Synthesis and Inhibitory Studies of Phosphonic Acid Analogues of Homophenylalanine and Phenylalanine towards Alanyl Aminopeptidases


Abstract
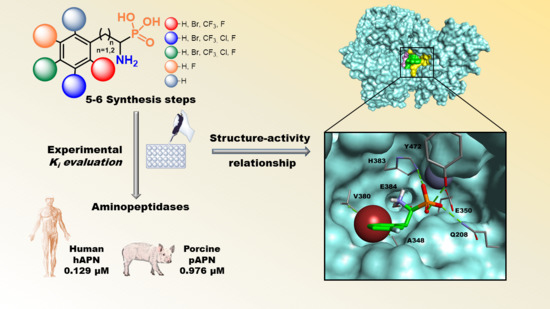
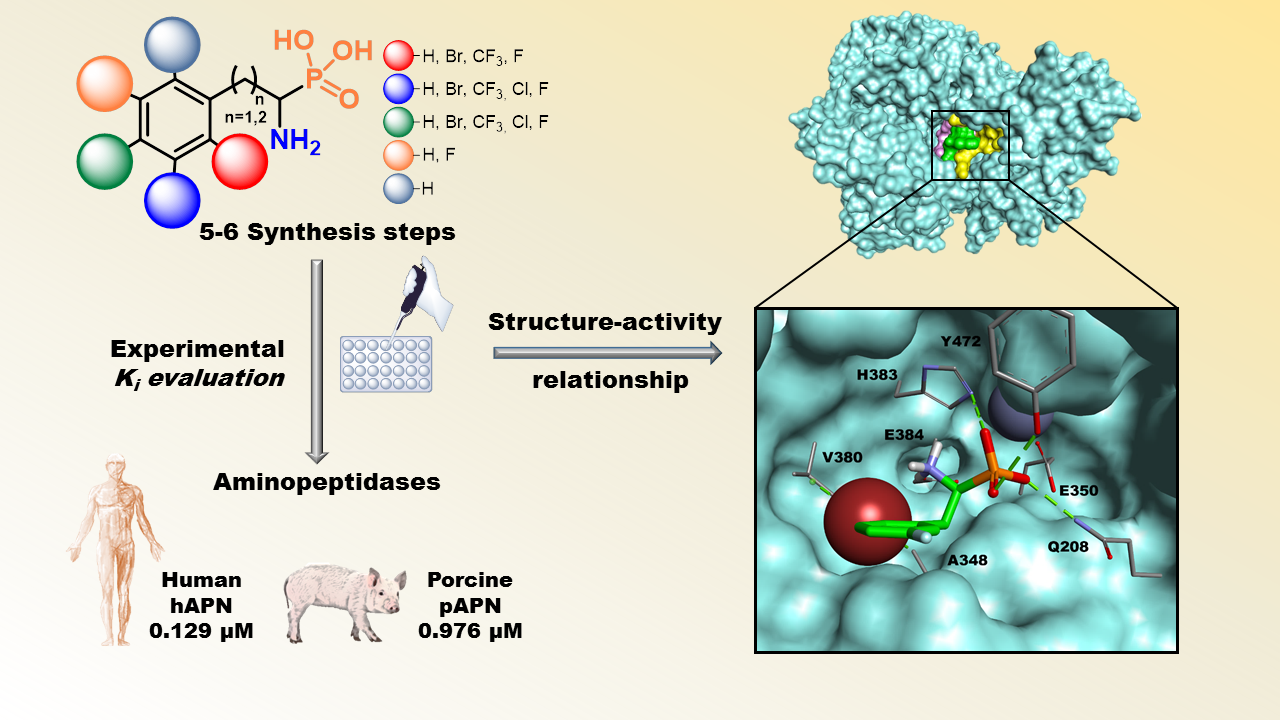
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. Synthesis of Substituted Methyl Phenylpropanoates/Methyl Phenylacetates (Compounds 2 and 8)
3-(2-Fluorophenyl)propionic Acid Methyl Ester (2b)
2.3. Synthesis of Substituted Phenylpropanols/Ethanols (Compounds 3 and 9)
3-(2-Fluorophenyl)propanol (3b)
2.4. Synthesis of Substituted Phenylpropionaldehydes/Phenylacetaldehydes by Using Pyridinium Chlorochromate (PCC) (Compounds 4 and 10)
2.4.1. 3-(4-Fluorophenyl)propanal (4d)
2.4.2. 3-(4-Fluorophenyl)propyl-3-(4-Fluorophenyl)propionate (5d)
2.5. Synthesis of Substituted Phenylacetaldehyde/Phenylpropionaldehyde by Using Dess-Martin Periodinane (DMP) (Compounds 4 and 10)
2.6. Diphenyl 1-{[(N-Benzyloxy)carbonyl]amino}alkylphosphonates (Compounds 6 and 13)
Diphenyl 1-{[(N-benzyloxy)carbonyl]amino}-3-phenylpropylphosphonate (6a)
2.7. Transesterification of Diphenyl 1-{[(N-Benzyloxy)carbonyl]amino}alkylphosphonates with Methanol (Compounds 14 and 16)
Dimethyl 1-{[(N-benzyloxy)carbonyl]amino}-3-(2-fluorophenyl)propylphosphonate (14b)
2.8. Hydrolysis of Diphenyl and Dimethyl 1-{[(N-Benzyloxy)carbonyl]amino}alkylphosphonates (Compounds 15 and 17)
2.8.1. 1-Amino-3-Phenylpropylphosphonic Acid (15a)
2.8.2. 1-Amino-3-(2-Fluorophenyl)propylphosphonic Acid (15b)
2.8.3. 1-Amino-3-(3-Fluorophenyl)propylphosphonic Acid (15c)
2.8.4. 1-Amino-3-(4-Fluorophenyl)propylphosphonic Acid (15d)
2.8.5. 1-Amino-3-(2,4-Difluorophenyl)propylphosphonic Acid (15e)
2.8.6. 1-Amino-3-(3,4-Difluorophenyl)propylphosphonic Acid (15f)
2.8.7. 1-Amino-3-(4-Trifluoromethylphenyl)propylphosphonic Acid (15g)
2.8.8. 1-Amino-3-(2-Trifluoromethylphenyl)propylphosphonic Acid (15h)
2.8.9. 1-Amino-3-(2-Bromo-4-Fluorophenyl)ethylphosphonic Acid (17a)
2.8.10. 1-Amino-3-(2-Bromo-5-Fluorophenyl)ethylphosphonic Acid (17b)
2.8.11. 1-Amino-3-(3-Bromo-4-Fluorophenyl)ethylphosphonic Acid (17c)
2.8.12. 1-Amino-3-(4-Bromo-2-Fluorophenyl)ethylphosphonic Acid (17d)
2.8.13. 1-Amino-3-(4-Bromo-3-Fluorophenyl)ethylphosphonic Acid (17e)
2.9. Diphenyl α-Aminoalkilphosphonates (Representative Example)
Diphenyl 1-Amino-3-(2-Trifluoromethyl)phenylpropylphosphonate (18h)
2.10. Bioassay
2.10.1. Enzymatic Studies
2.10.2. Molecular Modelling
2.10.3. Crystallography
3. Results and Discussion
3.1. Chemistry
3.2. Evaluation of Inhibitory Activity
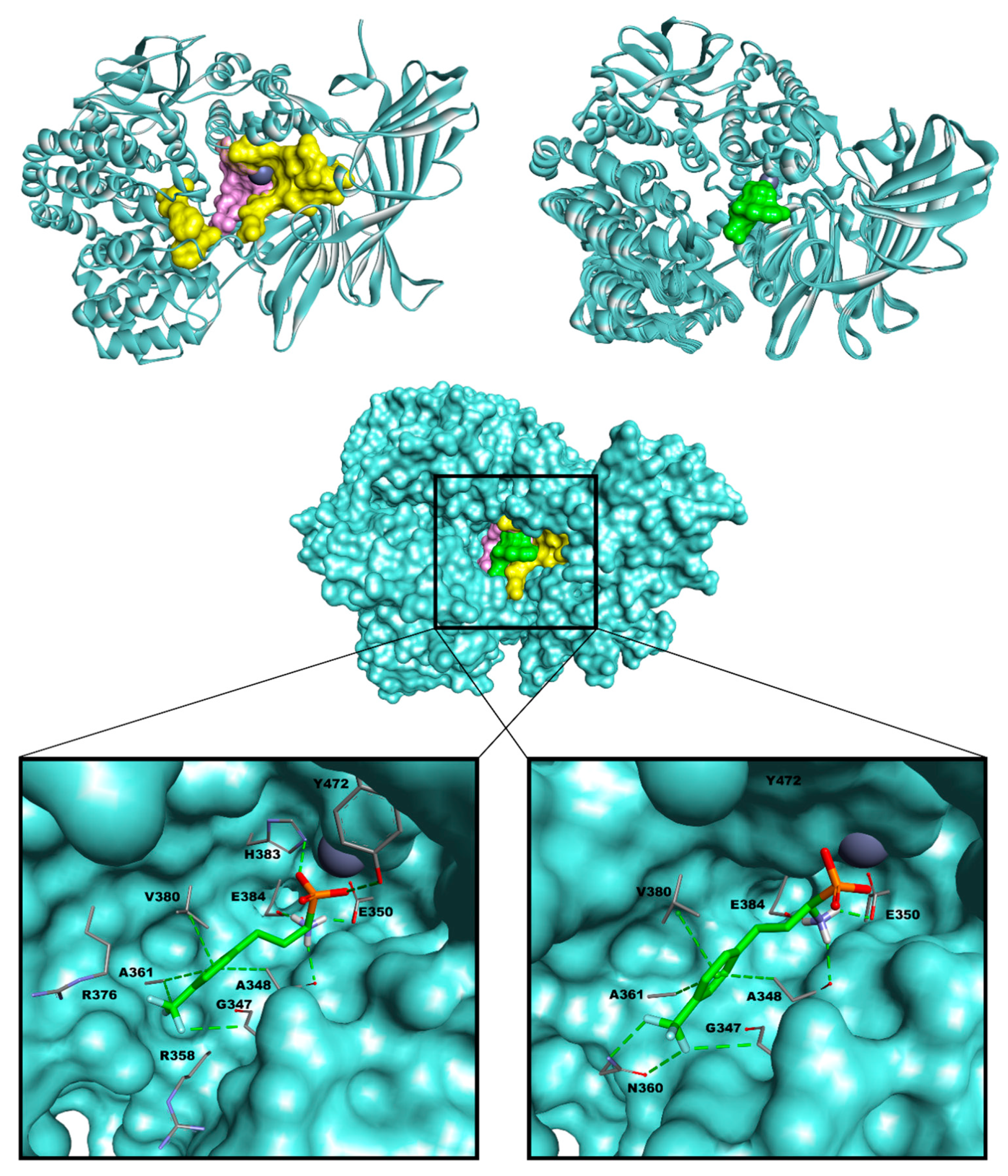
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Surowiak, P.; Drąg, M.; Materna, V.; Suchocki, S.; Grzywa, R.; Spaczyński, M.; Dietel, M.; Oleksyszyn, J.; Zabel, M.; Lage, H. Expression of aminopeptidase N/CD13 in human ovarian cancers. Int. J. Gynecol. Cancer 2006, 16, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Amin, M.A.; Fox, D.A. CD13/Aminopeptidase N Is a Potential Therapeutic Target for Inflammatory Disorders. J. Immunol. 2020, 204, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lin, Y.-L.; Peng, G.; Li, F. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef] [Green Version]
- Mina-Osorio, P. The moonlighting enzyme CD13: Old and new functions to target. Trends Mol. Med. 2008, 14, 361–371. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Adhikari, N.; Jha, T. Design of Aminopeptidase N Inhibitors as Anti-cancer Agents. J. Med. Chem. 2018, 61, 6468–6490. [Google Scholar] [CrossRef]
- Lee, J.; Vinh, N.B.; Drinkwater, N.; Yang, W.; Kannan Sivaraman, K.; Schembri, L.S.; Gazdik, M.; Grin, P.M.; Butler, G.S.; Overall, C.M.; et al. Novel Human Aminopeptidase N Inhibitors: Discovery and Optimization of Subsite Binding Interactions. J. Med. Chem. 2019, 62, 7185–7209. [Google Scholar] [CrossRef] [PubMed]
- Talma, M.; Mucha, A. P1′ Residue-Oriented Virtual Screening for Potent and Selective Phosphinic (Dehydro) Dipeptide Inhibitors of Metallo-Aminopeptidases. Biomolecules 2020, 10, 659. [Google Scholar] [CrossRef]
- Wanat, W.; Talma, M.; Pawełczak, M.; Kafarski, P. Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds. Pharmaceuticals 2019, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Wanat, W.; Talma, M.; Dziuk, B.; Pirat, J.-L.; Kafarski, P. Phosphonic Acid Analogs of Fluorophenylalanines as Inhibitors of Human and Porcine Aminopeptidases N: Validation of the Importance of the Substitution of the Aromatic Ring. Biomolecules 2020, 10, 579. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.H.M.; Zhou, D.; Rini, J.M. The X-ray Crystal Structure of Human Aminopeptidase N Reveals a Novel Dimer and the Basis for Peptide Processing. J. Biol. Chem. 2012, 287, 36804–36813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLong, M.A.; Amburgey, J.; Taylor, C.; Wos, J.A.; Soper, D.L.; Wang, Y.; Hicks, R. Synthesis and in vitro evaluation of human FP-receptor selective prostaglandin analogues. Bioorg. Med. Chem. Lett. 2000, 10, 1519–1522. [Google Scholar] [CrossRef]
- Leow, D.; Chen, Y.-H.; Hung, T.-H.; Su, Y.; Lin, Y.-Z. Photodriven Transfer Hydrogenation of Olefins. European J. Org. Chem. 2014, 2014, 7347–7352. [Google Scholar] [CrossRef]
- Hamilton, G.S.; Wu, Y.-Q.; Limburg, D.C.; Wilkinson, D.E.; Vaal, M.J.; Li, J.-H.; Thomas, C.; Huang, W.; Sauer, H.; Ross, D.T.; et al. Synthesis of N -Glyoxyl Prolyl and Pipecolyl Amides and Thioesters and Evaluation of Their In Vitro and In Vivo Nerve Regenerative Effects. J. Med. Chem. 2002, 45, 3549–3557. [Google Scholar] [CrossRef]
- Bailey, W.F.; Longstaff, S.C. Generation and Cyclization of a Benzyne-Tethered Alkyllithium: Preparation of 4-Substituted Indans. J. Org. Chem. 1998, 63, 432–433. [Google Scholar] [CrossRef]
- Fukuyama, T.; Nishikawa, T.; Ryu, I. Site-Selective C(sp 3)-H Functionalization of Fluorinated Alkanes Driven by Polar Effects Using a Tungstate Photocatalyst. European J. Org. Chem. 2020, 2020, 1424–1428. [Google Scholar] [CrossRef]
- Donslund, A.S.; Neumann, K.T.; Corneliussen, N.P.; Grove, E.K.; Herbstritt, D.; Daasbjerg, K.; Skrydstrup, T. Access to β-Ketonitriles through Nickel-Catalyzed Carbonylative Coupling of α-Bromonitriles with Alkylzinc Reagents. Chem. Eur. J. 2019, 25, 9856–9860. [Google Scholar] [CrossRef]
- Gan, Y.; Xu, W.; Liu, Y. Ligand-Controlled Regiodivergent Silylation of Allylic Alcohols by Ni/Cu Catalysis for the Synthesis of Functionalized Allylsilanes. Org. Lett. 2019, 21, 9652–9657. [Google Scholar] [CrossRef]
- Sieńczyk, M.; Kliszczak, M.; Oleksyszyn, J. Synthesis of isocyanide derivatives of α-aminoalkylphosphonate diphenyl esters. Tetrahedron Lett. 2006, 47, 4209–4211. [Google Scholar] [CrossRef]
- Moreno-Cinos, C.; Sassetti, E.; Salado, I.G.; Witt, G.; Benramdane, S.; Reinhardt, L.; Cruz, C.D.; Joossens, J.; Van der Veken, P.; Brötz-Oesterhelt, H.; et al. α-Amino Diphenyl Phosphonates as Novel Inhibitors of Escherichia coli ClpP Protease. J. Med. Chem. 2019, 62, 774–797. [Google Scholar] [CrossRef] [Green Version]
- Ryglowski, A.; Kafarski, P. Preparation of 1-aminoalkylphosphonic acids and 2-aminoalkylphosphonic acids by reductive amination of oxoalkylphosphonates. Tetrahedron 1996. [Google Scholar] [CrossRef]
- Karanewsky, D.S.; Petrillo, E.W. Phosphonyl Hydroxyacyl Amino Acid Derivatives as Antihypertensives. U.S. Patent US4616005 (A), 20 June 1986. [Google Scholar]
- Vassiliou, S.; Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-Guided, Single-Point Modifications in the Phosphinic Dipeptide Structure Yield Highly Potent and Selective Inhibitors of Neutral Aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef] [PubMed]
- Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Joachimiak, A.; Mucha, A. A structural insight into the P1 S1 binding mode of diaminoethylphosphonic and phosphinic acids, selective inhibitors of alanine aminopeptidases. Eur. J. Med. Chem. 2016, 117, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger LLC. Schrödinger Release 2018-4: LigPrep; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger LLC. Schrödinger Release 2018-4: Schrödinger Suite 2018-2 Induced Fit Docking Protocol; Glide, Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger LLC. Schrödinger Release 2018-4: Prime; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- CrysAlis CCD; Oxford Diffraction Ltd.: Abingdon, UK, 2002.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Kukhar, V.P.; Hudson, H.R. (Eds.) Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; Wiley: Chichester, UK, 2000; ISBN 978-0-471-89149-9. [Google Scholar]
- Soroka, M. The synthesis of 1-aminoalkylphosphonic acids. A revised mechanism of the reaction of phosphorus trichloride, amides and aldehydes or ketones in acetic acid (Oleksyszyn reaction). Liebigs Ann. Chem. 1990, 1990, 331–334. [Google Scholar] [CrossRef]
- Ziora, Z.; Kafarski, P. Amidoalkylation of Phosphorus Trichloride with Acetamide and Alkyl Oxocycloalkanecarboxylates. Phosphorus. Sulfur. Silicon Relat. Elem. 2009, 184, 1047–1053. [Google Scholar] [CrossRef]
- Dziuganowska, Z.A.; Andrasiak, I.; Ślepokura, K.; Kafarski, P. Amidoalkylation of Phosphorus Trichloride with Acetamide and Functionalized Cyclic Ketones-Evidence of Dominating Role of Side-Reactions. Phosphorus. Sulfur. Silicon Relat. Elem. 2014, 189, 1068–1075. [Google Scholar] [CrossRef]
- Oleksyszyn, J.; Tyka, R.; Mastalerz, P. Direct Synthesis of 1-Aminoalkanephosphonic and 1-Aminoalkanephosphinic Acids from Phosphorus Trichloride or Dichlorophosphines. Synthesis 1978, 1978, 479–480. [Google Scholar] [CrossRef]
- Oleksyszyn, J. An Amidoalkylation of Trivalent Phosphorus Compounds with P(O)H Functions Including Acetic Acid Solutions of PCl3, RPCl2 or R2PCl, Diesters of Phosphorus Acid and Phosphorus-III-Acids. J. Prakt. Chem. 1987, 329, 19–28. [Google Scholar] [CrossRef]
- Oleksyszyn, J.; Subotkowska, L.; Mastalerz, P. Diphenyl 1-Aminoalkanephosphonates. Synthesis (Stuttg) 1979, 1979, 985–986. [Google Scholar] [CrossRef]
- Oleksyszyn, J.; Tyka, R. An improved synthesis of 1-aminophosphonic acids. Tetrahedron Lett. 1977, 18, 2823–2824. [Google Scholar] [CrossRef]
- Birum, G.H. Urylenediphosphonates. General method for the synthesis of. alpha-ureidophosphonates and related structures. J. Org. Chem. 1974, 39, 209–213. [Google Scholar] [CrossRef]
- Li, J.; Sha, Y. A Convenient Synthesis of Amino Acid Methyl Esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, R.S.B.; Pinheiro, A.C.; da Silva, E.T.; da Costa, J.C.S.; Kaiser, C.R.; de Souza, M.V.N. Simple Methodology for the Preparation of Amino Alcohols from Amino Acid Esters Using NaBH 4–Methanol in THF. Synth. Commun. 2011, 41, 1276–1281. [Google Scholar] [CrossRef]
- Nystrom, R.F.; Brown, W.G. Reduction of Organic Compounds by Lithium Aluminum Hydride. II. Carboxylic Acids. J. Am. Chem. Soc. 1947, 69, 2548–2549. [Google Scholar] [CrossRef]
- Hoye, R.C. Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd edition (Seyden-Penne, Jacqueline). J. Chem. Educ. 1999, 76, 33. [Google Scholar] [CrossRef] [Green Version]
- Enders, D.; Schaumann, E. Category 5, Compounds with One Saturated Carbon Heteroatom Bond Amines and Ammonium Salts; Georg Thieme Verlag: New York, NY, USA, 2009; ISBN 9783131839510. [Google Scholar]
- Soai, K.; Oyamada, H.; Takase, M. The Preparation of N -Protected Amino Alcohols and N -Protected Peptide Alcohol by Reduction of the Corresponding Esters with Sodium Borohydride. An Improved Procedure Involving a Slow Addition of a Small Amount of Methanol. Bull. Chem. Soc. Jpn. 1984, 57, 2327–2328. [Google Scholar] [CrossRef]
- Omura, K.; Swern, D. Oxidation of alcohols by “activated” dimethyl sulfoxide. a preparative, steric and mechanistic study. Tetrahedron 1978, 34, 1651–1660. [Google Scholar] [CrossRef]
- Corey, E.J.; Suggs, J.W. Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett. 1975, 16, 2647–2650. [Google Scholar] [CrossRef]
- Al-Hamdany, A.J.; Jihad, T.W. Oxidation of Some Primary and Secondary Alcohols Using Pyridinium Chlorochromate. Tikrit J. Pure Sc. 2012, 17, 72–76. [Google Scholar]
- Hunsen, M. Pyridinium chlorochromate catalyzed oxidation of alcohols to aldehydes and ketones with periodic acid. Tetrahedron Lett. 2005, 46, 1651–1653. [Google Scholar] [CrossRef]
- Viana, H.; Carreiro, E.P.; Goth, A.; Bacalhau, P.; Caldeira, A.T.; do Rosário Martins, M.; Burke, A.J. Sequential alcohol oxidation/putative homo Claisen–Tishchenko-type reaction to give esters: A key process in accessing novel biologically active lactone macrocycles. RSC Adv. 2016, 6, 63214–63223. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Kumar, P. PCC-mediated novel oxidation reactions of homobenzylic and homoallylic alcohols. Tetrahedron Lett. 2003, 44, 1275–1278. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Kannan Sivaraman, K.; Paiardini, A.; Sieńczyk, M.; Ruggeri, C.; Oellig, C.A.; Dalton, J.P.; Scammells, P.J.; Drag, M.; McGowan, S. Synthesis and Structure–Activity Relationships of Phosphonic Arginine Mimetics as Inhibitors of the M1 and M17 Aminopeptidases from Plasmodium falciparum. J. Med. Chem. 2013, 56, 5213–5217. [Google Scholar] [CrossRef]
- vel Górniak, M.G.; Czernicka, A.; Młynarz, P.; Balcerzak, W.; Kafarski, P. Synthesis of fluorescent (benzyloxycarbonylamino)(aryl)methylphosphonates. Beilstein J. Org. Chem. 2014, 10, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Rudzińska, E.; Berlicki, Ł.; Kafarski, P.; Lämmerhofer, M.; Mucha, A. Cinchona alkaloids as privileged chiral solvating agents for the enantiodiscrimination of N-protected aminoalkanephosphonates—A comparative NMR study. Tetrahedron: Asymmetry 2009, 20, 2709–2714. [Google Scholar] [CrossRef]
- Lejczak, B.; Kafarski, P.; Szewczyk, J. Transesterification of Diphenyl Phosphonates using the Potassium Fluoride/Crown Ether/Alcohol System; Part 2. The Use of Diphenyl 1-Aminoalkanephosphonates in Phosphonopeptide Synthesis. Synthesis 1982, 412–414. [Google Scholar] [CrossRef]
- Drag, M.; Bogyo, M.; Ellman, J.A.; Salvesen, G.S. Aminopeptidase Fingerprints, an Integrated Approach for Identification of Good Substrates and Optimal Inhibitors. J. Biol. Chem. 2010, 285, 3310–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drąg, M.; Grembecka, J.; Pawełczak, M.; Kafarski, P. α-Aminoalkylphosphonates as a tool in experimental optimisation of P1 side chain shape of potential inhibitors in S1 pocket of leucine- and neutral aminopeptidases. Eur. J. Med. Chem. 2005, 40, 764–771. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Substituents | hAPN Ki [µM] | pAPN Ki [µM] | Cpd. | Substituents | hAPN Ki [µM] | pAPN Ki [µM] |
|---|---|---|---|---|---|---|---|
| 15a | all-H | 0.707 ± 0.176 | 7.64 ± 0.11 | 17a | 2-Br, 4-F | 14.0 ± 0.4 | 70.0 ± 6.6 |
| 15b | 2-F | 0.506 ± 0.038 | 12.7 ± 0.8 | 17b | 2-Br, 5-F | 41.7 ± 7.4 | 379 ± 126 |
| 15c | 3-F | 0.129 ± 0.025 | 2.63 ± 0.18 | 17c | 3-Br, 4-F | 0.248 ± 0.024 | 2.22 ± 0.14 |
| 15d | 4-F | 0.249 ± 0.025 | 1.69 ± 0.81 | 17t | 3-Cl, 4F | 0.542 ± 0.152 | 6.93 ± 0.78 |
| 15e | 2,4-diF | 0.661 ± 0.105 | 1.71 ± 0.46 | 17d | 4-Br, 2-F | 7.36 ± 0.21 | 82.5 ± 2.0 |
| 15f | 3,4-diF | 0.149 ± 0.011 | 1.02 ± 0.30 | 17oa | 4-Cl, 2F | 17.8 ± 2.09 | 160 ± 11 |
| 15g | 4-CF3 | 0.347 ± 0.062 | 0.976 ± 0.099 | 17e | 4-Br, 3-F | 0.670 ± 0.049 | 4.23 ± 0.44 |
| 15h | 2-CF3 | 0.804 ± 0.122 | 7.49 ± 0.58 | 17sa | 4-Cl, 3-F | 1.01 ± 0.16 | 14.8 ± 0.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanat, W.; Talma, M.; Dziuk, B.; Kafarski, P. Synthesis and Inhibitory Studies of Phosphonic Acid Analogues of Homophenylalanine and Phenylalanine towards Alanyl Aminopeptidases. Biomolecules 2020, 10, 1319. https://doi.org/10.3390/biom10091319
Wanat W, Talma M, Dziuk B, Kafarski P. Synthesis and Inhibitory Studies of Phosphonic Acid Analogues of Homophenylalanine and Phenylalanine towards Alanyl Aminopeptidases. Biomolecules. 2020; 10(9):1319. https://doi.org/10.3390/biom10091319
Chicago/Turabian StyleWanat, Weronika, Michał Talma, Błażej Dziuk, and Paweł Kafarski. 2020. "Synthesis and Inhibitory Studies of Phosphonic Acid Analogues of Homophenylalanine and Phenylalanine towards Alanyl Aminopeptidases" Biomolecules 10, no. 9: 1319. https://doi.org/10.3390/biom10091319