112, E1414–E1422. [Google Scholar] [CrossRef] [Green Version]
- Bolger, G.; Michaeli, T.; Martins, T.; St John, T.; Steiner, B.; Rodgers, L.; Riggs, M.; Wigler, M.; Ferguson, K. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol. Cell. Biol. 1993, 13, 6558–6571. [Google Scholar] [CrossRef]
- Card, G.L.; England, B.P.; Suzuki, Y.; Fong, D.; Powell, B.; Lee, B.; Luu, C.; Tabrizizad, M.; Gillette, S.; Ibrahim, P.N. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 2004, 12, 2233–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.X.; Hassell, A.M.; Vanderwall, D.; Lambert, M.H.; Holmes, W.D.; Luther, M.A.; Rocque, W.J.; Milburn, M.V.; Zhao, Y.; Ke, H. Atomic structure of PDE4: Insights into phosphodiesterase mechanism and specificity. Science 2000, 288, 1822–1825. [Google Scholar] [CrossRef]
- Wong, K.Y.; Gao, J. Insight into the phosphodiesterase mechanism from combined QM/MM free energy simulations. FEBS J. 2011, 278, 2579–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosdocimi, T.; Mollica, L.; Donini, S.; Semrau, M.S.; Lucarelli, A.P.; Aiolfi, E.; Cavalli, A.; Storici, P.; Alfei, S.; Brullo, C. Molecular bases of PDE4D inhibition by memory-enhancing GEBR library compounds. Biochemistry 2018, 57, 2876–2888. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Ora, J.; Puxeddu, E. Dual bronchodilation and exacerbations of COPD. J. Thorac. Dis. 2016, 8, 2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dozier, L.; Bartos, G.; Kerdel, F. Apremilast and psoriasis in the real world: A retrospective case series. J. Am. Acad. Dermatol. 2020, 83, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Woo, T.E.; Kuzel, P. Crisaborole 2% ointment (Eucrisa) for atopic dermatitis. Skin Ther. Lett. 2019, 24, 4–6. [Google Scholar]
- Rolan, P.; Hutchinson, M.; Johnson, K. Ibudilast: A review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin. Pharmacother. 2009, 10, 2897–2904. [Google Scholar] [CrossRef]
- Gong, B.; Vitolo, O.V.; Trinchese, F.; Liu, S.; Shelanski, M.; Arancio, O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Investig. 2004, 114, 1624–1634. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; D’Amato, E.C.; Burgin, A.B. Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics 2015, 12, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Mori, F.; Pérez-Torres, S.; De Caro, R.; Porzionato, A.; Macchi, V.; Beleta, J.; Gavalda, A.; Palacios, J.M.; Mengod, G. The human area postrema and other nuclei related to the emetic reflex express cAMP phosphodiesterases 4B and 4D. J. Chem. Neuroanat. 2010, 40, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.-L.C.; Goya, S.; Nakae, S.; Wang, D.; Bruss, M.; Hou, C.; Umetsu, D.; Conti, M. Phosphodiesterase 4B is essential for TH2-cell function and development of airway hyperresponsiveness in allergic asthma. J. Allergy Clin. Immunol. 2010, 126, 1252–1259.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaud, A.; Stamatiou, P.B.; Jin, S.-L.C.; Lachance, N.; MacDonald, D.; Laliberté, F.; Liu, S.; Huang, Z.; Conti, M.; Chan, C.-C. Deletion of phosphodiesterase 4D in mice shortens α 2-adrenoceptor–mediated anesthesia, a behavioral correlate of emesis. J. Clin. Investig. 2002, 110, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Schmiechen, R.; Schneider, H.H.; Wachtel, H. Close correlation between behavioural response and binding in vivo for inhibitors of the rolipram-sensitive phosphodiesterase. Psychopharmacology 1990, 102, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Jacobitz, S.; McLaughlin, M.M.; Livi, G.P.; Burman, M.; Torphy, T.J. Mapping the functional domains of human recombinant phosphodiesterase 4A: Structural requirements for catalytic activity and rolipram binding. Mol. Pharmacol. 1996, 50, 891–899. [Google Scholar] [PubMed]
- Borroni, E.; Bohrmann, B.; Grueninger, F.; Prinssen, E.; Nave, S.; Loetscher, H.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Siddiqui, A. Sembragiline: A novel, selective monoamine oxidase type B inhibitor for the treatment of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2017, 362, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuchi, R.; Hishikawa, N.; Kurata, T.; Sato, K.; Kono, S.; Yamashita, T.; Deguchi, K.; Abe, K. Clinical and demographic predictors of mild cognitive impairment for converting to Alzheimer’s disease and reverting to normal cognition. J. Neurol. Sci. 2014, 346, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, Z.; Huang, Y.-Y.; Wu, D.; Luo, H.-B. Novel Phosphodiesterase Inhibitors for Cognitive Improvement in Alzheimer’s Disease: Miniperspective. J. Med. Chem. 2018, 61, 5467–5483. [Google Scholar] [CrossRef]
- Teich, A.F.; Nicholls, R.E.; Puzzo, D.; Fiorito, J.; Purgatorio, R.; Arancio, O. Synaptic therapy in Alzheimer’s disease: A CREB-centric approach. Neurotherapeutics 2015, 12, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Y.; Chowdhary, A.; Fox, D.; Gurney, M.E.; Zhang, H.-T.; Auerbach, B.D.; Salvi, R.J.; Yang, M.; Li, G. Memory enhancing effects of BPN14770, an allosteric inhibitor of phosphodiesterase-4D, in wild-type and humanized mice. Neuropsychopharmacology 2018, 43, 2299–2309. [Google Scholar] [CrossRef] [PubMed]
- Bruno, O.; Fedele, E.; Prickaerts, J.; Parker, L.; Canepa, E.; Brullo, C.; Cavallero, A.; Gardella, E.; Balbi, A.; Domenicotti, C. GEBR-7b, a novel PDE4D selective inhibitor that improves memory in rodents at non-emetic doses. Br. J. Pharmacol. 2011, 164, 2054–2063. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Brullo, C.; Prickaerts, J.; Arancio, O.; Villa, C.; Rebosio, C.; Calcagno, E.; Balbi, M.; Van Hagen, B.T.; Argyrousi, E.K. Memory-enhancing effects of GEBR-32a, a new PDE4D inhibitor holding promise for the treatment of Alzheimer’s disease. Sci. Rep. 2017, 7, 46320. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Y.; Zhang, H.-T.; Gurney, M.E.; O’Donnell, J.M. Comparison of the pharmacological profiles of selective PDE4B and PDE4D inhibitors in the central nervous system. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Prickaerts, J.; Heckman, P.R.; Blokland, A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 1033–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical trials for BPN14770. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03817684?term=BPN14770&rank=1 (accessed on 5 January 2021).
- Clinical trials for MK0952. Available online: https://clinicaltrials.gov/ct2/show/NCT00362024?term=MK-0952 (accessed on 5 January 2021).
- Lešnik, S.; Furlan, V.; Bren, U. Rosemary (Rosmarinus officinalis L.): Extraction techniques, analytical methods and health-promoting biological effects. Phytochem. Rev. 2021. Available online: https://link.springer.com/article/10.1007/s11101-021-09745-5#citeas (accessed on 26 February 2021). [CrossRef]
- Furlan, V.; Konc, J.; Bren, U. Inverse molecular docking as a novel approach to study anticarcinogenic and anti-neuroinflammatory effects of curcumin. Molecules 2018, 23, 3351. [Google Scholar] [CrossRef] [Green Version]
- Furlan, V.; Bren, U. Protective effects of [6]-gingerol against chemical carcinogens: Mechanistic insights. Int. J. Mol. Sci. 2020, 21, 695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kores, K.; Lešnik, S.; Bren, U.; Janežič, D.; Konc, J. Discovery of novel potential human targets of resveratrol by inverse molecular docking. J. Chem. Inf. Model. 2019, 59, 2467–2478. [Google Scholar] [CrossRef] [PubMed]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction methods, antioxidative action, bioavailability and anticarcinogenic effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef]
- Lee, W.-H.; Loo, C.-Y.; Bebawy, M.; Luk, F.; Mason, R.S.; Rohanizadeh, R. Curcumin and its derivatives: Their application in neuropharmacology and neuroscience in the 21st century. Curr. Neuropharmacol. 2013, 11, 338–378. [Google Scholar] [CrossRef] [Green Version]
- Veldman, E.R.; Jia, Z.; Halldin, C.; Svedberg, M.M. Amyloid binding properties of curcumin analogues in Alzheimer’s disease postmortem brain tissue. Neurosci. Lett. 2016, 630, 183–188. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [Green Version]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Abusnina, A.; Keravis, T.; Zhou, Q.; Justiniano, H.; Lobstein, A.; Lugnier, C. Tumour growth inhibition and anti-angiogenic effects using curcumin correspond to combined PDE2 and PDE4 inhibition. Thromb. Haemost. 2015, 114, 319–328. [Google Scholar] [CrossRef] [PubMed]
- DiSilvestro, R.A.; Joseph, E.; Zhao, S.; Bomser, J. Diverse effects of a low dose supplement of lipidated curcumin in healthy middle aged people. Nutr. J. 2012, 11, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, E.A.; Zhang, Y.; Xu, C.; Wakita, R.; Emala, C.W. Active components of ginger potentiate β-agonist–induced relaxation of airway smooth muscle by modulating cytoskeletal regulatory proteins. Am. J. Respir. Cell Mol. Biol. 2014, 50, 115–124. [Google Scholar] [CrossRef]
- Silva, I.X.; de Oliveira, M.G.; da Conceicao, E.G.; Taft, C.A.; da Silva, C.H.T.P.; da Silva, V.B. Binding model of capsaicin is able to reach the peripheral anionic site of acetylcholinesterase. Curr. Bioact. Compd. 2017, 13, 152–156. [Google Scholar] [CrossRef]
- Xu, W.; Liu, J.; Ma, D.; Yuan, G.; Lu, Y.; Yang, Y. Capsaicin reduces Alzheimer-associated tau changes in the hippocampus of type 2 diabetes rats. PLoS ONE 2017, 12, e0172477. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, L.; Pan, X.; Chen, J.; Wang, L.; Wang, W.; Cheng, R.; Wu, F.; Feng, X.; Yu, Y. The effect of resveratrol on beta amyloid-induced memory impairment involves inhibition of phosphodiesterase-4 related signaling. Oncotarget 2016, 7, 17380. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, J.; Konc, J.; Samudrala, R.; Chopra, G. CANDOCK: Chemical atomic network-based hierarchical flexible docking algorithm using generalized statistical potentials. J. Chem. Inf. Model. 2020, 60, 1509–1527. [Google Scholar] [CrossRef]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Åqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213–225. [Google Scholar] [CrossRef]
- Cerutti, D.S.; Swope, W.C.; Rice, J.E.; Case, D.A. ff14ipq: A self-consistent force field for condensed-phase simulations of proteins. J. Chem. Theory Comput. 2014, 10, 4515–4534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Florian, J.; Goodman, M.F.; Warshel, A. Theoretical investigation of the binding free energies and key substrate-recognition components of the replication fidelity of human DNA polymerase β. J. Phys. Chem. B 2002, 106, 5739–5753. [Google Scholar] [CrossRef]
- Jeon, Y.; Heo, Y.-S.; Kim, C.; Hyun, Y.-L.; Lee, T.; Ro, S.; Cho, J. Phosphodiesterase: Overview of protein structures, potential therapeutic applications and recent progress in drug development. Cell. Mol. Life Sci. 2005, 62, 1198–1220. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, Y.S.; Liao, Q.; Petrović, D.A.; Krüger, D.M.; Strodel, B.; Amyes, T.L.; Richard, J.P.; Kamerlin, S.C. Enzyme architecture: Modeling the operation of a hydrophobic clamp in catalysis by triosephosphate isomerase. J. Am. Chem. Soc. 2017, 139, 10514–10525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, Y.S.; Amyes, T.L.; Richard, J.P.; Kamerlin, S.C. Uncovering the role of key active-site side chains in catalysis: An extended Brønsted relationship for substrate deprotonation catalyzed by wild-type and variants of triosephosphate isomerase. J. Am. Chem. Soc. 2019, 141, 16139–16150. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33 (Suppl. 2), W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, P. Geometry of interaction of metal ions with histidine residues in protein structures. Protein Eng. Des. Sel. 1990, 4, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Klvana, M.; Jerabek, P.; Goodman, M.F.; Florián, J. An abridged transition state model to derive structure, dynamics, and energy components of DNA polymerase β fidelity. Biochemistry 2011, 50, 7023–7032. [Google Scholar] [CrossRef] [Green Version]
- Klvana, M.; Bren, U. Aflatoxin B1–formamidopyrimidine DNA adducts: Relationships between structures, free energies, and melting temperatures. Molecules 2019, 24, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, F.S.; Warshel, A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 1992, 97, 3100–3107. [Google Scholar] [CrossRef]
- King, G.; Warshel, A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 1989, 91, 3647–3661. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL. 2002. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations; Los Alamos National Lab.(LANL): Los Alamos, NM, USA, 2019; pp. 2575–9752.
- Lee, F.S.; Chu, Z.-T.; Bolger, M.B.; Warshel, A. Calculations of antibody-antigen interactions: Microscopic and semi-microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng. Des. Sel. 1992, 5, 215–228. [Google Scholar] [CrossRef]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef]
- Åqvist, J.; Luzhkov, V.B.; Brandsdal, B.O. Ligand binding affinities from MD simulations. Acc. Chem. Res. 2002, 35, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Bren, U.; Lah, J.; Bren, M.; Martínek, V.; Florián, J. DNA duplex stability: The role of preorganized electrostatics. J. Phys. Chem. B 2010, 114, 2876–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åqvist, J.; Medina, C.; Samuelsson, J.-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. Des. Sel. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Åqvist, J.; Hansson, T. On the validity of electrostatic linear response in polar solvents. J. Phys. Chem. 1996, 100, 9512–9521. [Google Scholar] [CrossRef]
- Cleves, A.E.; Jain, A.N. Knowledge-guided docking: Accurate prospective prediction of bound configurations of novel ligands using Surflex-Dock. J. Comput. Aided Mol. Des. 2015, 29, 485–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations: A cross-docking study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.R.; Voelker, T.; Zhang, H.-T.; O’Donnell, J.M. Dual inhibitors of phosphodiesterase-4 and serotonin reuptake. J. Med. Chem. 2009, 52, 1530–1539. [Google Scholar] [CrossRef] [Green Version]






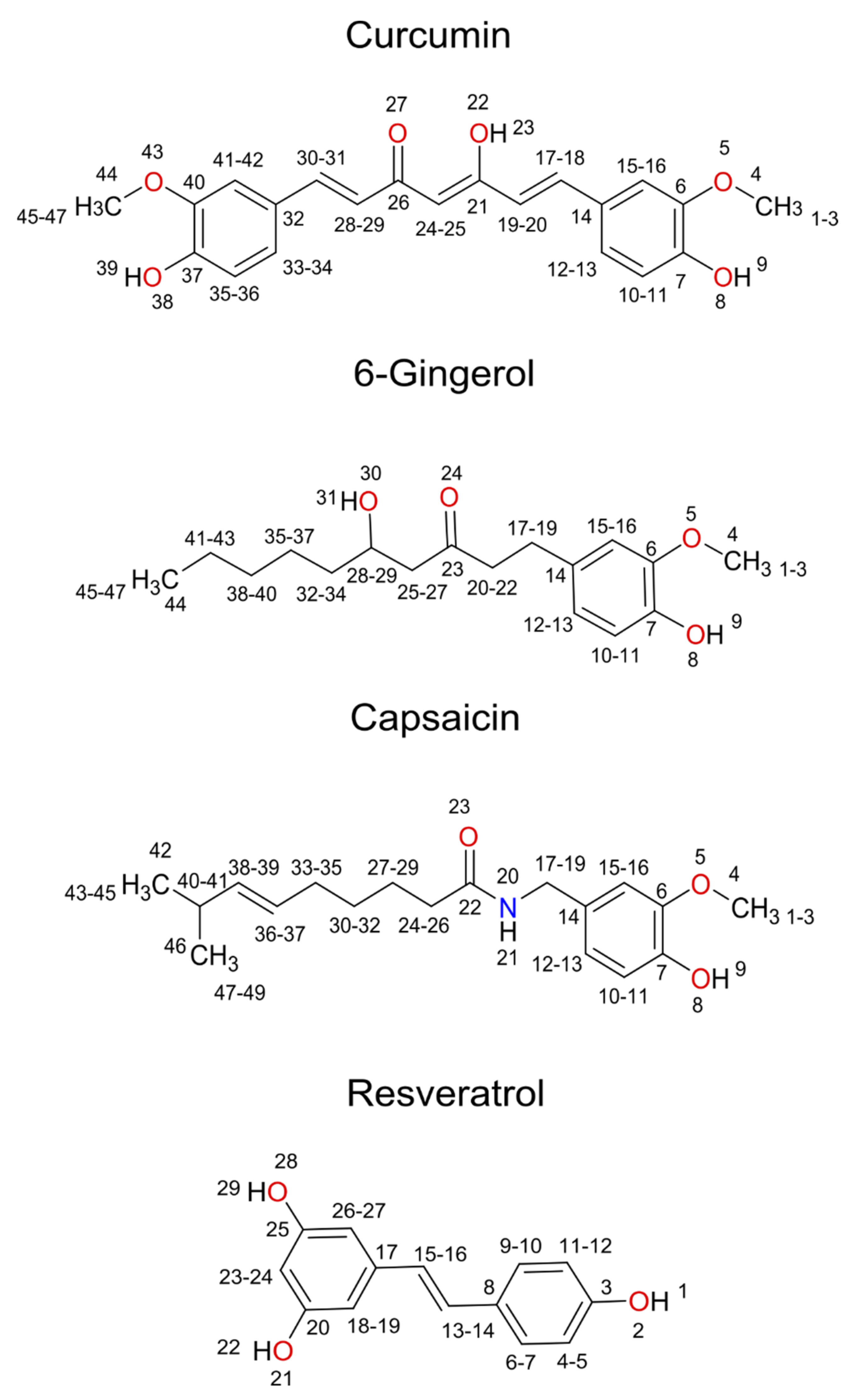{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pocket Name | Features | Important Residues |
|---|---|---|
| M-pocket | Contains metal ions Zn2+ and Mg2+ | His330, His366, Asp367, His370, Asn375, Gln376, Leu395, Glu396, Asp438, Met439, Ser440, Asp484 |
| S-pocket | Contains polar residues and water molecules | Gly372, Ser374, Glu505, Arg508, Ser521, Cys524 |
| Q-pocket | Divided into two hydrophobic micro-pockets Q1 and Q2, separated by a glutamine saddle (Gln535) | Q1-pocket: Tyr325, Asn487, Thr499, Tyr495 Q2-pocket: Ile502, Met503, Phe506, Met523, Phe538 |
| CR3 | α-helix consisting of 12 amino-acid residues | Gln595, Val596, Ser597, Glu598, Phe599, Ile600, Ser601, Asn602, Thr603, Phe604, Leu605, Asp606 |
| Ligands | Docking Score Values (Arbitrary Units) |
|---|---|
| Curcumin | −62.24 |
| 6-Gingerol | −50.16 |
| Capsaicin | −41.58 |
| Resveratrol | −30.18 |
| MD Simulation Run | Production Run 1 | Production Run 2 | Production Run 3 | Production Run 4 | Average of All Runs |
|---|---|---|---|---|---|
| Curcumin | |||||
| Average ligand RMSD (Å) | 0.93 | 0.87 | 0.88 | 0.99 | 0.92 ± 0.08 |
| Average backbone RMSD (Å) | 0.69 | 0.75 | 0.68 | 0.72 | 0.71 ± 0.08 |
| 6-Gingerol | |||||
| Average ligand RMSD (Å) | 1.53 | 1.82 | 1.72 | 1.90 | 1.74±0.16 |
| Average backbone RMSD (Å) | 0.76 | 0.68 | 0.72 | 0.71 | 0.72 ± 0.11 |
| Capsaicin | |||||
| Average ligand RMSD (Å) | 1.95 | 1.83 | 2.10 | 2.08 | 1.99 ± 0.14 |
| Average backbone RMSD (Å) | 0.71 | 0.82 | 0.81 | 0.81 | 0.78 ± 0.11 |
| Resveratrol | |||||
| Average ligand RMSD (Å) | 0.47 | 0.35 | 0.36 | 0.43 | 0.40 ± 0.04 |
| Average backbone RMSD (Å) | 0.58 | 0.63 | 0.67 | 0.64 | 0.63 ± 0.07 |
| MD Simulation Run | Production Run 1 | Production Run 2 | Production Run 3 | Production Run 4 | Average of All Runs |
|---|---|---|---|---|---|
| Curcumin | |||||
| Average ligand RMSF (Å) | 1.08 | 1.07 | 0.92 | 0.85 | 0.98±0.09 |
| 6-Gingerol | |||||
| Average ligand RMSF (Å) | 1.30 | 0.97 | 1.07 | 1.10 | 1.11±0.14 |
| Capsaicin | |||||
| Average ligand RMSF (Å) | 1.71 | 1.91 | 1.67 | 1.86 | 1.79±0.12 |
| Resveratrol | |||||
| Average ligand RMSF (Å) | 1.00 | 0.88 | 0.95 | 1.05 | 0.97±0.07 |
| Energies | (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) | ** (kcal/mol) |
|---|---|---|---|---|---|---|
| Curcumin | ||||||
| Production run 1 | −53.72 | −33.43 | −60.10 | −40.34 | −0.66 | −10.83 |
| Production run 2 | −52.38 | −33.41 | −62.07 | −40.55 | −0.73 | −11.24 |
| Production run 3 | −52.33 | −33.52 | −61.04 | −39.43 | −0.69 | −11.22 |
| Production run 4 | −52.94 | −33.41 | −60.43 | −40.23 | −0.69 | −10.85 |
| Average * | −52.84 ± 0.64 | −33.44 ± 0.05 | −60.66 ± 1.15 | −40.14 ± 0.49 | −0.69 ± 0.03 | −11.03 ± 0.25 |
| 6-Gingerol | ||||||
| Production run 1 | −49.25 | −24.38 | −49.89 | −43.84 | −0.49 | −6.97 |
| Production run 2 | −50.71 | −24.49 | −49.37 | −43.16 | −0.51 | −7.27 |
| Production run 3 | −49.11 | −25.40 | −50.58 | −44.17 | −0.47 | −6.85 |
| Production run 4 | −48.15 | −24.36 | −49.98 | −43.53 | −0.44 | −6.85 |
| Average * | −49.56 ± 1.52 | −24.66 ± 0.50 | −49.96 ± 0.50 | −43.67 ± 0.43 | −0.48 ± 0.03 | −6.99 ± 0.39 |
| Capsaicin | ||||||
| Production run 1 | −50.33 | −28.20 | −38.69 | −35.67 | −0.38 | −5.36 |
| Production run 2 | −52.33 | −27.72 | −37.29 | −35.81 | −0.35 | −5.27 |
| Production run 3 | −52.62 | −27.74 | −37.39 | −35.76 | −0.36 | −5.37 |
| Production run 4 | −50.67 | −28.30 | −38.37 | −36.70 | −0.38 | −4.96 |
| Average * | −51.49 ± 1.15 | −27.99 ± 0.31 | −37.93 ± 0.70 | −35.99 ± 0.48 | −0.37 ± 0.02 | −5.24 ± 0.35 |
| Resveratrol | ||||||
| Production run 1 | −31.64 | −16.53 | −51.15 | −47.18 | −0.53 | −4.57 |
| Production run 2 | −30.18 | −16.44 | −51.90 | −47.04 | −0.50 | −4.58 |
| Production run 3 | −29.80 | −16.46 | −52.68 | −47.61 | −0.48 | −4.56 |
| Production run 4 | −30.58 | −16.49 | −52.48 | −47.59 | −0.49 | −4.65 |
| Average * | −30.55 ± 0.39 | −16.48 ± 0.04 | −52.05 ± 0.65 | −47.36 ± 0.35 | −0.51 ± 0.02 | −4.59 ± 0.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furlan, V.; Bren, U. Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations. Biomolecules 2021, 11, 479. https://doi.org/10.3390/biom11030479
Furlan V, Bren U. Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations. Biomolecules. 2021; 11(3):479. https://doi.org/10.3390/biom11030479
Chicago/Turabian StyleFurlan, Veronika, and Urban Bren. 2021. "Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations" Biomolecules 11, no. 3: 479. https://doi.org/10.3390/biom11030479