
Differential MicroRNA Expression Involved in Endometrial Receptivity of Goats

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animal and Tissue Collection
2.2. Small RNA (sRNA) Library Construction and Sequencing
2.3. Analysis of Sequencing Data
2.4. Identification of Differentially Expressed miRNAs (DEMs)
2.5. Potential Transcription Factors Prediction of DEMs
2.6. Target Gene Prediction of DEMs
2.7. Functional Analysis of DEMs
2.8. Validation of miRNA Expression by Stem-Loop Quantitative RT-PCR
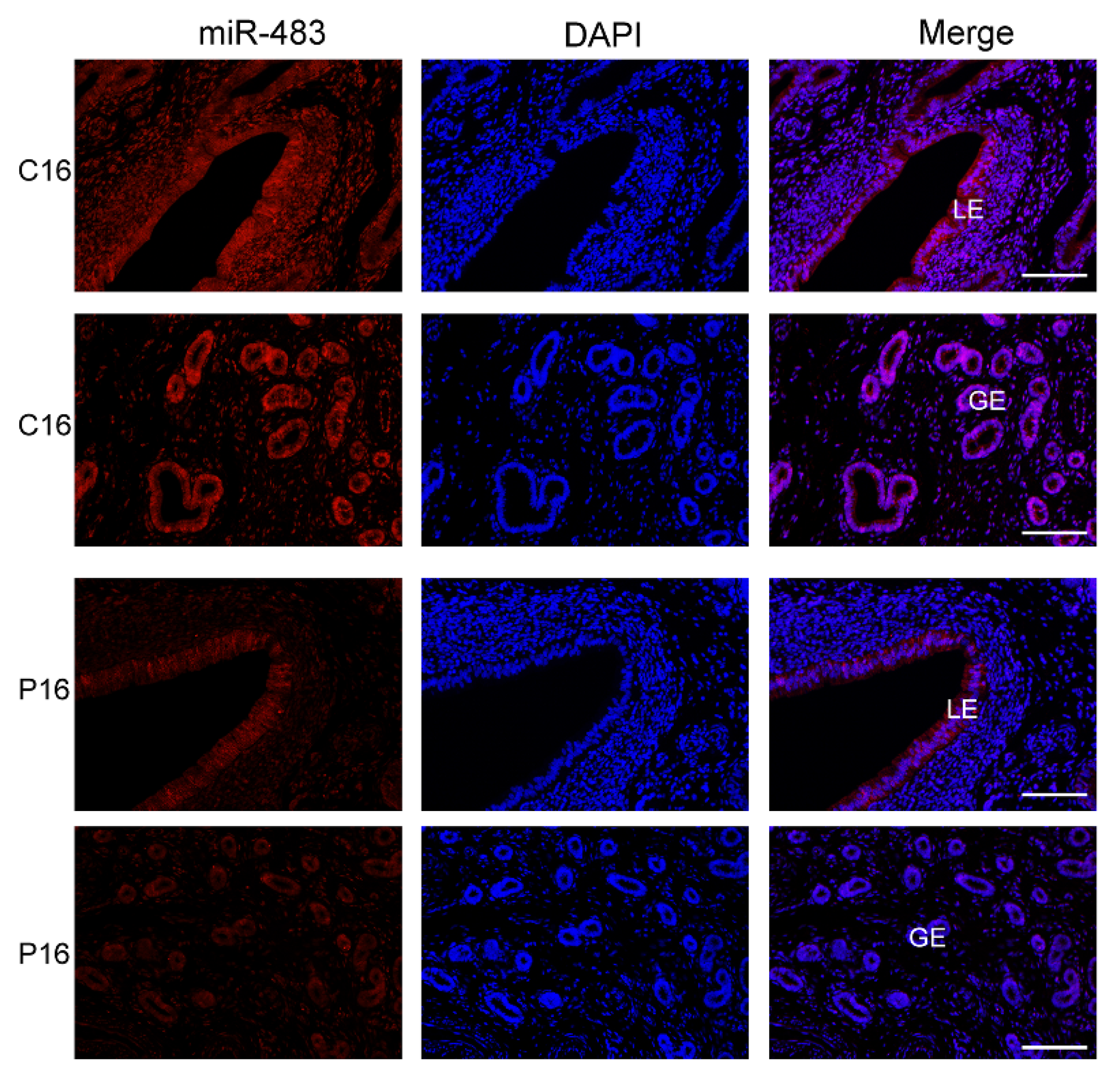
2.9. FISH Analysis
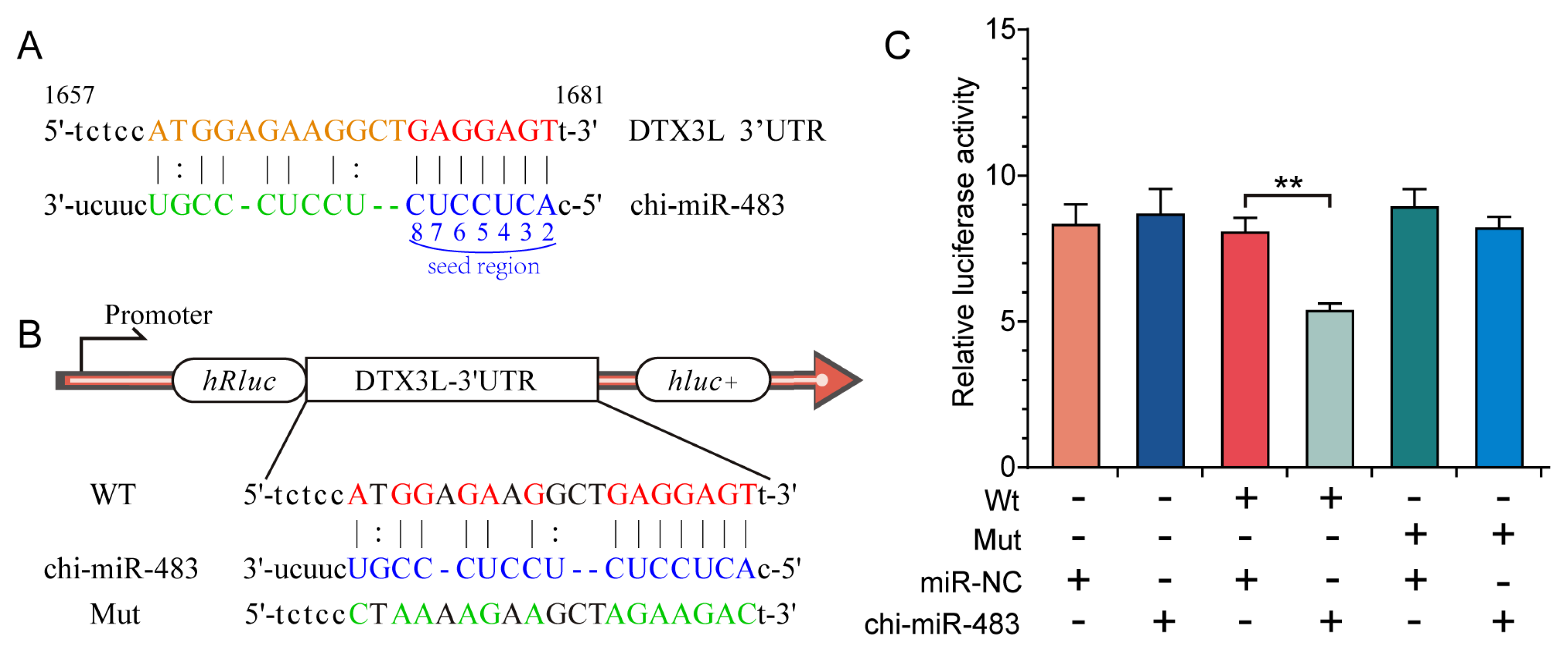
2.10. Dual-Luciferase Reporter Assay
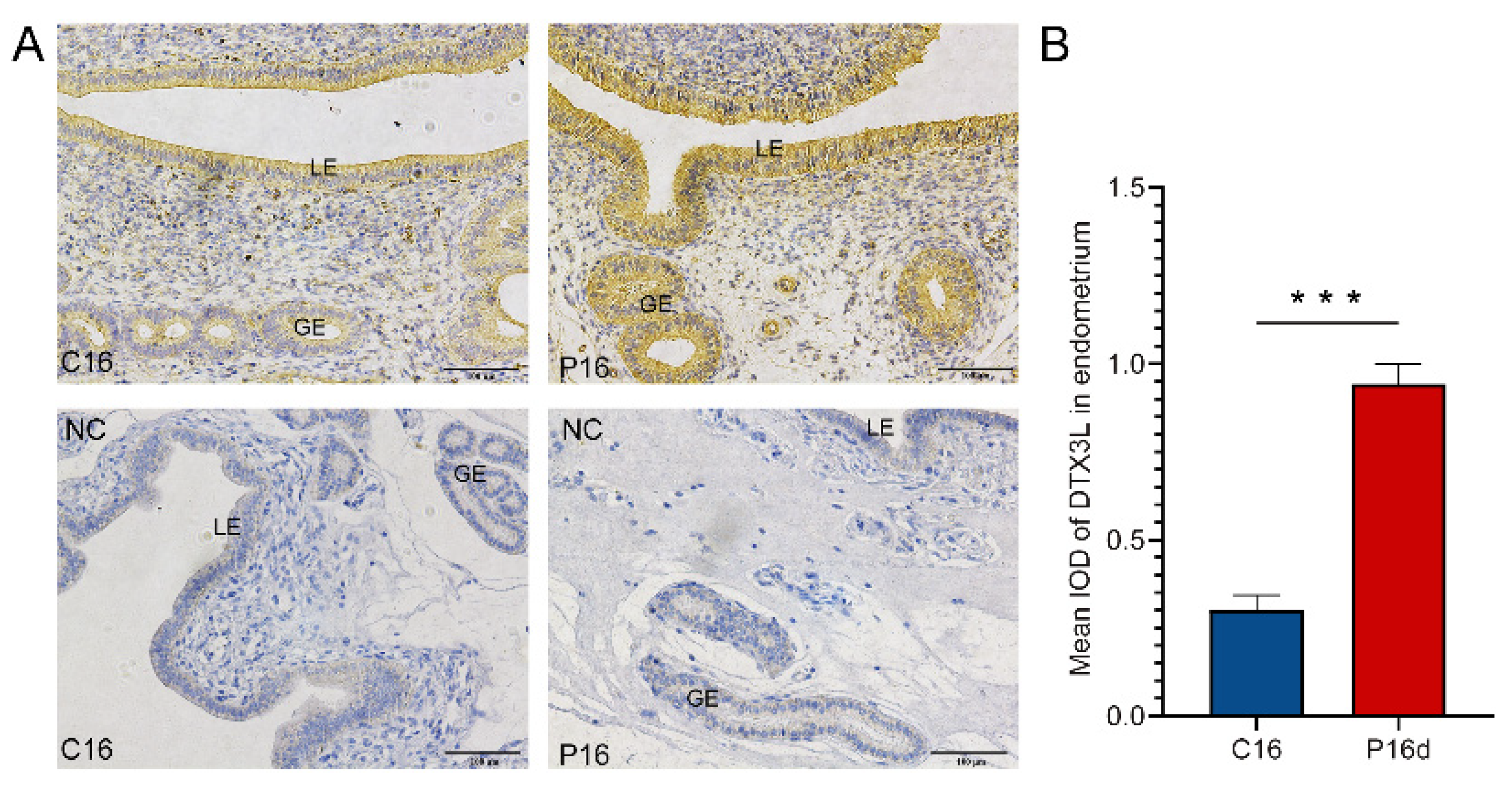
2.11. IHC Analysis
2.12. Statistical Analysis
3. Results
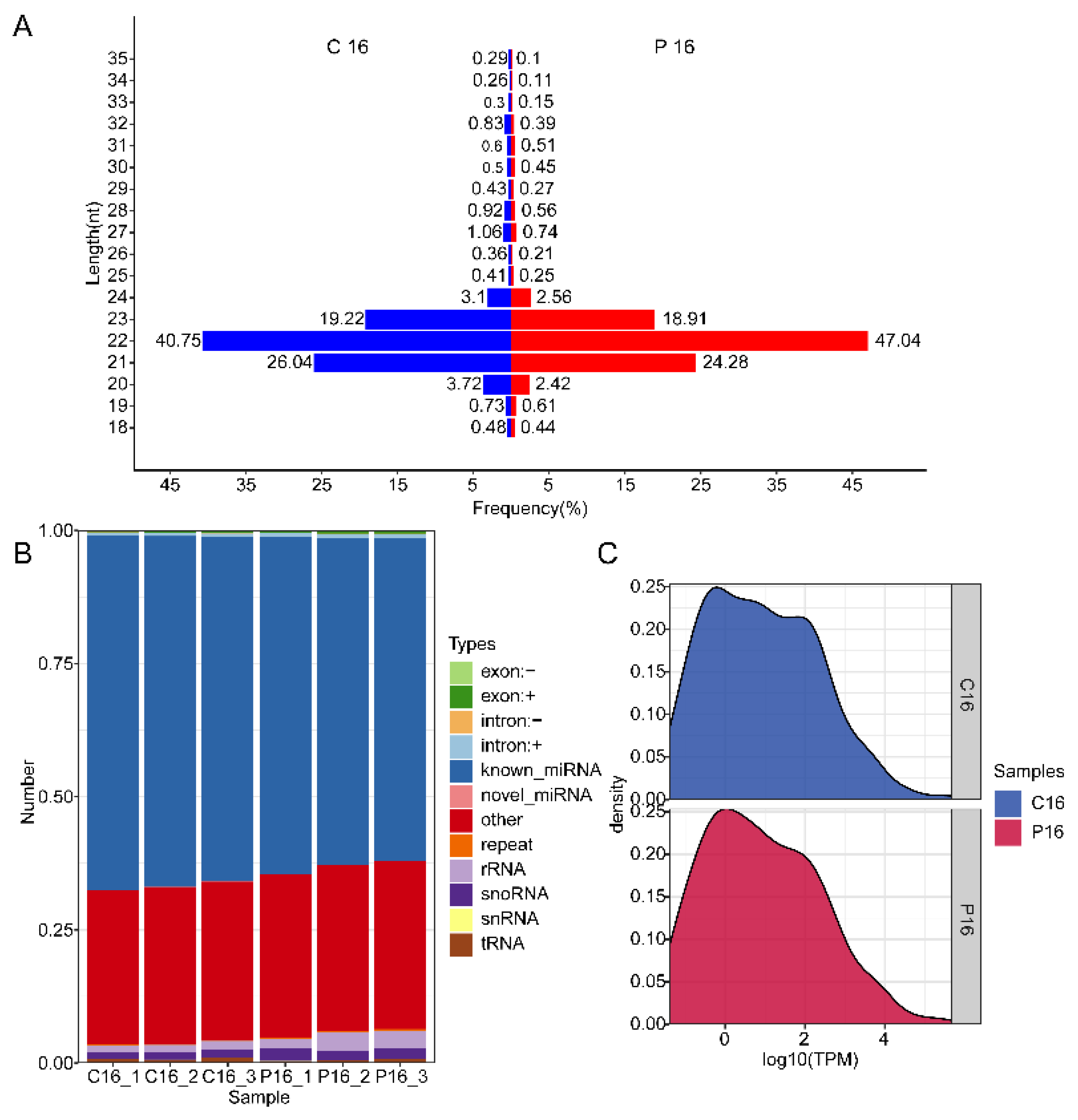
3.1. Overview of the Sequencing Data
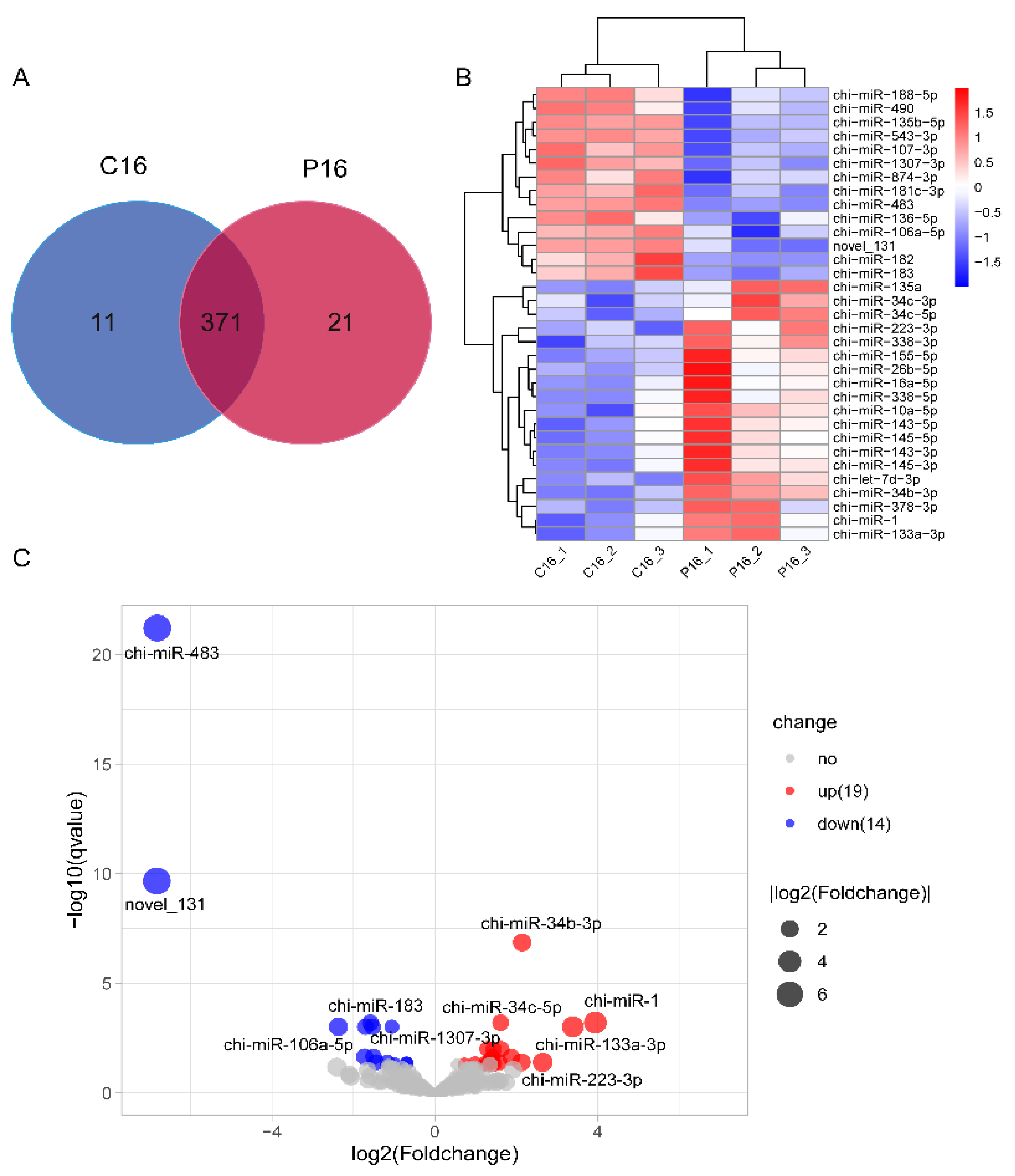
3.2. Analysis of Differentially Expressed miRNAs in C16 and P16 Endometrial Samples
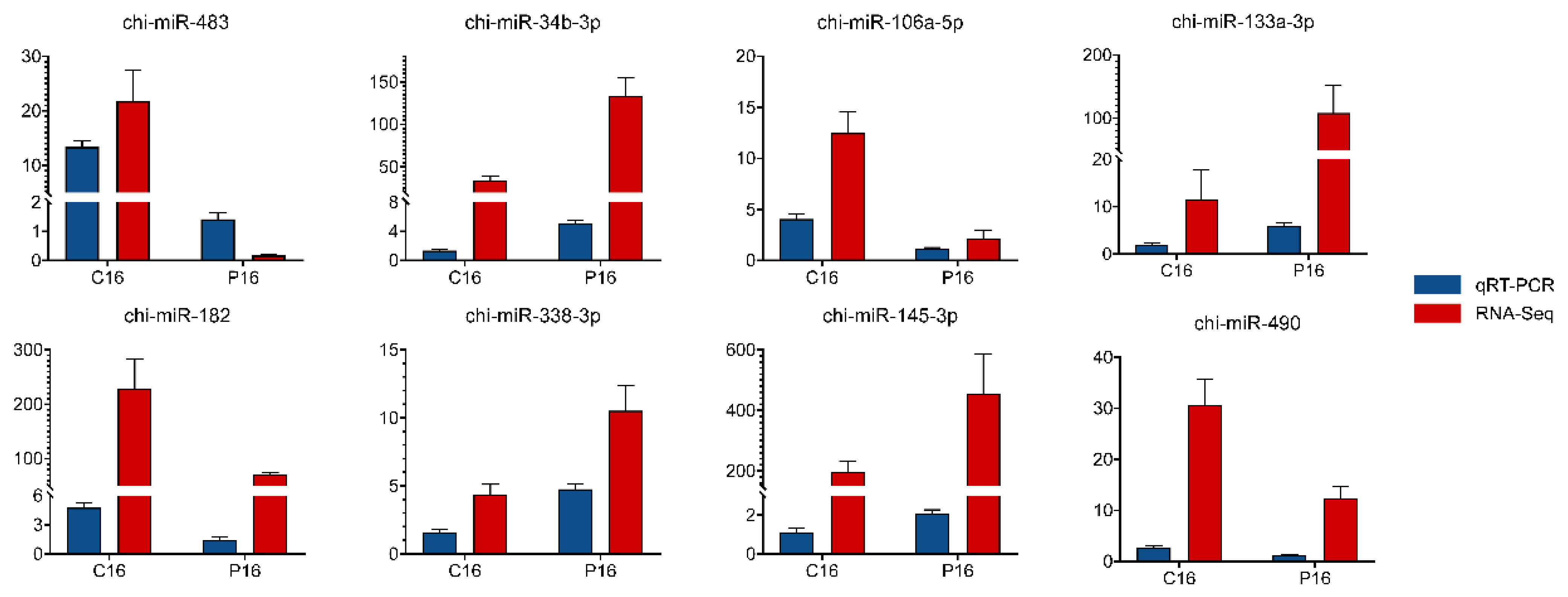
3.3. Validation of Sequencing Results by qRT-PCR
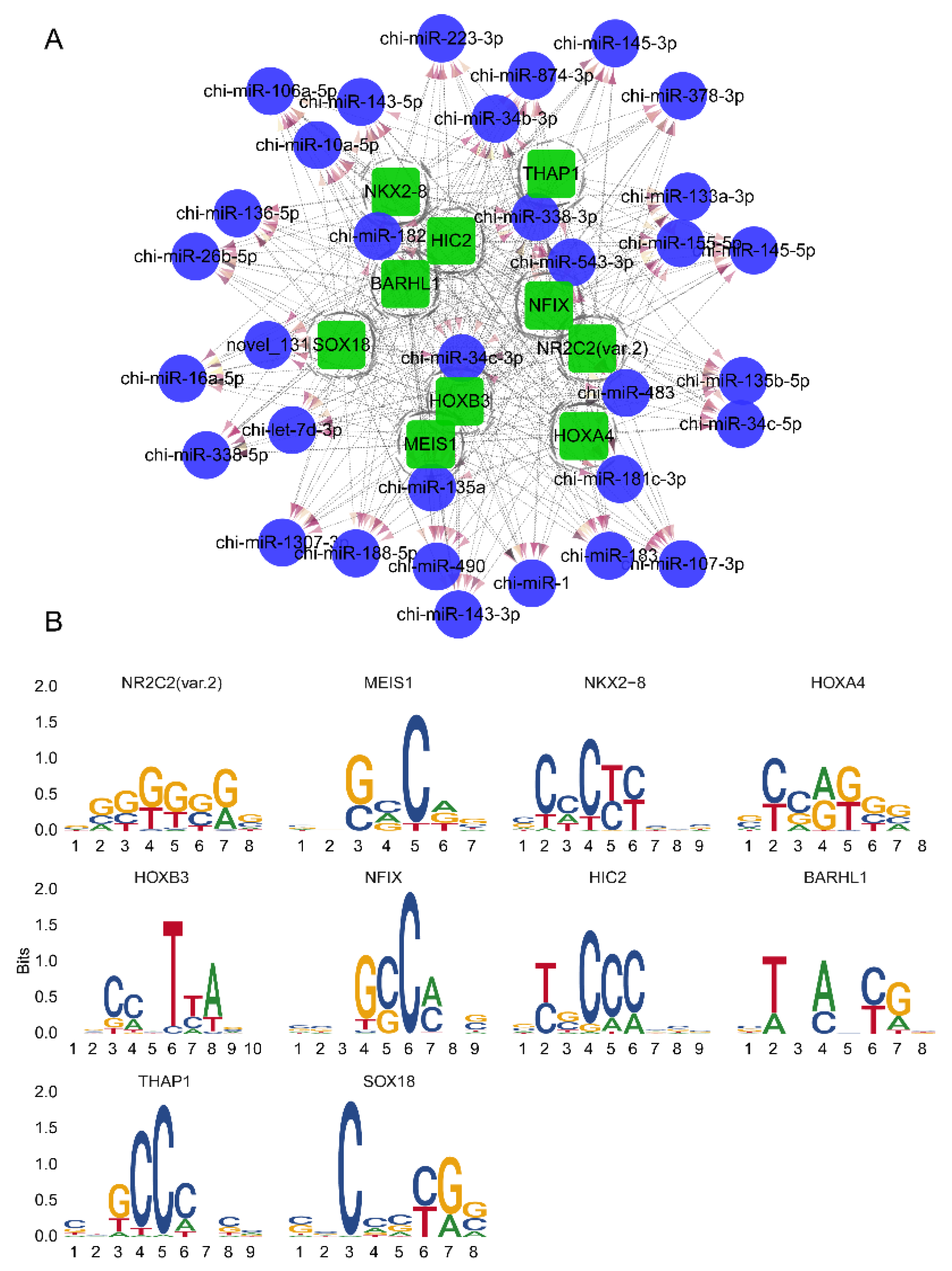
3.4. Potential Transcription Factors Prediction for DEMs
3.5. Target Gene Predictions for DEMs
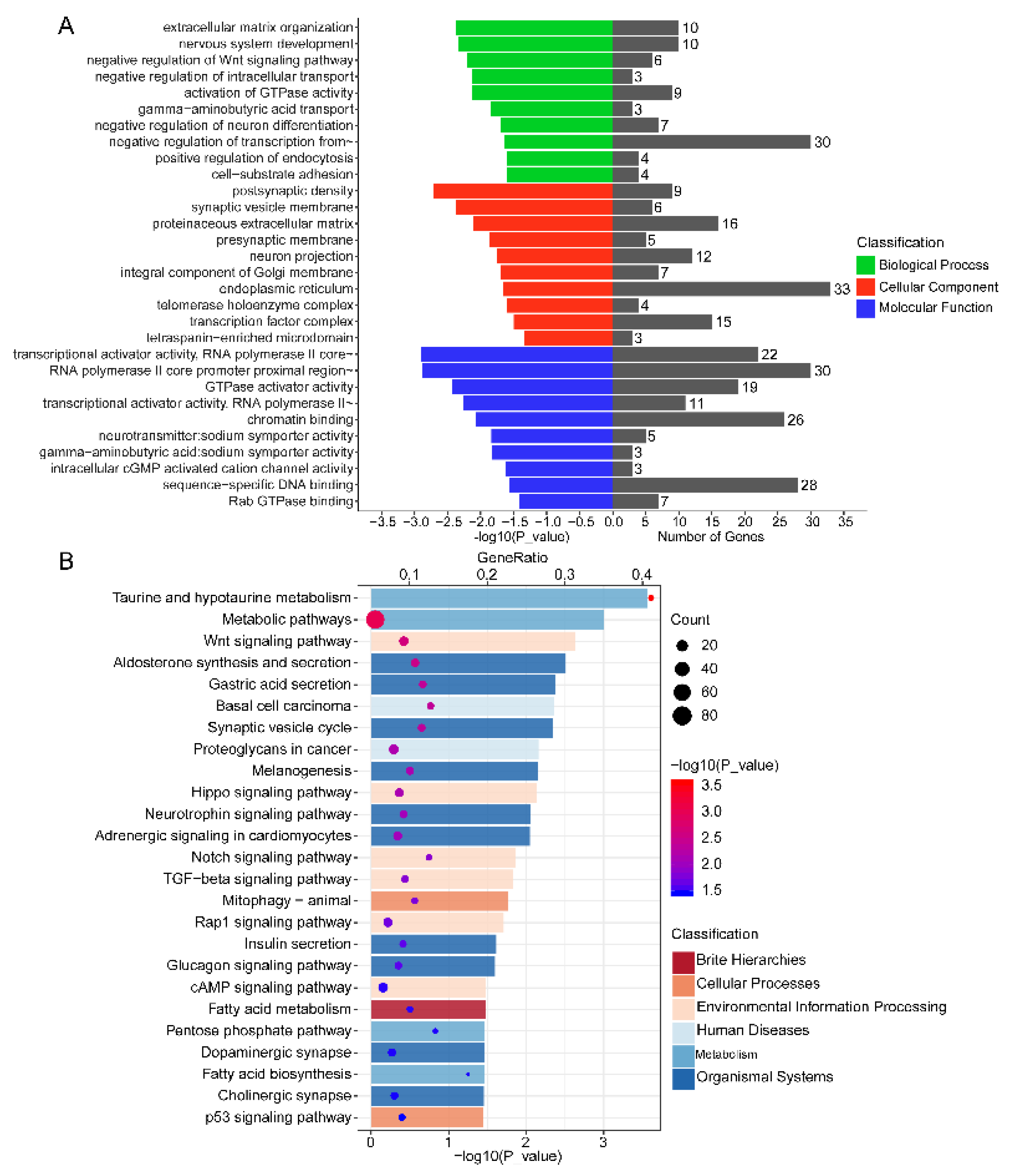
3.6. Functional Annotation of DEMs in Endometrium
3.7. Chi-miR-483 Can Directly Target the 3′-UTR of DTX3L to Reduce the Expression Level of DTX3L
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guillomot, M. Cellular interactions during implantation in domestic ruminants. J. Reprod. Fertil. Suppl. Only 1995, 49, 39–52. [Google Scholar] [CrossRef]
- Geisert, R.; Schmitt, R. Early embryonic survival in the pig: Can it be improved? J. Anim. Sci. 2002, 80, E54–E65. [Google Scholar]
- Simmons, D.G.; Kennedy, T.G. Uterine sensitization-associated gene-1: A novel gene induced within the rat endometrium at the time of uterine receptivity/sensitization for the decidual cell reaction. Biol. Reprod. 2002, 67, 1638–1645. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Luo, X.; Toloubeydokhti, T.; Chegini, N. The expression profile of micro-RNA in endometrium and endometriosis and the influence of ovarian steroids on their expression. Mol. Hum. Reprod. 2007, 13, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Altmäe, S.; Martinez-Conejero, J.A.; Esteban, F.J.; Ruiz-Alonso, M.; Stavreus-Evers, A.; Horcajadas, J.A.; Salumets, A. MicroRNAs miR-30b, miR-30d, and miR-494 regulate human endometrial receptivity. Reprod. Sci. 2013, 20, 308–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miravet-Valenciano, J.A.; Rincon-Bertolin, A.; Vilella, F.; Simon, C. Understanding and improving endometrial receptivity. Curr. Opin. Obstet. Gynecol. 2015, 27, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Igwebuike, U. A review of uterine structural modifications that influence conceptus implantation and development in sheep and goats. Anim. Reprod. Sci. 2009, 112, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.; Laufer, N. Repeated implantation failure: Clinical approach. Fertil. Steril. 2012, 97, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Latreille, M.; Hausser, J.; Stützer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L. MicroRNA-7a regulates pancreatic β cell function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.R.; Georges, S.A.; Seay, H.R.; Tapscott, S.J.; McManus, M.T.; Goldhamer, D.J.; Swanson, M.S.; Harfe, B.D. Essential role for Dicer during skeletal muscle development. Dev. Biol. 2007, 311, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, R.J.; Egan, B.; Katayama, M.; Zierath, J.R.; Krook, A. Temporal analysis of reciprocal miRNA-mRNA expression patterns predicts regulatory networks during differentiation in human skeletal muscle cells. Physiol. Genom. 2015, 47, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Zhang, D.; Yang, Y.; Cui, X.; Sun, J.; Liang, C.; Qin, H.; Yang, X.; Liu, S.; Yan, Q. MicroRNA-200c impairs uterine receptivity formation by targeting FUT4 and α 1,3-fucosylation. Cell Death Differ. 2017, 24, 2161–2172. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, X.; Liu, J.; Ma, X.; Zhou, Z.; Song, Y.; Cao, B. MiR-26a promoted endometrial epithelium cells (EECs) proliferation and induced stromal cells (ESCs) apoptosis via the PTEN-PI3K/AKT pathway in dairy goats. J. Cell. Physiol. 2018, 233, 4688–4706. [Google Scholar] [CrossRef] [PubMed]
- Ponsuksili, S.; Tesfaye, D.; Schellander, K.; Hoelker, M.; Hadlich, F.; Schwerin, M.; Wimmers, K. Differential expression of miRNAs and their target mRNAs in endometria prior to maternal recognition of pregnancy associates with endometrial receptivity for in vivo-and in vitro-produced bovine embryos. Biol. Reprod. 2014, 91, 135. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.; Lewis, S.; Avendano, J.; Williams, E.; Ribeiro, F.; Nuti, L.; Foxworth, W.; Ing, N. Fucosyltransferase gene expression in goat endometrium during the estrous cycle and early pregnancy. Theriogenology 2019, 132, 118–127. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; Van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Davidson, E.H. Genomic Regulatory Systems: In Development and Evolution; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Chien, C.-H.; Chiang-Hsieh, Y.-F.; Tsou, A.-P.; Weng, S.-L.; Chang, W.-C.; Huang, H.-D. Large-scale investigation of human TF-miRNA relations based on coexpression profiles. BioMed Res. Int. 2014, 2014, 623078. [Google Scholar] [CrossRef]
- Tan, G.; Lenhard, B. TFBSTools: An R/bioconductor package for transcription factor binding site analysis. Bioinformatics 2016, 32, 1555–1556. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [Green Version]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R. Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Lu, T.; Zhao, Z.; Liu, G.; Lian, Z.; Guo, Y.; Sun, B.; Liu, D.; Li, Y. Comprehensive analysis of mRNAs and miRNAs in the ovarian follicles of uniparous and multiple goats at estrus phase. BMC Genom. 2020, 21, 267. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.R.; Vallet, J.L.; Freking, B.A.; Nonneman, D.J. Molecular cloning and characterisation of heparanase mRNA in the porcine placenta throughout gestation. Reprod. Fertil. Dev. 2009, 21, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, L.; Hou, C.; Li, X.; Li, C.; Zhao, S.; Yu, M. Expression of heparanase is associated with breed-specific morphological characters of placental folded bilayer between Yorkshire and Meishan pigs. Biol. Reprod. 2014, 90, 56. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- Lu, J.; Clark, A.G. Impact of microRNA regulation on variation in human gene expression. Genome Res. 2012, 22, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Revel, A.; Achache, H.; Stevens, J.; Smith, Y.; Reich, R. MicroRNAs are associated with human embryo implantation defects. Hum. Reprod. 2011, 26, 2830–2840. [Google Scholar] [CrossRef]
- Liu, W.; Niu, Z.; Li, Q.; Pang, R.T.; Chiu, P.C.; Yeung, W.S.B. Micro RNA and embryo implantation. Am. J. Reprod. Immunol. 2016, 75, 263–271. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chen, Z.; Martin, M.; Zhang, J.; Sangwung, P.; Woo, B.; Tremoulet, A.H.; Shimizu, C.; Jain, M.K.; Burns, J.C. miR-483 targeting of CTGF suppresses endothelial-to-mesenchymal transition: Therapeutic implications in Kawasaki disease. Circ. Res. 2017, 120, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Yang, Z.; Dong, D.; Xu, J.; Wang, J.; Li, H.; Huang, Y.; Lan, X.; Lei, C.; Ma, Y. miR-483 inhibits bovine myoblast cell proliferation and differentiation via IGF1/PI3K/AKT signal pathway. J. Cell. Physiol. 2019, 234, 9839–9848. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Zhang, H.; Wang, G. MiR-483 suppresses cell proliferation and promotes cell apoptosis by targeting SOX3 in breast cancer. Eur. Rev. Med. Pharm. Sci. 2019, 23, 2069–2074. [Google Scholar]
- Yu, F.; Zhou, C.; Liu, Y.; Wang, B.; Mao, L.; Li, Y. miR-483 is down-regulated in gastric cancer and suppresses cell proliferation, invasion and protein O-GlcNAcylation by targeting OGT. Neoplasma 2018, 65, 406. [Google Scholar] [CrossRef] [Green Version]
- Niu, Z.-Y.; Li, W.-L.; Jiang, D.-L.; Li, Y.-S.; Xie, X.-J. Mir-483 inhibits colon cancer cell proliferation and migration by targeting TRAF1. Kaohsiung J. Med. Sci. 2018, 34, 479–486. [Google Scholar] [CrossRef]
- Xu, P.; Tao, X.; Zhao, C.; Huang, Q.; Chang, H.; Ban, N.; Bei, Y.; Xia, X.; Shen, C.; Wang, K. DTX3L is upregulated in glioma and is associated with glioma progression. Int. J. Mol. Med. 2017, 40, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Bauersachs, S.; Ulbrich, S.E.; Einspanier, R.; Meyer, H.H.; Schmidt, S.E.; Reichenbach, H.-D.; Vermehren, M.; Sinowatz, F.; Blum, H. Monozygotic twin model reveals novel embryo-induced transcriptome changes of bovine endometrium in the preattachment period. Biol. Reprod. 2006, 74, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.-Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, R13. [Google Scholar] [CrossRef] [Green Version]
- Wüst, S.; Dröse, S.; Heidler, J.; Wittig, I.; Klockner, I.; Franko, A.; Bonke, E.; Günther, S.; Gärtner, U.; Boettger, T. Metabolic maturation during muscle stem cell differentiation is achieved by miR-1/133a-mediated inhibition of the Dlk1-Dio3 mega gene cluster. Cell Metab. 2018, 27, 1026–1039.e1026. [Google Scholar] [CrossRef] [Green Version]
- Besser, J.; Malan, D.; Wystub, K.; Bachmann, A.; Wietelmann, A.; Sasse, P.; Fleischmann, B.K.; Braun, T.; Boettger, T. MiRNA-1/133a clusters regulate adrenergic control of cardiac repolarization. PLoS ONE 2014, 9, e113449. [Google Scholar]
- Mullany, L.E.; Herrick, J.S.; Wolff, R.K.; Stevens, J.R.; Samowitz, W.; Slattery, M.L. MicroRNA-transcription factor interactions and their combined effect on target gene expression in colon cancer cases. Genes Chromosomes Cancer 2018, 57, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Taylor, H.S. The role of HOX genes in human implantation. Hum. Reprod. Update 2000, 6, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Troy, P.J.; Daftary, G.S.; Bagot, C.N.; Taylor, H.S. Transcriptional repression of peri-implantation EMX2 expression in mammalian reproduction by HOXA10. Mol. Cell. Biol. 2003, 23, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botti, G.; De Chiara, A.; Di Bonito, M.; Cerrone, M.; Malzone, M.G.; Collina, F.; Cantile, M. Noncoding RNAs within the HOX gene network in tumor pathogenesis and progression. J. Cell. Physiol. 2019, 234, 395–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Guillomot, M. Changes in extracellular matrix components and cytokeratins in the endometrium during goat implantation. Placenta 1999, 20, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.P.; Redmer, D.A. Growth and microvascular development of the uterus during early pregnancy in ewes. Biol. Reprod. 1992, 47, 698–708. [Google Scholar] [CrossRef]
- Salamonsen, L.; Nagase, H.; Woolley, D. Matrix metalloproteinases and their tissue inhibitors at the ovine trophoblast-uterine interface. J. Reprod. Fertil. Suppl. Only 1995, 49, 29–38. [Google Scholar] [CrossRef]
- Gillingham, A.K.; Bertram, J.; Begum, F.; Munro, S. In vivo identification of GTPase interactors by mitochondrial relocalization and proximity biotinylation. Elife 2019, 8, e45916. [Google Scholar] [CrossRef]
- Scheffzek, K.; Shivalingaiah, G. Ras-Specific GTPase-Activating Proteins—Structures, Mechanisms, and Interactions. Cold Spring Harb. Perspect. Med. 2019, 9, a031500. [Google Scholar] [CrossRef]
- Dunlap, K.A.; Filant, J.; Hayashi, K.; Rucker, E.B., III; Song, G.; Deng, J.M.; Behringer, R.R.; DeMayo, F.J.; Lydon, J.; Jeong, J.-W. Postnatal deletion of Wnt7a inhibits uterine gland morphogenesis and compromises adult fertility in mice. Biol. Reprod. 2011, 85, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.L.; Kaspar, P.; Brunet, L.J.; Bhatt, H.; Gadi, I.; Köntgen, F.; Abbondanzo, S.J. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 1992, 359, 76–79. [Google Scholar] [CrossRef]
- Li, Q.; Kannan, A.; Wang, W.; DeMayo, F.J.; Taylor, R.N.; Bagchi, M.K.; Bagchi, I.C. Bone morphogenetic protein 2 functions via a conserved signaling pathway involving Wnt4 to regulate uterine decidualization in the mouse and the human. J. Biol. Chem. 2007, 282, 31725–31732. [Google Scholar] [CrossRef] [Green Version]
- Chow, R.; Wessels, J.; Foster, W. Brain-derived neurotrophic factor (BDNF) expression and function in the mammalian reproductive Tract. Hum. Reprod. Update 2020, 26, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.J.; Lessey, B.A. Embryo implantation and tumor metastasis: Common pathways of invasion and angiogenesis. In Seminars in Reproductive Endocrinology; Thieme Medical Publishers: New York, NY, USA, 1999; pp. 275–290. [Google Scholar]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Lupse, B.; Maedler, K. Hippo signaling: Key emerging pathway in cellular and whole-body metabolism. Trends Endocrinol. Metab. 2018, 29, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Strakova, Z.; Reed, J.; Ihnatovych, I. Human transcriptional coactivator with PDZ-binding motif (TAZ) is downregulated during decidualization. Biol. Reprod. 2010, 82, 1112–1118. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads | N% > 10% | Low Quality | 5′ Adapter Contaminant | 3′ Adapter Null or Insert Null | With Poly A/T/G/C | Clean Reads |
|---|---|---|---|---|---|---|---|
| C16 | 12,151,788 | 18 | 11,569 | 679 | 151,535 | 21,570 | 11,966,417 |
| (100.00%) | (0.00%) | (0.09%) | (0.01%) | (1.25%) | (0.18%) | (98.47%) | |
| P16 | 13,520,599 | 52 | 15,103 | 1628 | 265,879 | 10,864 | 13,227,072 |
| (100%) | (0.00%) | (0.11%) | (0.01%) | (1.97%) | (0.08%) | (97.83%) |
| miRNA | Average TPM in C16 | miRNA | Average TPM in P16 |
|---|---|---|---|
| chi-miR-148a-3p | 470,173 | chi-miR-148a-3p | 358,978 |
| chi-miR-99a-5p | 243,974 | chi-miR-99a-5p | 239,837 |
| chi-miR-143-3p | 46,655 | chi-miR-143-3p | 110,970 |
| chi-miR-21-5p | 38,853 | chi-miR-26a-5p | 53,046 |
| chi-miR-26a-5p | 33,294 | chi-miR-21-5p | 37,842 |
| chi-miR-126-3p | 19,458 | chi-miR-10b-5p | 20,501 |
| chi-miR-10b-5p | 15,653 | chi-miR-126-3p | 17,344 |
| chi-let-7i-5p | 9218 | chi-let-7f-5p | 12,137 |
| chi-let-7g-5p | 7995 | chi-let-7g-5p | 11,049 |
| chi-let-7f-5p | 7944 | chi-let-7i-5p | 10,918 |
| chi-miR-199a-3p | 6749 | chi-let-7a-5p | 7798 |
| chi-let-7a-5p | 5646 | chi-miR-27b-3p | 7696 |
| chi-miR-200a | 5589 | chi-miR-1 | 7249 |
| chi-let-7c-5p | 5033 | chi-miR-199a-3p | 6988 |
| chi-let-7b-5p | 4965 | chi-let-7c-5p | 6668 |
| chi-miR-27b-3p | 4601 | chi-let-7b-5p | 6219 |
| chi-miR-151-3p | 4501 | chi-miR-10a-5p | 5917 |
| chi-miR-200b | 4439 | chi-miR-151-3p | 5227 |
| chi-miR-125b-5p | 3800 | chi-miR-200a | 5189 |
| chi-miR-10a-5p | 3327 | chi-miR-200b | 4371 |
| miRNA | Average Readcount in C16 | Average Readcount in P16 | log2 Fold-Change | p-Value | q-Value |
|---|---|---|---|---|---|
| novel_131 | 39.85 | 0.35 | −6.83 | 1.29 × 10−12 | 2.18 × 10−10 |
| chi-miR-483 | 152.43 | 1.35 | −6.82 | 1.83 × 10−24 | 6.16 × 10−22 |
| chi-miR-106a-5p | 87.16 | 16.88 | −2.37 | 2.71 × 10−5 | 9.50 × 10−4 |
| chi-miR-188-5p | 39.37 | 11.93 | −1.72 | 1.12 × 10−3 | 2.21 × 10−2 |
| chi-miR-135b-5p | 51.37 | 15.79 | −1.70 | 2.16 × 10−5 | 9.50 × 10−4 |
| chi-miR-183 | 932.05 | 311.26 | −1.58 | 9.34 × 10−6 | 6.13 × 10−4 |
| chi-miR-182 | 1595.76 | 553.28 | −1.53 | 3.13 × 10−5 | 9.58 × 10−4 |
| chi-miR-181c-3p | 32.80 | 11.56 | −1.50 | 1.03 × 10−3 | 2.17 × 10−2 |
| chi-miR-874-3p | 40.21 | 14.78 | −1.44 | 2.94 × 10−3 | 3.96 × 10−2 |
| chi-miR-490 | 212.90 | 94.84 | −1.17 | 2.57 × 10−3 | 3.94 × 10−2 |
| chi-miR-1307-3p | 454.00 | 218.84 | −1.05 | 2.82 × 10−5 | 9.50 × 10−4 |
| chi-miR-136-5p | 1387.52 | 693.62 | −1.00 | 4.80 × 10−3 | 4.90 × 10−2 |
| chi-miR-543-3p | 441.79 | 272.33 | −0.70 | 3.46 × 10−3 | 4.32 × 10−2 |
| chi-miR-107-3p | 457.79 | 284.97 | −0.68 | 3.46 × 10−3 | 4.32 × 10−2 |
| chi-miR-135a | 269.47 | 446.58 | 0.73 | 4.78 × 10−3 | 4.90 × 10−2 |
| chi-miR-10a-5p | 23116.13 | 45875.93 | 0.99 | 3.90 × 10−3 | 4.53 × 10−2 |
| chi-miR-26b-5p | 9990.30 | 23746.46 | 1.25 | 3.82 × 10−3 | 4.53 × 10−2 |
| chi-let-7d-3p | 31.10 | 75.94 | 1.29 | 3.38 × 10−4 | 9.49 × 10−3 |
| chi-miR-16a-5p | 1380.89 | 3376.81 | 1.29 | 4.57 × 10−3 | 4.90 × 10−2 |
| chi-miR-145-3p | 1364.07 | 3536.32 | 1.37 | 1.51× 10−3 | 2.68 × 10−2 |
| chi-miR-143-3p | 325393.81 | 862304.74 | 1.41 | 2.13 × 10−3 | 3.59 × 10−2 |
| chi-miR-338-3p | 30.42 | 81.38 | 1.42 | 9.74 × 10−4 | 2.17 × 10−2 |
| chi-miR-378-3p | 1175.13 | 3190.20 | 1.44 | 4.14 × 10−4 | 9.97 × 10−3 |
| chi-miR-143-5p | 1553.50 | 4241.32 | 1.45 | 4.04 × 10−3 | 4.54 × 10−2 |
| chi-miR-145-5p | 7862.77 | 23587.26 | 1.58 | 2.81 × 10−3 | 3.96 × 10−2 |
| chi-miR-34c-5p | 1040.08 | 3191.46 | 1.62 | 9.38 × 10−6 | 6.13 × 10−4 |
| chi-miR-155-5p | 297.21 | 916.87 | 1.63 | 4.04 × 10−4 | 9.97 × 10−3 |
| chi-miR-34c-3p | 19.36 | 71.16 | 1.88 | 1.27 × 10−3 | 2.38 × 10−2 |
| chi-miR-338-5p | 12.77 | 55.77 | 2.13 | 2.84 × 10−3 | 3.96 × 10−2 |
| chi-miR-34b-3p | 233.76 | 1032.78 | 2.14 | 1.23 × 10−9 | 1.38 × 10−7 |
| chi-miR-223-3p | 1.88 | 11.77 | 2.65 | 2.54 × 10−3 | 3.94 × 10−2 |
| chi-miR-133a-3p | 80.34 | 842.29 | 3.39 | 2.71 × 10−5 | 9.50 × 10−4 |
| chi-miR-1 | 3678.63 | 56458.03 | 3.94 | 1.09 × 10−5 | 6.13 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zang, X.; Zhou, C.; Wang, W.; Gan, J.; Li, Y.; Liu, D.; Liu, G.; Hong, L. Differential MicroRNA Expression Involved in Endometrial Receptivity of Goats. Biomolecules 2021, 11, 472. https://doi.org/10.3390/biom11030472
Zang X, Zhou C, Wang W, Gan J, Li Y, Liu D, Liu G, Hong L. Differential MicroRNA Expression Involved in Endometrial Receptivity of Goats. Biomolecules. 2021; 11(3):472. https://doi.org/10.3390/biom11030472
Chicago/Turabian StyleZang, Xupeng, Chen Zhou, Wenjing Wang, Jianyu Gan, Yaokun Li, Dewu Liu, Guangbin Liu, and Linjun Hong. 2021. "Differential MicroRNA Expression Involved in Endometrial Receptivity of Goats" Biomolecules 11, no. 3: 472. https://doi.org/10.3390/biom11030472