1. Introduction
Inherited metabolic disorders (IMDs) are genetic diseases causing the impaired function of enzymes or transporters involved in intermediary metabolism. The mutational spectrum of the majority of IMDs is dominated by missense mutations (50–80%), frequently leading to protein variants with deficient catalytic activity or impaired folding. The latter results in premature protein degradation and consequently, lower cellular protein steady-state levels. Attractive therapeutic approaches for these conditions are based on the use of small molecules acting as “pharmacological chaperones” rescuing variant proteins’ folding, or as “activity chaperones” protecting enzyme activity [
1,
2]. Small molecules acting as pharmacological chaperones are already approved for the treatment of genetic diseases associated with protein misfolding such as Sapropterin dihydrochloride (Kuvan) from Biomarin (phenylketonuria), Tafamidis, Pfizer (specific TTR variants responsible for familial amyloid polyneuropathy), Lumacaftor, Orkambi (F508del CFTR in cystic fibrosis) and Migalastat (Galafold) from Amicus Therapeutics (responsive variant forms of α-galactosidase A causing Fabry disease) [
3].
Phenylketonuria (PKU; OMIM 261600) is the most common IMD of amino acid metabolism (1:10,000 newborns) and is caused by mutations in the PAH gene, encoding phenylalanine hydroxylase (hPAH; EC 1.14.16.1). When untreated, PKU can lead to severe psychomotor developmental delay. To date, more than 1000 different mutations have been identified in PAH (BIOPKU database;
http://www.biopku.org; last accessed on 13 October 2020) ≈62% being classified as missense mutations. As such, PKU is a suitable target for the development of activity chaperone-and pharmacological chaperone-based therapies. Functional hPAH assembles as a homotetramer (dimer of dimers) and harbors a non-heme iron in its catalytic site, where hydroxylation of
l-phenylalanine (
l-Phe) into
l-tyrosine (
l-Tyr) takes place in the presence of molecular oxygen and the cofactor (6R)-
l-erythro-5,6,7,8-tetrahydrobiopterin (BH
4) (
Figure 1A).
Each hPAH monomer is organized into three structural domains, namely an N-terminal regulatory domain, a catalytic domain, and a C-terminal tetramerization domain. The active site where
l-Phe and the BH
4 cofactor bind is located in the catalytic domain, in a pocket composed of both hydrophobic and negatively charged residues, where a 2-His-1-carboxylate facial triad (His285, His290 and Glu330) binds to mononuclear ferric iron, which is also coordinated to three water molecules. The enzyme alternates between a so-called ‘resting’ state, where the N-terminal regulatory domain covers the active site and leads to lower substrate affinity and lower catalytic activity, and an ‘activated’ state where a rotating displacement of this domain facilitates
l-Phe access to the active site leading to an enzyme with higher catalytic activity. High levels of circulating substrate stabilize the ‘activated’ state by the allosteric
l-Phe binding to the interface established by the dimerization of the regulatory domains of adjacent monomers [
4,
5,
6,
7,
8]. In the absence of
l-Phe, BH
4 binding has an inhibitory effect resulting from locking the protein in a non-activated state with less mobile regulatory domains, presenting increased resistance to unfolding and degradation [
7,
9,
10]. In fact, this mechanism underlies the rationale to use the synthetic form of BH
4 (sapropterin dihydrochloride) as a pharmacological chaperone that stabilizes some misfolded hPAH variants [
11,
12]. Importantly, despite BH
4 being used in high amounts per dose (20 mg/kg body weight) and patients with more severe PKU phenotypes often being unresponsive to this molecule, the BH
4 mode of action remains a leading strategy in the design of pharmacological chaperones to promote the enzyme stabilization and structural integrity.
Our groups previously reported an alternative strategy to design hPAH modulators based on the
l-Phe structure by preparing and evaluating a series of
l-Phe-iminoboronates that increased hPAH activity by a pre-activation mechanism similar to the one involving
l-Phe [
13]. Interestingly, to our knowledge, the catalytic iron center has never been explored to design hPAH modulators. Thus, given the ability of 3-hydroxyquinolin-2(1H)-one (3HQ) to complex metals [
14], in the present study, we evaluated an in-house library of 3HQ derivatives, recently developed as anticancer agents [
15]. In addition to potentially stabilizing the hPAH catalytic non-heme iron, these compounds contain a BH
4 cofactor-like fused six membered core and offer the possibility of installing
l-Phe-like motifs as substituents to improve affinity and functionality (
Figure 1B). This combination of structural features of both the cofactor and the substrate along with iron coordinative functionalities should provide an excellent scaffold to promote an interaction specifically directed to the protein’s catalytic domain. We rationalized that this approach is likely to favor the discovery of compounds stabilizing the protein structure (pharmacological chaperones) and/or protecting enzyme activity (activity chaperones), that ultimately sustains cell function. These compounds should bind with enough affinity to ensure specificity without preventing
l-Phe binding. Herein, we describe the biochemical and biophysical characterization of 3HQ derivatives as pharmacological/activity chaperones of hPAH, as well as their efficiency and toxicity in a cellular context.
2. Materials and Methods
2.1. Compounds Library
The 20 3HQ derivatives (
Figure 2) were synthesized according to protocols already reported by our group [
15] and described in detail in the
Supplementary Material for compounds 1 and 2. The synthesized 3HQ derivatives were solubilized to 10 mM in DMSO and further used at final concentrations of 50 to 200 μM in 0.5 to 2% DMSO.
2.2. Production and Purification of Full-Length and Truncated Recombinant hPAH
Recombinant full-length wild-type (WT) hPAH was expressed in
E. coli Top10 cells in fusion with a hexa-histidyl peptide to allow purification by immobilized metal affinity chromatography (IMAC) using a Ni-NTA agarose (Qiagen; Hilden, Germany) essentially as described previously [
16]. After IMAC purification, hPAH tetramers were isolated by size-exclusion chromatography (SEC) on a HiLoad Superdex 200 HR column (1.6 × 60 cm, GE Healthcare; Chicago, IL, USA) equilibrated with 20 mM Na-HEPES buffer containing 200 mM NaCl, pH 7.0 (SEC buffer), at a flow rate of 0.7 mL/min, at 4 °C (
Supplementary Figure S1, peak 3) [
13].
The truncated form of hPAH corresponding to the N-terminal regulatory domain (hPAH-RD
1–120) was expressed in E. coli TB1 cells in fusion with maltose binding protein (MBP) and a sequence recognized by Factor Xa (FXa) (MBP-FXa-hPAH-RD
1–120) to allow further removal of the MBP tag, as described previously [
17] (
Supplementary Figure S2, peak 2). The MBP-FXa-hPAH-RD
1–120 protein was purified by affinity chromatography using an amylose resin (New England Biolabs; Ipswich, MA, USA). SEC was employed to isolate the hPAH-RD
1–120 dimers as described above. The protein concentrations were determined by the Bradford method, using bovine serum albumin as the standard.
2.3. Enzymatic Activity Assays
The hPAH activity was measured essentially as previously described [
13]. Briefly, the enzymatic reaction was performed in a 200 μL final volume, containing 250 mM Na-HEPES, pH 7, 0.1 mg/mL catalase (Sigma-Aldrich; St. Louis, MO, USA), 5 μg (final concentration of 0.112 μM of tetramer) of recombinant hPAH tetramers and 100 μM (NH
4)
2Fe(II)SO
4. Using 100 μM
l-Phe and 100 μM of tested compound, three experimental conditions were used to study the compounds’ modulatory effect, namely: non activated (NA), 3HQ-activated (3HQ-A) and substrate/3HQ-activated (Phe/3HQ-A) [
13] (
Supplementary Figure S3). The reaction was started by addition of 75 μM BH
4 (Schircks Laboratories; Bauma, Switzerland) prepared in 5 mM ascorbic acid. In the NA assay, the
l-Phe substrate and tested compounds were added together with 75 µM BH
4 at the start of the hydroxylation reaction. In the 3HQ-A condition, hPAH was pre-incubated with the tested compound for 4 min at 25 °C and the reaction was started by the addition of BH
4 together with
l-Phe. In the Phe/3HQ-A condition, hPAH was pre-incubated simultaneously with
l-Phe and the tested compound (4 min, 25 °C). Appropriate control reactions were performed where the compounds were omitted and 1% DMSO was used. Kinetic parameters were determined using variable concentrations of
l-Phe (25 to 2500 μM) and fixed concentrations of BH
4 (75 μM) and tested compound (100 μM). After a 1-min incubation, the reaction was stopped, and the amount of
l-Tyr produced was quantified by HPLC as previously described [
13]. The specific hPAH enzymatic activity is expressed as nmol of
l-Tyr produced during 1 min per mg of protein (nmol
l-Tyr.min
−1.mg
−1). The steady-state kinetic data were analyzed by nonlinear regression analysis using GraphPad Prism (La Jolla, CA, USA). Data was fitted to the modified Hill equation accounting for cooperative substrate binding as well as substrate inhibition [
18] (Equation (1)), to the modified Michaelis–Menten equation which accounts for substrate inhibition (Equation (2)) and to the non-modified Michaelis–Menten equation:
The hPAH time-dependent loss of activity at a fixed temperature was assessed in a period of 120 min, in the absence and presence of compounds. In these assays, the enzymatic reaction was performed essentially as described above for the Phe/3HQ-A condition. Compounds (100 μM) were pre-incubated with the enzyme at 42 °C. After a 30, 45, 60, 90 and 120 min period, an aliquot of the protein (5 μg) was incubated, at 37 °C, with 100 μM
l-Phe for 4 min, and the reaction proceeded as above at 37 °C [
19].
For the above-described activity assays, three independent assays were always performed.
2.4. Differential Scanning Fluorimetry
Thermal denaturation profiles of recombinant hPAH were obtained by differential scanning fluorimetry (DSF) in a C1000 Touch thermal cycler equipped with a CFX96 optical reaction module (Bio-Rad; Hercules, CA, USA) as previously described [
13]. Briefly, DSF assays (
n = 3) were performed using 100 μg/mL hPAH tetramers (final concentration of 0.45 μM of tetramer), SYPRO Orange (5000-fold commercially available stock solution; Invitrogen; Carlsbad, CA, USA) at a 2.5-x final concentration and ramping the temperature from 20 to 90 °C, at 1 °C/min. Fluorescence data were acquired using the FRET channel. Data were processed using CFX Manager Software V3.0 (Bio-Rad) and the GraphPad Prism. Temperature scan curves were fitted to a biphasic dose–response function and the
Tm values were obtained from the midpoint of the first (
Tm1) and second transitions (
Tm2) corresponding, respectively, to the thermal denaturation of the regulatory and catalytic domains [
20,
21]. The
Tm values in the presence of 3HQ were compared to those obtained in the presence of 1% DMSO (vehicle control) and a change in the
Tm was considered significant when the |Δ
Tm| ≥ 2 °C [
22].
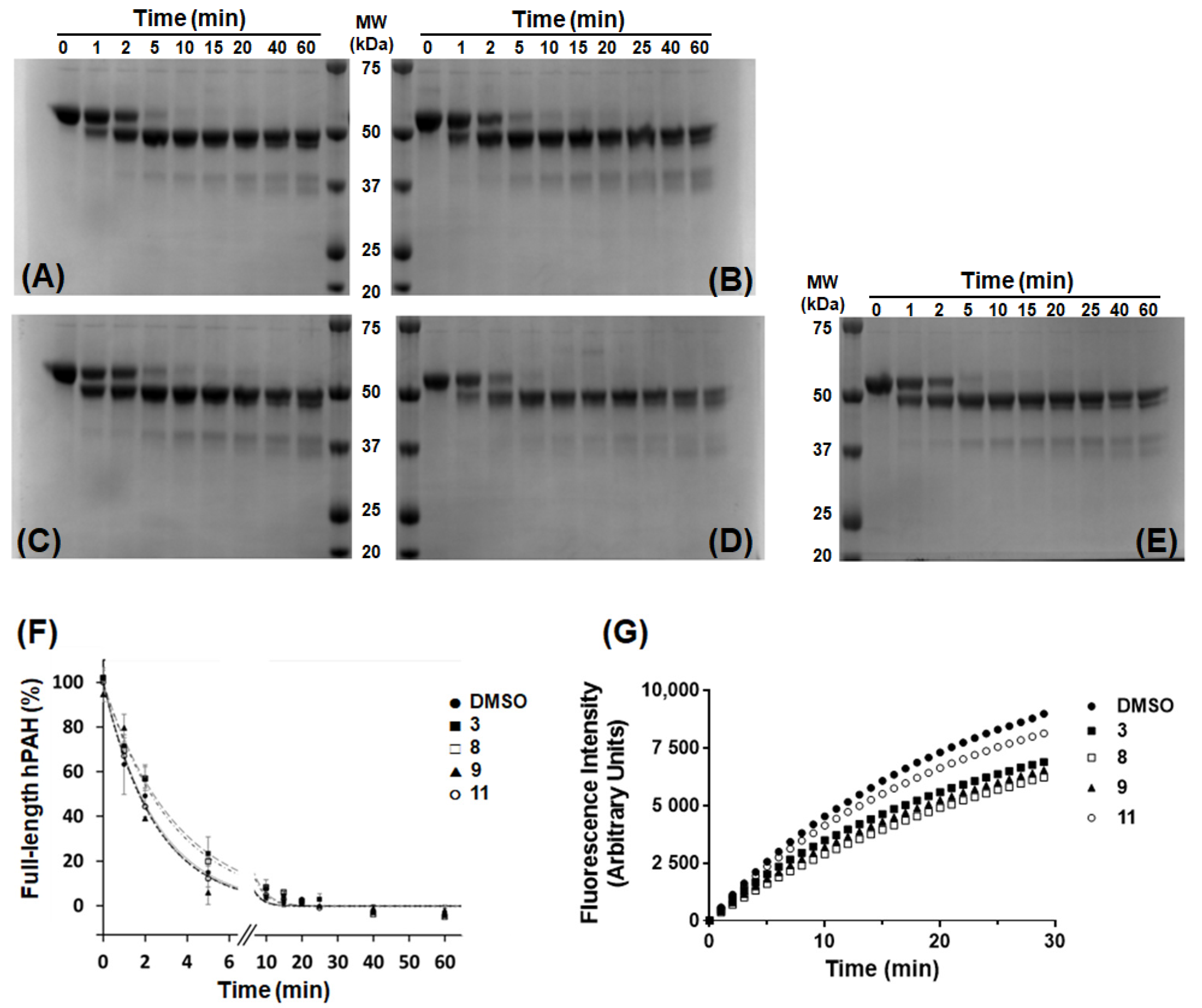
2.5. Limited Proteolysis by Trypsin
Limited proteolysis of purified hPAH was performed as previously described [
23], at 25 °C in SEC buffer and with a trypsin (Sigma-Aldrich) to hPAH ratio of 1:200 (by mass; final hPAH protein concentration 300 μg/mL), in the presence of 100 µM of 3HQ derivatives or 1% DMSO. Control assays were also performed in the presence of 100 μM and 1 mM
l-Phe. At different time points (0 to 60 min), aliquots of the reaction were mixed with soybean trypsin inhibitor (Sigma-Aldrich) at a protease to inhibitor ratio of 1:1.5 (by mass) and analyzed by SDS-PAGE (10% polyacrylamide). The band corresponding to full-length hPAH was quantified by densitometric analysis using the Quantity One 1D software v4.6.3 (Bio-Rad). Data were fitted to a single exponential decay equation to obtain the decay constant of proteolysis (
kP). Assays were performed in duplicate and data represent the mean ± SEM.
To rule out compounds that inhibit/inactivate trypsin, changes in the cleavage rate of N-(Benzyloxy)carbonyl-glycylglycylarginyl-7-amido-4-methylcoumarin (Z–Gly–Gly–Arg–AMC; Bachem, Bubendorf, Switzerland), a synthetic substrate of trypsin, were monitored by fluorimetric measurements in a microplate reader (FLUOstar Omega; BMGLabtech, Ortenberg, Germany). The assays (n = 3) were performed in a 96-well plate, in SEC buffer and in a final volume of 200 μL containing 1.5 μg/mL of trypsin, 50 μM of Z–Gly–Gly–Arg–AMC and 100 μM of 3HQs derivatives. Fluorescence was measured in real-time, with λexc 360 nm and λem 460 nm, during 30 min at 25 °C. Controls were performed using substrate alone with 1% DMSO (negative control) and trypsin with no compounds and 1% DMSO (positive control).
2.6. Electron Paramagnetic Resonance Spectroscopy
EPR spectra of hPAH (at ≈100 μM monomer) in the absence or presence of equimolar amounts of 3HQs were recorded at 4K in a Bruker EMX spectrometer (Billerica, MA, USA) equipped with an ESR-900 continuous flow helium cryostat from Oxford Instruments (Abingdon, Johnson, UK). Spectra were recorded for control samples containing 100 μM FeCl3 solutions incubated with equimolar amounts of 3HQs to evaluate direct binding of the compounds to free Fe3+ in solution. Microwave frequency: 9.39 GHz; microwave power, 2 mW; modulation amplitude, 1 mT.
2.7. Surface Plasmon Resonance
The affinity of hPAH for 3HQs was evaluated by surface plasmon resonance (SPR) using a Biacore 4000 (GE Healthcare) instrument. All assays were carried out at 25 °C. Optimization of hPAH immobilization on a carboxymethylated dextran (CM5) matrix of the sensor chip was initiated by performing a pH scouting, using as buffers 10 mM sodium acetate (pH 5.0, 5.2, 5.5 and 5.8), 10 mM Bis-Tris (pH 6.0, 6.2 and 6.5), and 10 mM sodium phosphate (pH 7.0). hPAH was diluted to 10 µg/mL in the best immobilization buffer (10 mM sodium acetate pH 5.5) and immobilized onto the CM5 sensor chip using the standard amine coupling procedure. The HBS-N buffer, consisting of 10 mM HEPES pH 7.4 and 150 mM NaCl, was used as background buffer. Prior to immobilization, the carboxymethylated surface of the chip was activated with a 1:1 ratio of 400 mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide and 100 mM N-hydroxysuccinimide for 10 min. The hPAH was coupled to the surface with a 1 to 2 min injection time at a flow rate of 10 μL/min in order to reach 2000 to 5000 RU. The remaining activated carboxymethylated groups were blocked with a 7 min injection of 1 M ethanolamine pH 8.5.
The 3HQs were directly diluted in running buffer (10 mM HEPES, 150 mM NaCl, 5 mM MgCl
2, 0.1 mM EDTA, 0.05% (
v/
v) Tween-20, 1 mM DTT, pH 7.2, and 2% DMSO) and injected at ten different concentrations using two-fold dilutions series, from 200 µM to ≈ 0.4 μM. The interaction between hPAH and
l-Phe in the ≈ 2–1000 μM range was also analyzed, for comparison with literature values [
24,
25]. Moreover,
l-Phe (at 1 mM) was used as a positive control to collect information on surface integrity and activity throughout the experiment. For competition experiments, 1 mM
l-Phe was added to the running buffer. All sensorgrams were processed by first subtracting the binding response recorded from the control surface (reference spot), followed by subtracting an average of the buffer blank injections from the reaction spot. Interactions were assessed from the obtained plots of steady-state SPR response levels against 3HQs concentration, employing the provided Biacore 4000 evaluation software.
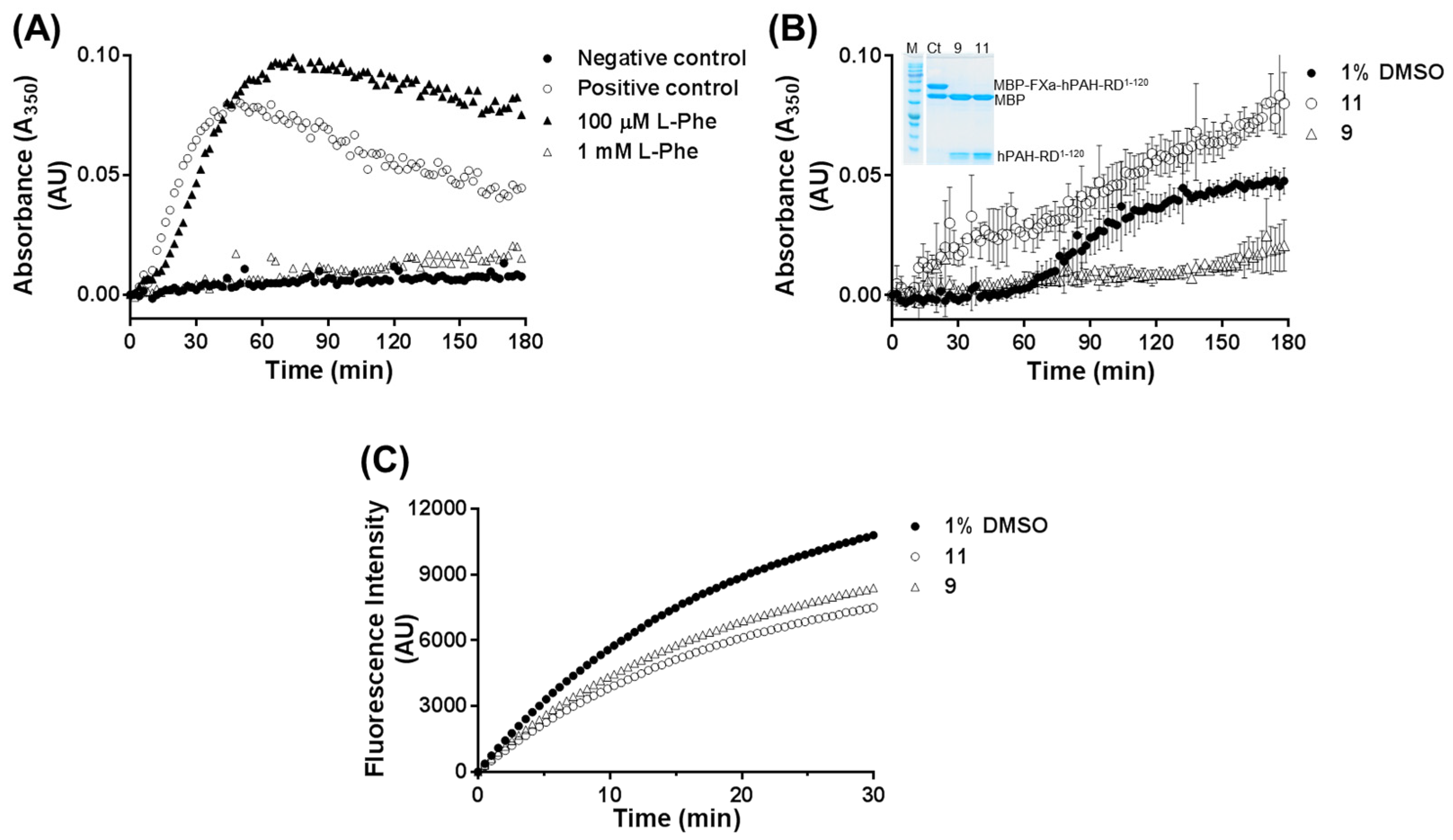
2.8. Light Scattering Assay
Self-assembly of the dimeric forms of hPAH-RD
1–120 was followed in real-time at 25 °C upon cleavage of MBP-FXa-hPAH-RD
1–120 with FXa (New England Biolabs) to release the MBP fusion partner. Assays (
n = 3) were performed in the absence or presence of tested compounds as described in [
17]. Briefly, FXa (at a final ratio 1:150 (
w/
w) relative to the fusion protein) was added to the reaction mixture containing the MBP-FXa-hPAH-RD
1–120 fusion protein (0.74 mg/mL), 20 mM Na-HEPES, 100 mM NaCl, pH 7.0 and tested 3HQ (100 μM). The increase in absorbance at 350 nm was followed for 180 min on a FLUOstar Omega (BMG Labtech) microplate reader. A shaking step was introduced before each measurement. Appropriate control assays were performed and included: (i) a positive control consisting of MBP-FXa-hPAH-RD
1–120 in the presence of FXa, 1% DMSO and in the absence of compounds; and (ii) a negative control consisting of MBP-FXa-hPAH-RD
1–120 in the absence of FXa and compounds. For all the assays, aliquots of the aggregation reaction were collected at the end of the reaction (180 min) and analyzed by SDS-PAGE to confirm the extent of MBP cleavage by FXa. In addition, the effect of tested compounds on FXa activity was monitored using the synthetic FXa substrate Boc–Ile–Glu-Gly–Arg–AMC (t-(Butyloxycarbonyl)-isoleucylglutamylglycylarginyl-7-amido-4-methylcoumarin; from Bachem). Assays (
n = 3) were performed in a 96-well plate, in a final volume of 200 μL containing 20 mM Na-HEPES, 100 mM NaCl, pH 7.0, 5 μg/mL of FXa, 50 μM of Boc–Ile-Glu–Gly–Arg–AMC and 100 μM of 3HQ derivatives. Fluorescence was measured in real-time (λ
exc 360 nm and λ
em 460 nm) during 30 min, at 25 °C. Controls were performed using substrate alone with 1% DMSO (negative control) and FXa with no compounds and 1% DMSO (positive control).
2.9. Studies in Eukaryotic Cells
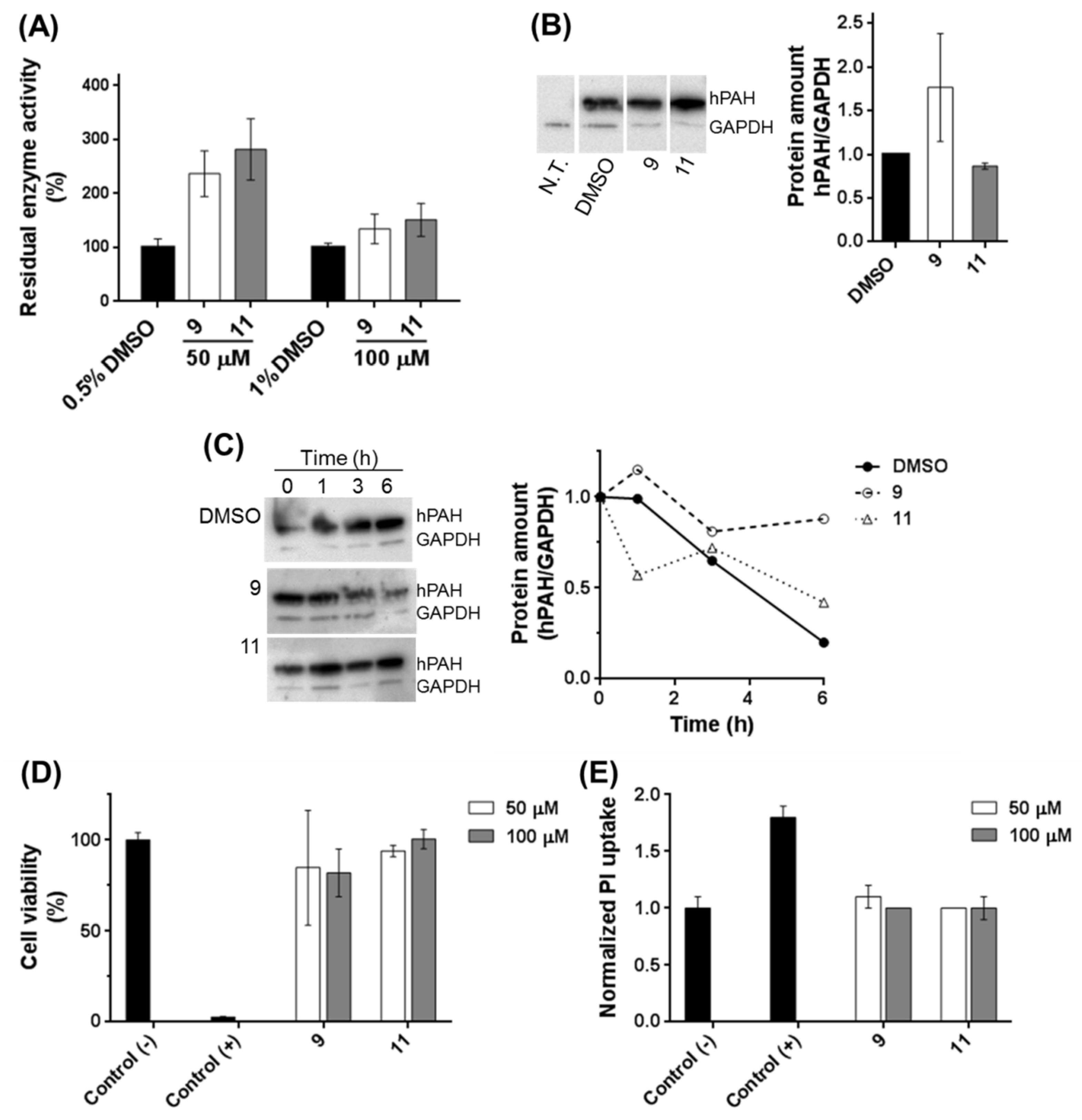
HEK-293T cells (ATCC CRL-11268) were grown in RPMI 1640 medium (Gibco Life Technologies¸ Carlsbad, CA, USA) and supplemented with heat inactivated (60 °C for 30 min) fetal bovine serum (10%; v/v), penicillin (100 U/mL) and streptomycin (0.1 mg/mL) at 37 °C, in 5% CO2 in a humidified atmosphere. 1 × 105 cells were transfected with 750 ng of DNA (eukaryotic vector expressing hPAH; pEF-DEST51-PAH-WT) using lipofectamine (Invitrogen) as the transfection agent. Compounds (50 and 100 μM) or DMSO (0.5 and 1%) were added 4 h after transfection, and cells were further incubated for 24 h. For sample collection cells were trypsinized with 500 μL of TripLE Express (Gibco Life Technologies), centrifuged at 200× g for 5 min, at 4 °C, and washed with PBS. The pellet was resuspended in 50 μL of PBS with 200 μM PMSF and cells were disrupted by passage (≈20×) through a fine needle. The cell debris were removed by centrifugation at 14,000× g for 5 min at 4 °C and the obtained cell lysates were used for enzymatic activity assays and western blot analysis. Three independent cell cultures were performed.
For the stability assays, after 24 h incubation with compounds, protein translation was stopped by the addition of 10 μg/mL puromycin (Sigma-Aldrich). Cells were collected at 0, 1, 3, and 6 h after blockage of translation. Two independent cell cultures were performed.
The hPAH activity of cell lysates was measured essentially as described in [
26] in a final volume of 200 μL containing 1 mM
l-Phe, 250 mM Na-Hepes, pH 7, 0.1 mg/mL catalase and 20 μL of cell lysate. After 4 min of pre-incubation, 100 μM (NH
4)
2Fe(II)SO
4 was added to the mixture. Two min after ferrous ammonium sulphate addition, the reaction was initiated by the injection of BH
4 (75 μM). Tyrosine production was assessed by measuring the increase in fluorescence intensity (λ
exc 274 nm and λ
em 304 nm) in a FLUOstar Omega microplate reader (BMG Labtech), at 25 °C, during 30 min. Enzyme activity was determined as the amount of
l-Tyr produced per min per mg of total protein. For each cell culture enzymatic assays were performed in triplicate.
For western blot analysis of cell lysates, 4 μg of total protein were applied on an 8 to 16% mini-protean TGX precast polyacrylamide gel (Bio-Rad), and the proteins were transferred to a polyvinylidene fluoride (PVDF; GE Healthcare) membrane and visualized by immunodetection using mouse anti-PAH (MAB 5278; Millipore; Burlington, MA, USA) and mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH (loading control); sc-365062, Santa Cruz Biotechnology; Dallas, TX, USA) as the primary antibodies and the anti-mouse IgG-HRP (W402B, Promega; Madison, WI, USA) as the secondary antibody. Immunoblots were developed with enhanced chemiluminescent agents (ECL Prime; GE Healthcare) and the images were acquired in a charge-coupled device imager ChemiDoc XRS (Bio-Rad) using the Quantity One software (Bio-Rad).
Cytotoxicity of selected 3HQs was assessed in HEK-293T cells using the Alamar Blue assay as a general cell viability endpoint method that monitors the cellular metabolic state, and the propidium iodide (PI) exclusion assay was used to evaluate membrane integrity essentially as described [
27,
28]. The 3HQs were tested at a final concentration of 50 and 100 μM (two independent assays; five replicates each). Control assays were performed using 1% DMSO (negative control), or SDS at 1 mg/mL as a positive control of cell death in the PI assay. HEK-293T cells were seeded in sterile flat bottom 96-well tissue culture plates, in RPMI 1640 culture medium, supplemented with 10% fetal bovine serum, 100 units/mL of penicillin G (sodium salt), 100 μg/mL of streptomycin sulphate and 2 mM L-glutamine, at a cell density of 2 × 10
4 cells per well. Cells were incubated at 37 °C and 5% CO
2. The culture medium was replaced by fresh medium (supplemented as above) containing the different 3HQs to be analyzed. After 24 h incubation, the culture medium was replaced by 0.3 mM PI (Sigma-Aldrich) in fresh medium (stock solution 1.5 mM in DMSO) and fluorescence was measured (λ
exc 485 nm, λ
em 590 nm) in a FLUOstar Omega microplate reader. The Alamar Blue assay was performed by replacing the culture medium with fresh medium containing 5 mM resazurin (Invitrogen). The cells were further incubated for 3 h and fluorescence was measured (λ
exc 530 nm, λ
em 590 nm) in a FLUOstar Omega microplate reader (BMG Labtech). The relative PI uptake and cell viability (%) were calculated using Equations (3) and (4), respectively.
2.10. Molecular Modelling Studies
To study and rationalize the influence of different 3-hydroxyquinolin-2(1H)-one substituents on hPAH molecular recognition, compounds
3,
8,
9, and
11 were subjected to molecular docking simulations. These compounds were built and protonated at pH 7.4, and geometry was optimized using the Molecular Operating Environment (MOE) software, version 2019.01 suite (Chemical Computing Group Inc;
http://www.chemcomp.com (accessed on 30 December 2019); Montreal, QC, Canada). All molecules were submitted to a conformational search and energy minimization as implemented in MOE 2019.1. Conformers were created by a systematic search generating rotatable bond angle combinations. Each dihedral angle combination was subject to an energy minimization by means of the Amber12: EHT force field. The global minimum of each molecule was submitted to docking simulations. For the docking simulation studies, the target models of hPAH were designed starting from Protein Data Bank (PDB) crystallographic structures 1MMT and 3PAH. The original PDB entries were selected taking into account the source organism (
Homo sapiens), the best available X-ray resolution and the co-crystallized ligands. Before being used in the docking simulations, all the structures were submitted to the protein preparation tool of the MOE 2019.01 software package, to remove the co-crystallized inhibitors, add hydrogen atoms at pH 7.4, assign the correct protonation and tautomeric states, missing residues, and to remove all crystallographic water molecules.
All the docking simulations (non-covalent) were performed using GOLD 5.2 program (CCDC; Cambridge, UK) [
29]. The hPAH binding site was defined at the iron center in the protein catalytic site, and the binding site radius was set to 20 Å. No constraints were used in any calculation. Flexible ligand sampling was considered in the docking procedure. All other parameters were set to defaults for the GOLD docking process. Molecular docking studies were then performed using the GoldScore scoring function from GOLD 5.2 software package and each ligand was subjected to 1000 docking runs. The docking protocol was validated for all the four structures prepared re-docking the crystallographic ligands and their poses were reproducible with root-mean-square deviations (RMSD’s) below 1.5 Å.
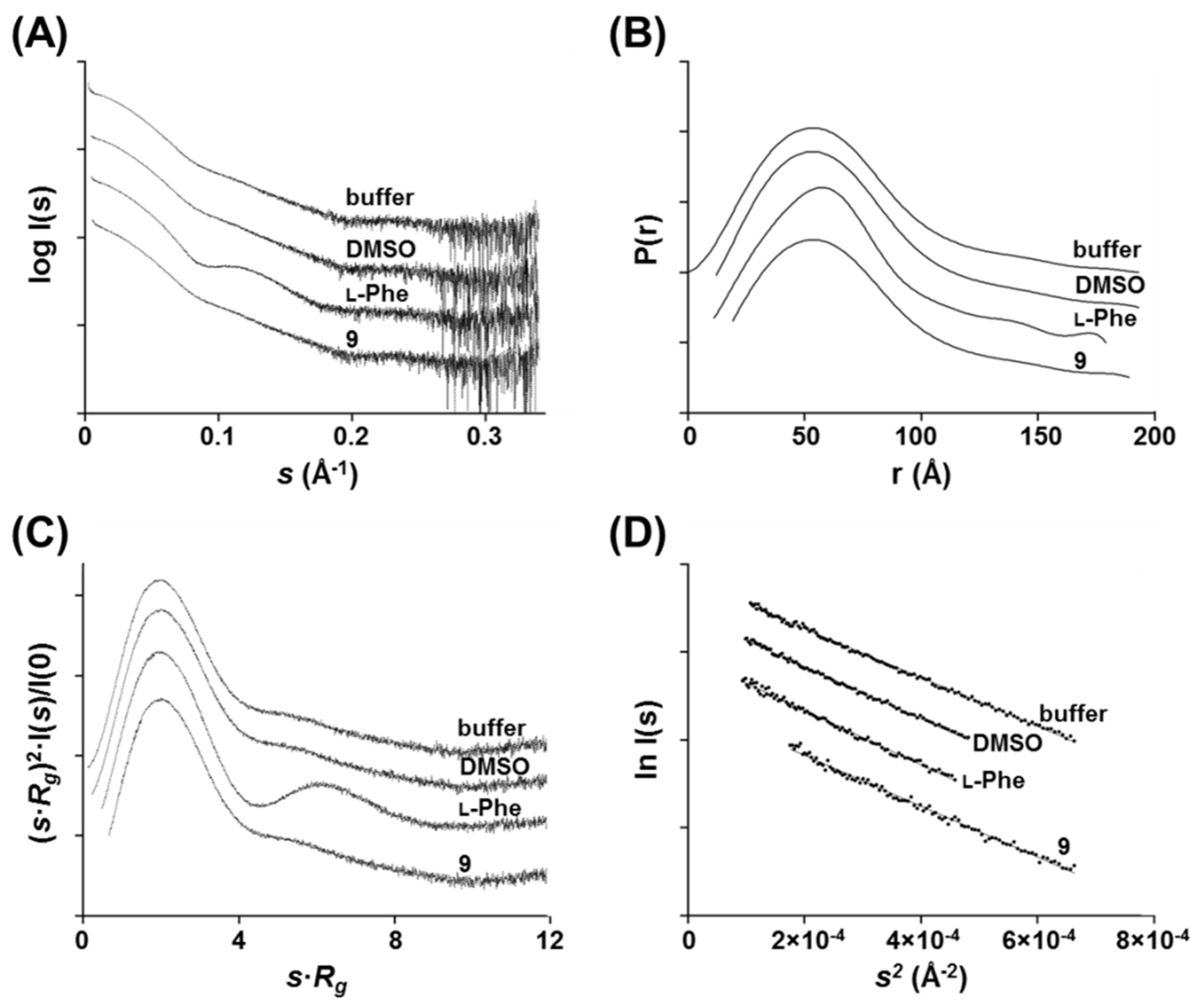
2.11. Small-Angle X-ray Scattering (SAXS)
Small-angle X-ray scattering experiments were performed at the Diamond Light Source B21 [
30] beamline at a wavelength of 0.95 Å. Tetrameric hPAH was thawed on ice, centrifuged at 12,000×
g for 8 min at 4 °C, and quantitated using a NanoDrop (molar extinction coefficient: 10.09 mg/mL/cm) prior to data acquisition. Measurements were performed in static mode with the automated sample changer at 20 °C using a protein concentration of 86 μM (monomer) in 20 mM Na-HEPES, 200 mM NaCl, pH 7.0 in the absence or presence of 2% DMSO.
l-Phe and compound 9 were tested at a final concentration of 200 μM in 2% DMSO. Data were recorded using a Pilatus 2M detector covering a momentum transfer 0.0031 < s < 0.380 Å-1 (s = 4π sin θ/λ, where 2θ is the scattering angle). The data were processed and analyzed using the ATSAS program suite [
31]. The sample frames were monitored for radiation damage, the selected frames were averaged, and the buffer contribution was subtracted using PRIMUS [
32]. The radius of gyration (
Rg) and the maximum particle dimension (
Dmax) were estimated from the Guinier approximation using PRIMUS and from the pair-distribution function
P(r) using the GNOM package [
33], respectively. Fitting of the experimental curves and the theoretical curves computed from SASBDB models was performed using CRYSOL [
31].
4. Discussion
Due to the high number of different pathogenic mutations identified in the PAH gene (>1000), development of a ‘universal’ molecule aiming at correcting the activity and/or stability of the entire spectrum of protein variants appears to be unrealistic. Therefore, the discovery of different classes of small molecules modulators of hPAH folding and/or activity will contribute to the development of pharmacological strategies to treat a larger cohort of PKU patients. Random searches of large compound libraries using high-throughput screenings (HTS) or shape-focused virtual screenings have been used to identify small molecules targeting hPAH [
34,
38]. Herein, a different approach was applied based on compounds inspired by functional and/or structural characteristics of the enzymes’s catalytic site, namely its substrate
l-Phe, the BH
4 cofactor and the catalytic non-heme ferric center. Using this rationale, we evaluated a library of 20 compounds with 3HQ as the core structure displaying the ability to complex iron, a structural similarity to BH
4 and, for some molecules,
l-Phe-like motifs as substituent groups were present. Our goal was to identify 3HQ derivatives able to potentiate/stabilize enzyme structure and activity without significantly inhibiting the enzyme by outcompeting the natural substrate, and still allowing further activation by
l-Phe in response to a potentially neurotoxic circulating substrate overload.
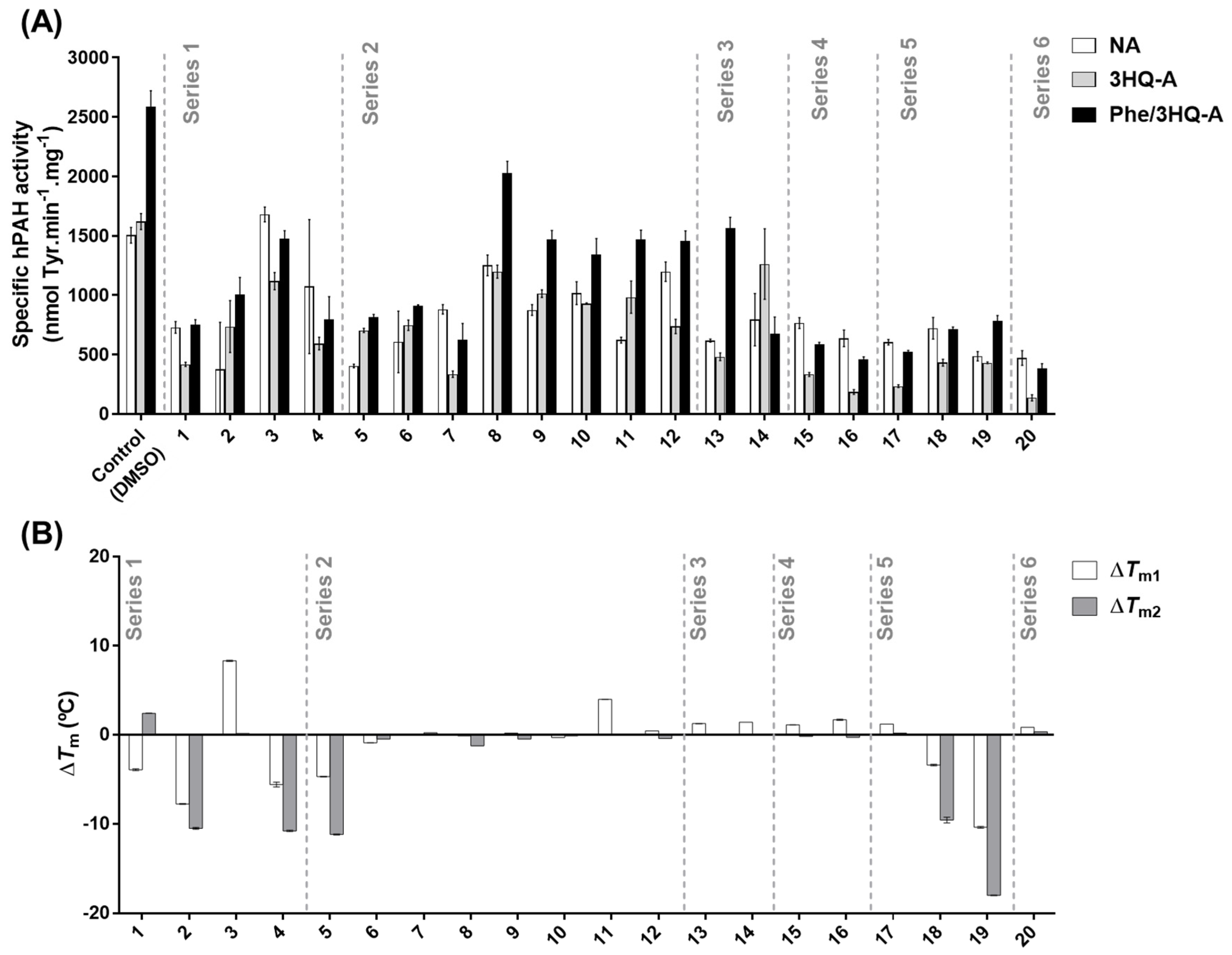
Among the 20 3HQ derivatives,
3,
8,
9, and
11 were selected as the most promising candidate molecules. None of these compounds presented inhibitory effects on hPAH activity in the non-activated condition, while compound
3 even afforded a mild increase. Importantly, they all allowed further activation by
l-Phe with respect to the pre-activated condition control. This is an especially relevant characteristic since often active variants with close-to-WT activity exhibit disturbed allosteric regulation as the underlying pathogenic mechanism [
21], both in PKU and other IMD (e.g., in [
39,
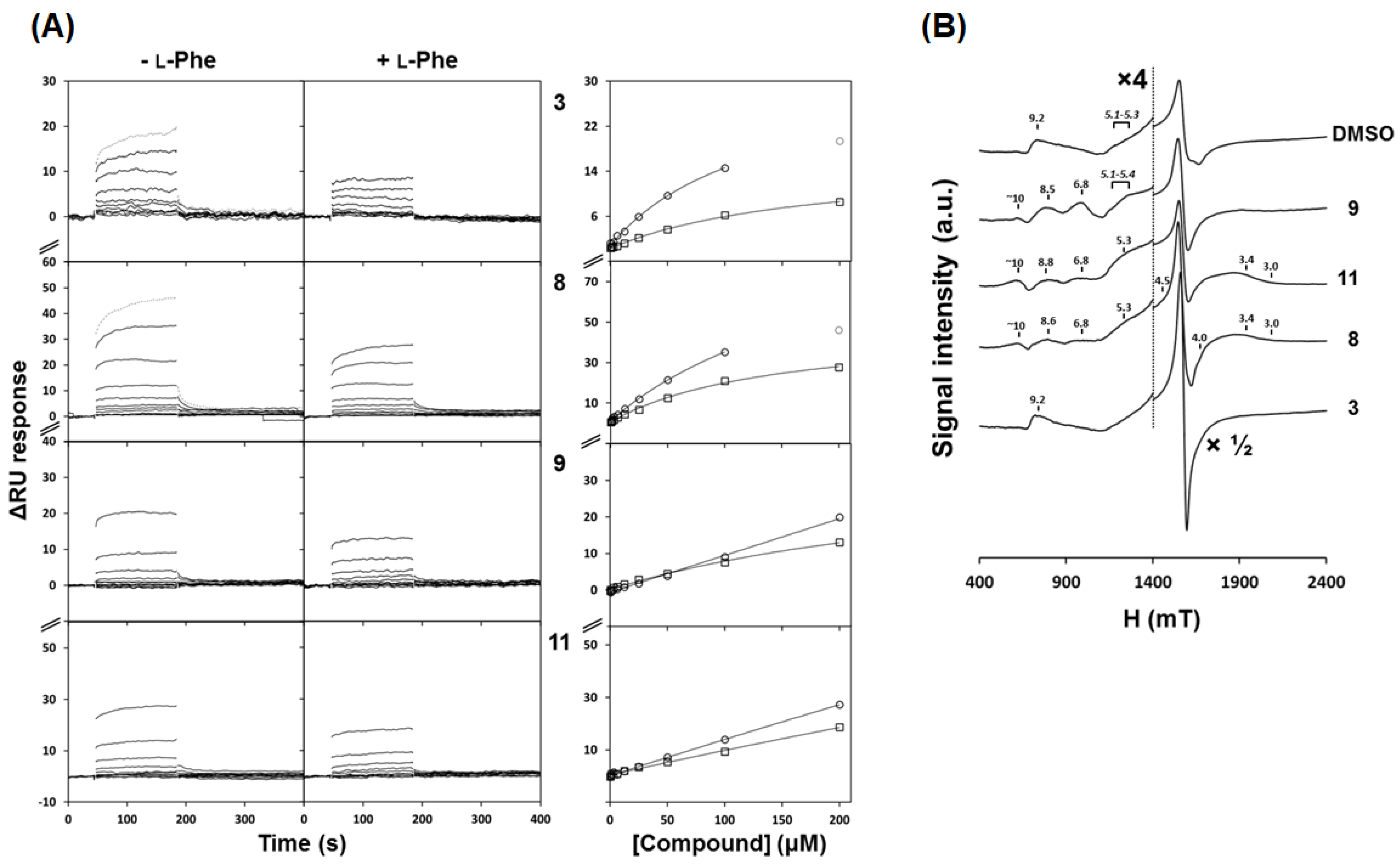
40]). The changes observed in the EPR spectra provided evidence that these four compounds establish interactions with the catalytic iron. However, the modest decrease in enzymatic activity does not foresee a tight binding of the 3HQs to the iron center as that observed for catecholamines such as dopamine, noradrenaline, and adrenaline [
36]. These molecules are potent inhibitors of hPAH activity with a high affinity binding (
KD < 1 μM) and their effect has been associated with a tight direct coordination of Fe
3+ [
36]. As for the selected 3HQs derivatives, the EPR studies suggest different modes of iron interaction, as judged from the shape and intensity of the main signal centered at g = 4.25 and from the additional resonances that appeared as a consequence of compound incubation. Compound
3 promoted an increase in the g = 4.25 signal intensity, in contrast to the decrease promoted by catecholamines [
39]. While for all compounds the resonances observed at g ≈ 9.2 and at g ≈ 5.1–5.3 are possibly related with some degree of flexibility of the iron coordination geometry (which includes three waters) [
35], a less rhombic symmetry was observed for compounds
8,
9, and
11, suggesting the exclusion of an iron-bound water molecule from iron coordination. Similar observations have been previously reported for hPAH incubated with its cofactor BH
4, dopamine [
35], noradrenaline (in the absence or presence of oxygen), or prepared in the hydroxyl moiety rich Tris buffer (shown to compete with the cofactor analogue 6-methyl-5,6,7,8-tetrahydrobiopterin for the active site) [
41]. Structural studies have shown that dopamine and other catecholamines eliminate two of the three iron-coordinating waters, in line with the increase in less rhombic signals at higher g values observed by EPR [
35,
36,
41]. Herein, the EPR studies suggest that the 3HQ derivatives may exert a similar effect of replacing water ligands from the iron center through the hydroxyl moiety from the hydroxyquinoline ring system. The comparative analysis of the estimated
KD (in the absence of
l-Phe) obtained by SPR suggests higher affinities for compounds
3 and
8 than for
9 and
11. In addition, steady-state kinetics studies characterized the effect of compounds
3 and
8 as competitive inhibitors towards the
l-Phe substrate. We posit that the phenylalanine moiety present in the structures of compounds
3 and
8 contributes to the higher affinity for the protein and direct competition with the substrate
l-Phe, as compared to those of compounds
9 and
11, which contain instead the carbomethoxy derivatives of L-glutamate and L-glycine, respectively.
Evaluation of the time-dependence of full-length hPAH degradation by trypsin has been a valuable tool to study changes in hPAH global conformation occurring upon pre-incubation with the substrate or in the presence of the BH
4 cofactor [
4,
8,
42]. Allosteric
l-Phe binding protects rat PAH (rPAH) from trypsin digestion [
43] as the rotational movement of the N-terminal domain, necessary for their dimerization, and
l-Phe allosteric binding decreases the accessibility of the C-terminal part of this domain that is highly prone to trypsin digestion. A similar protective effect has been reported for BH
4 binding to the hPAH catalytic center [
42] by a mechanism involving H-bonding between the two hydroxyl groups of BH
4 and Ser23 located in the regulatory domain and formation of a more closed conformation, which is less prone to trypsin digestion [
4]. The protective effect of compounds
3 and
8 against hPAH proteolysis may be caused by a tighter binding to the catalytic pocket, thus promoting the stabilization of a more closed structure rather than inducing dimerization of the N-terminal domain, as these compounds did not show any ability to pre-activate the enzyme.
The absence of
kP proteolytic rates variations in the presence of compounds
9 or
11 argues against strong global conformational changes of hPAH. The steady-state kinetic parameters (absence of competitive inhibition) obtained for these compounds suggests that, besides binding to the catalytic domain (EPR assays), compounds
9 and
11 will also bind to another region of the protein. Of note is the fact that only compounds
9 and
11 were able per se to pre-activate the enzyme (
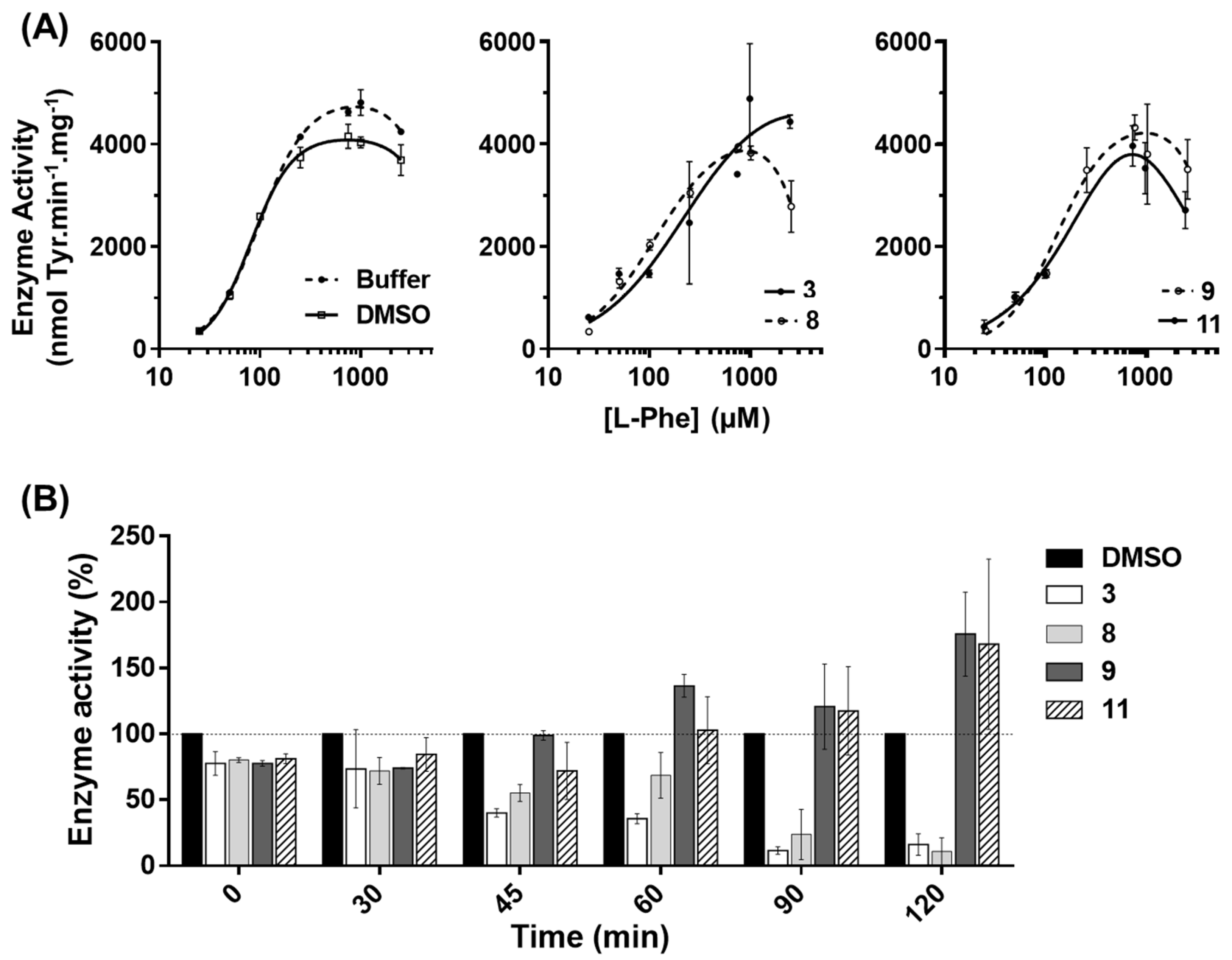
Figure 3A; NA vs. 3HQ-A), indicating a higher exposure of the catalytic pocket in the presence of these compounds. Corroborating this hypothesis, the data obtained for compound
9 on the aggregation behavior of the N-terminal regulatory domain (hPAH-RD
1–120) strongly suggests that this molecule is able to bind to this domain, probably in a region different from the
l-Phe allosteric site. Indeed, in the presence of compound
9, hPAH allostery was maintained (
Table 1) and the protein was able to be further activated by
l-Phe (
Figure 3A). Moreover, solution scattering data of hPAH in the presence of compound
9 reveals no gross structural changes consistent with domain dimerization as in the
l-Phe allosteric activation mechanism [
8,
37], suggesting a global conformation similar to the enzyme’s ‘resting state’. Concerning compound
11, no direct evidence of a direct binding to hPAH-RD
1–120 was obtained, although binding to other regions of the protein besides the active site could not be excluded. Indeed, among the selected 3HQ derivatives, only compounds
3 and
11 afforded a slight stabilization of the regulatory domain. This may result from the secondary binding to another site within the protein, which may propagate and indirectly affect the regulatory domain stability. Upon exposure of isolated hPAH to a thermal insult, compounds
9 and
11 were those that most contributed to retaining the enzymatic activity at long incubation times. Further studies on HEK-293T cells showed that, also in a cellular environment, compounds
9 and
11 were able to stabilize the protein leading to a higher hPAH content and activity. The preservation of enzyme activity also suggests a proper assembly of the biologic tetrameric forms. Taken together, these data indicate that an appropriate screening strategy to identify hPAH stabilizers should rely both on hPAH activity protection and conformational and thermal stability.
From our series of 3HQ derivatives, compounds
9 and
11 could be regarded as promising hit molecules for the development of a new class of molecules acting as activity chaperones protecting hPAH activity. The majority of the experimental approaches aiming at the identification of pharmacological chaperones target the enzyme catalytic center. From this perspective, enzyme inhibitors are often included in the group of molecules that upon binding to the active site promote protein stabilization, thus acting as pharmacological chaperones [
44,
45], alike sapropterin dihydrochloride being employed for hPAH clinical variants. Upon hPAH binding, BH
4 establishes interactions with residues of the N-terminal regulatory domain, promoting a less flexible and more closed structure with a lower enzymatic activity but less prone to unfolding and degradation [
11]. In addition, enzyme inhibitors have also been described as protectors of enzyme activity of human tyrosine hydroxylase [
46,
47], a protein that shares with hPAH several structural and catalytic properties. Binding of
l-Phe to the allosteric site stabilizes an ‘activated’ state necessary for the physiological response to toxic levels of
l-Phe. Therefore, the ideal stabilizing compound should bind to a protein region other than the active site and the allosteric site [
7]. In this respect, compound
9 is a starting structure for the discovery of such a molecule. It is well-established that compounds envisaged to be used as stabilizers of protein structure/activity should bind specifically to the target molecule but should also easily dissociate from the enzyme in the presence of the substrate, allowing the protein to exert its biological function [
3]. In line with this observation, the selected 3HQs presented lower affinities than those obtained for the substrate
l-Phe. Finally, an important feature of a molecule intended to be used as a pharmacological therapy is their effect in a cellular context and their biocompatibility. Compounds
9 and
11 were devoid of toxicity and, importantly, preserved the hPAH content and concomitantly enzyme activity. These compounds, besides representing strong candidates for structure optimization aiming at improved properties, also provided proof-of-concept for the utilized strategy of compound design directed to a catalytic center that can be applied to other deficient enzymes responsible for IMDs.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}