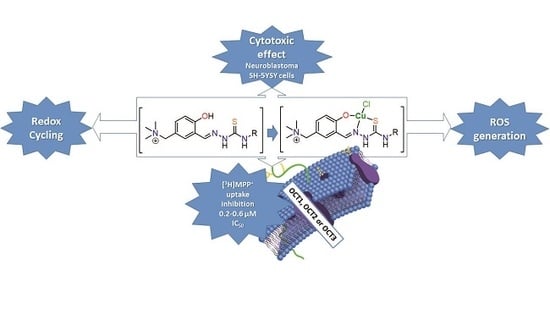
Insight into the Anticancer Activity of Copper(II) 5-Methylenetrimethylammonium-Thiosemicarbazonates and Their Interaction with Organic Cation Transporters

 , , , , , , ,
, , , , , , ,  , ,
, ,
Abstract
:
1. Introduction
2. Experimental Section
2.1. Chemicals
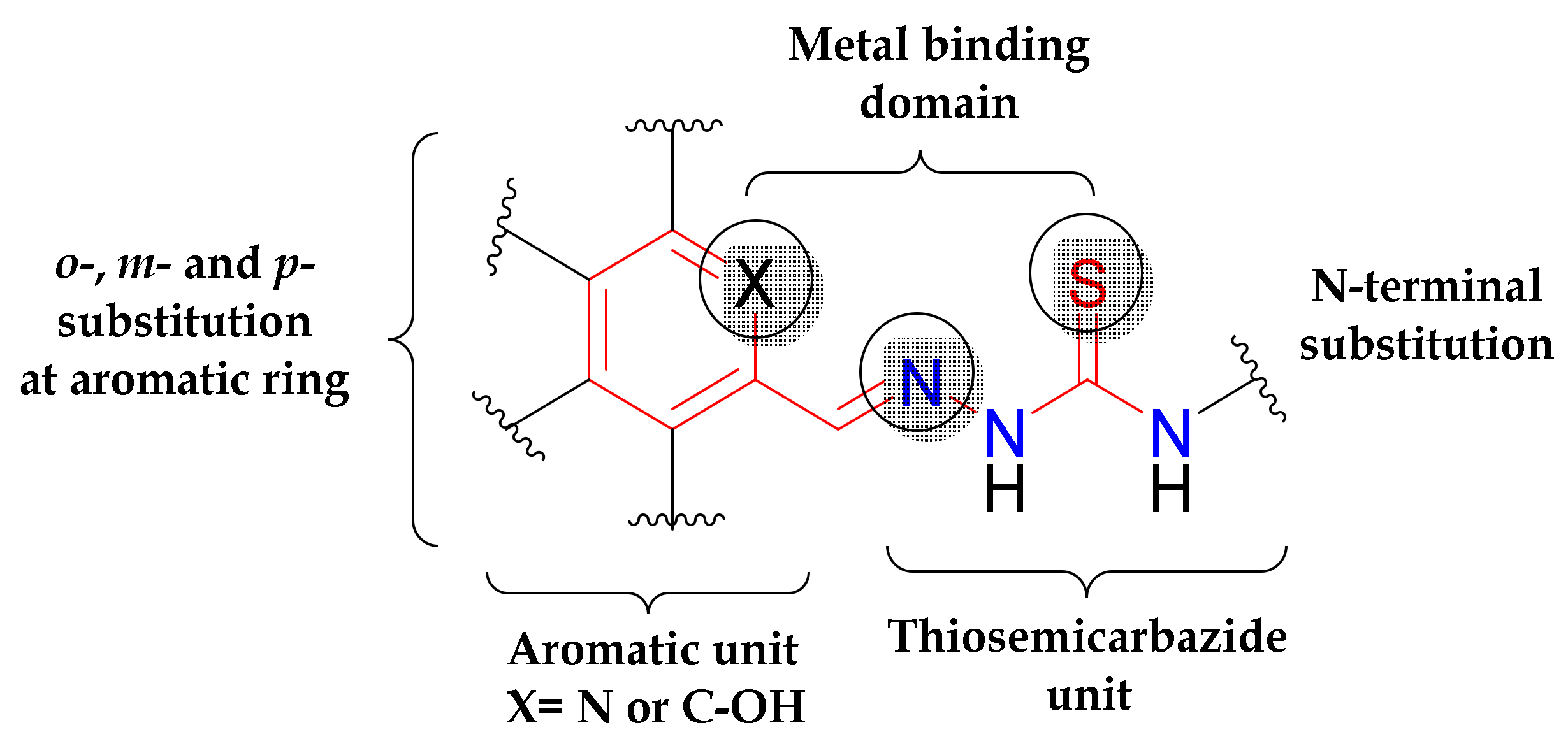
2.2. Synthesis of the Proligands
- [H2LH]Cl·1.4H2O (E-isomer). Yield: 85.0%; E-isomer 1H NMR (500 MHz, [D6]DMSO, 25 °C): δ = 11.51 (s, 1H; N2′H), 10.67 (s, 1H; C2-OH), 8.36 (s, 1H; C11H), 8.30 (s, 1H; N3′-H), 8.10 (s, 1H; C6H), 7.81 (s, 1H; N3′-H), 7.35 (dd, 1H, J = 8.4 Hz, 2.2 Hz; C4-H), 7.05 (d, 1H, J = 8.4 Hz; C3H), 4.41 (s, 2H; C7H2), 3.01 ppm (m, 9H; 3× C8–10H3); 13C NMR (126 MHz, [D6]DMSO, 25 °C): δ = 177.8 (C12), 157.9 (C2), 138.2 (C11), 135.0 (C4), 130.9 (C6), 120.8 (C1), 119.0 (C5), 116.5 (C3), 67.8 (C7), 51.6 ppm (C8–10); IR (ATR): ῦ = 3365, 3322, 3240, 3153, 1593, 1521, 1445, 1363, 1249, 1167, 1087, 968, 870, 844, 822, 751 cm−1; MS (ESI): m/z (%): 267 (25) [M]+, 208 (100) [M-NMe3]+; elemental analysis calcd (%) for C12H19ClN4OS·1.4H2O: C 43.94, H 6.70, N 17.08, S 9.77; found: C 44.26, H 6.55, N 16.88, S 9.98.
- [H2LMe]Cl·H2O. Predominant E-isomer (ca. 85%). Yield: 70.3%; 1H NMR (500 MHz, [D6]DMSO, 25 °C): δ = 11.55 (s, 1H; N2′-H), 10.61 (s, 1H; C2-OH), 8.44 (s, 1H; N3′-H), 8.35 (s, 1H; C11-H), 8.20 (s, 1H; C6-H), 7.35 (dd, 1H, J = 8.4 Hz, 2.2 Hz; C4-H), 7.05 (d, 1H, J = 8.3 Hz; C3-H), 4.44 (s, 2H; C7H2), 3.05–3.00 ppm (m, 12H; 3× C8–10H3 and C13H3). 13C NMR (126 MHz, [D6]DMSO, 25 °C): δ = 177.6 (C12), 157.8 (C2), 137.9 (C11), 134.8 (C4), 131.4 (C6), 120.9 (C1), 119.4 (C5), 116.5 (C3), 67.7 (C7), 51.6 (C8–10), 30.8 ppm (C13). IR (ATR): ῦ = 3285, 3138, 2998, 1613, 1532, 1243, 1083, 1040, 975, 879, 828, 762, 640 cm−1; MS (ESI): m/z (%): 281 (15) [M]+, 222 (100) [M-NMe3]+; elemental analysis calcd (%) for C13H21ClN4OS·H2O: C 46.63, H 6.92, N 16.73, S 9.58; found: C 47.02, H 6.87, N 16.71, S 9.81.
- [H2LEt]Cl·0.6H2O. Predominant E-isomer (ca. 87%). Yield: 77.1%. 1H NMR (500 MHz, [D6]DMSO, 25 °C): δ = 11.49 (s, 1H; N1′-H), 10.66 (s, 1H; OH), 8.47 (s, 1H; N3′-H), 8.35 (s, 1H; C11-H), 8.22 (s, 1H; C6-H), 7.35 (dd, 1H, J = 8.4 Hz, 2.1 Hz; C4-H), 7.09-7.03 (m, 1H; C3-H), 4.47 (s, 2H; C7H2), 3.62 (dq, 2H, C13H2), 3.03 (s, 9H; 3× C8–10H3), 1.16 ppm (t, 3H, J = 7.1 Hz; C14H3); 13C NMR (126 MHz, [D6]DMSO, 25 °C): δ = 176.6 (C12), 157.8 (C2), 138.1 (C11), 134.8 (C4), 131.4 (C6), 120.8 (C1), 119.1 (C5), 116.5 (C3), 67.4 (C7), 51.5 (C8–10), 38.3 (C13), 14.6 ppm (C14); IR (ATR): ῦ = 3355, 3197, 1612, 1531, 1486, 1271, 1222, 1077, 971, 921, 878, 843, 804, 752 cm−1; MS (ESI): m/z (%): 295 (16) [M]+ 236 (100) [M-NMe3]+; elemental analysis calcd (%) for C14H23ClN4OS·0.6H2O: C 49.21, H 7.14, N 16.40, S 9.38; found: C 48.93, H 6.84, N 16.64, S 10.00.
- [H2LPh]Cl·H2O (E-isomer). Yield: 78.4%. 1H NMR (500 MHz, [D6]DMSO, 25 °C): δ = 11.94 (s, 1H; N2′-H), 10.81 (s, 1H; C2-OH), 9.94 (s, 1H; N3′-H), 8.49 (s, 1H; C11-H), 8.20 (s, 1H; C6-H), 7.64 (d, 2H; C14-H and C18-H ), 7.43-7.33 (m, 3H; C4-H, C15-H and C17-H), 7.20 (t, 1H, J = 7.3 Hz; C16-H), 7.11 (d, 1H, J = 7.2 Hz; C3-H), 4.45 (s, 2H; C7H2), 3.03 ppm (s, 9H; 3× C8–10H3); 13C NMR (126 MHz, [D6]DMSO, 25 °C): δ = 175.73 (C12), 158.2 (C2), 139.2 (C11), 138.9 (C13), 135.2 (C4), 131.3 (C6), 128.1 (C15 and C17), 125.4 (C16), 125.2 (C14 and C18), 120.5 (C1), 119.1 (C5), 116.5 (C3), 67.6 (C7), 51.5 ppm (C8–10); IR (ATR): ῦ = 3552, 3454, 3277, 2949, 2854, 2715, 1612, 1531, 1500, 1441, 1268, 1203, 1070, 971, 872, 833, 746, 699, 645, 583 cm−1; MS (ESI): m/z (%): 343 (100) [M]+, 284 (30) [M-NMe3]+; elemental analysis calcd (%) for C18H23ClN4OS·H2O: C 54.47, H 6.35, N 14.11, S 8.08; found: C 54.55, H 6.04, N 13.94, S 8.03.
2.3. Synthesis of Copper(II) Complexes
- [Cu(HLH)Cl]Cl·2.5H2O (1). Yield: 71.9%. IR (ATR): ῦ = 3277, 3071, 2815, 2691, 1625, 1534, 1474, 1351, 1178, 967, 875, 825, 727, 677 cm−1; UV/Vis (H2O): λmax (ε) = 623 (125), 370 (11,466), 316 (18,475), 309 nm (18,401 mol−1dm3cm−1); MS (ESI): m/z (%): 328 (100) [Cu(LH)]+, 268 (80) [Cu(LH–NMe3)]+; elemental analysis calcd (%) for C12H18Cl2CuN4OS·2.5H2O: C 32.33, H 5.20, N 12.56, S 7.20; found: C 32.46, H 4.94, N 12.28, S 7.20.
- [Cu(HLMe)Cl]Cl·2.5H2O (2). Yield: 65.7%. IR (ATR): ῦ = 3334, 3235, 3020, 2978, 2885, 2796, 1607, 1528, 1476, 1349, 1175, 1032, 921, 876, 826, 760, 710, 616 cm−1; UV/Vis (H2O): λmax (ε) = 621 (208), 368 (16,980), 316 (24,249), 307 nm (25,137 mol−1dm3cm−1); MS (ESI): m/z (%): 342 (100) [Cu(LMe)]+, 283 (98) [Cu(LMe–NMe3)]+; elemental analysis calcd (%) for C13H20Cl2CuN4OS·2.5H2O: C 33.95, H 5.48, N 12.18, S 6.97; found: C 33.96, H 5.30, N 11.78, S 6.86. Single crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of EtOH/H2O (3:1) solution of 2 at room temperature.
- [Cu(HLEt)Cl]Cl·2.5H2O (3). Yield: 66.7%. IR (ATR): ῦ = 3352, 3215, 3024, 2978, 2850, 1586, 1530, 1476, 1352, 1174, 1049, 921, 878, 829, 621 cm−1; UV/Vis (H2O): λmax (ε) = 616 (188), 368 (12,159), 318 (17,433), 308 nm (17,715 mol−1dm3cm−1); MS (ESI): m/z (%): 356 (100) [Cu(LEt)]+, 297 (90) [Cu(LEt–NMe3)]+; elemental analysis calcd (%) for C14H22Cl2CuN4OS·2.5H2O: C 35.48, H 5.74, N 11.82, S 6.77; found: C 35.67, H 5.41, N 11.60, S 6.97. The X-ray diffraction quality single crystals were obtained by slow evaporation of aqueous solution of 3 at room temperature.
- [Cu(HLPh)Cl]Cl·5H2O (4). Yield: 73.2%; IR (ATR): ῦ = 3437, 3364, 3119, 2955, 2849, 2798, 1584, 1542, 1479, 1443, 1325, 1257, 1176, 970, 923, 874, 827, 758, 640 cm−1; UV/Vis (H2O): λmax (ε) = 601 (115), 382 (16,534), 323 nm (20,281 mol−1dm3cm−1); MS (ESI): m/z (%): 404 (100) [Cu(LPh)]+, 345 (80) [Cu(LPh–NMe3)]+; elemental analysis calcd (%) for C18H22Cl2CuN4OS·5H2O: C 38.13, H 5.69, N 9.88, S 5.66; found: C 38.17, H 5.48, N 9.70, S 5.81. The X-ray diffraction quality single crystals were obtained by slow evaporation of EtOH/H2O (3:1) solution of 4 at room temperature.
2.4. Spectroscopic Studies (UV/Vis and 1H NMR Titrations, Kinetic Measurements and Lipophilicity Determination)
2.5. X-ray Crystallography
2.6. Electrochemical and Spectroelectrochemical Studies
2.7. EPR Spectroscopy, ROS Generation and Antioxidative Activity
2.8. Cytotoxic Activity
2.8.1. Cell Lines
2.8.2. Cytotoxicity Assay
2.8.3. Antiproliferation Assay
[3H]-MPP+ Uptake Inhibition Assay
3. Results and Discussion
3.1. Synthesis and Characterisation of Proligands and Copper(II) Complexes
3.2. Single Crystal X-ray Diffraction Analysis
3.3. A Comparative Solution Equilibrium Study on Proligands and Their Copper(II) Complexes
3.3.1. Proton Dissociation Processes and Lipophilicity of the Proligands
3.3.2. Solution Stability of Copper(II) Complexes and Their Reduction by GSH
3.4. Cyclic Voltammetry and Spectroelectrochemistry
3.5. Electron Paramagnetic Resonance Spectroscopy, ROS Generation and Antioxidant Activity in Cell-Free Media
3.6. Cytotoxicity
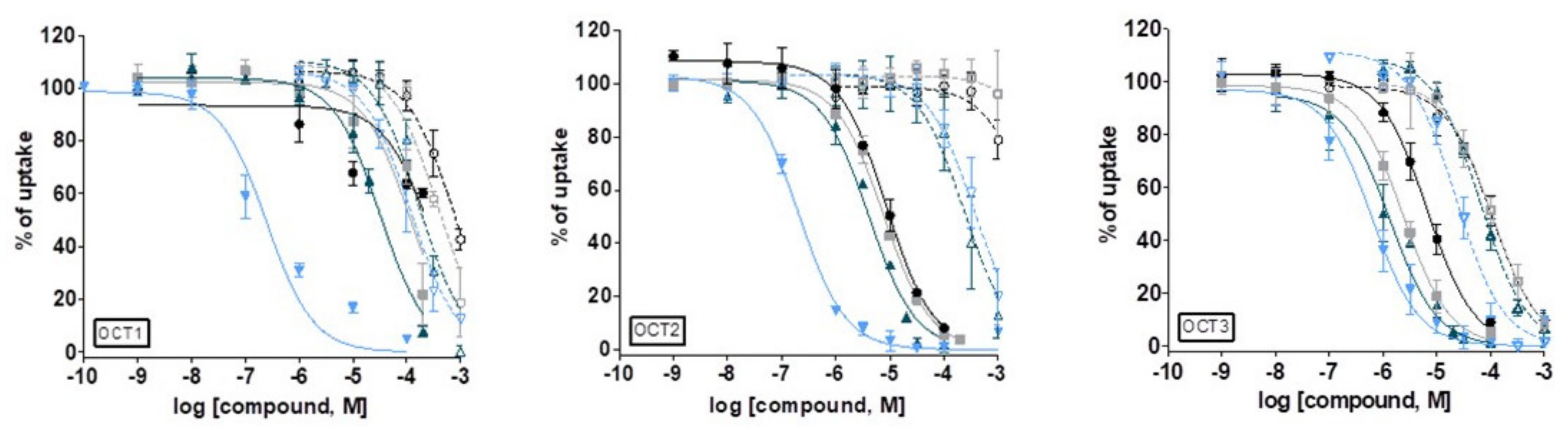
3.7. Interaction with OCT1, 2 and 3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brockman, R.W.; Thomson, J.R.; Bell, M.J.; Skipper, H.E. Observations on the antileukemic activity of pyridine-2-carboxaldehyde thiosemicarbazone and thiocarbohydrazone. Cancer Res. 1956, 16, 167–170. [Google Scholar] [PubMed]
- French, F.A.; Blanz, E.J.; DoAmaral, J.R.; French, D.A. Carcinostatic activity of thiosemicarbazones of formyl heteroaromatic compounds. VII. 2-Formylpyridine derivatives bearing additional ring substituents. J. Med. Chem. 1970, 13, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, K.C.; Schenkman, J.B.; Denk, H.; Mooney, P.D.; Moore, C.E.; Wodinsky, I.; Sartorelli, A.C. 4-Methyl-5-amino-1-formylisoquinoline Thiosemicarbazone, a Second-Generation Antineoplastic Agent of the a-(N) Heterocyclic Carboxaldehyde Thiosemicarbazone Series. Cancer Res. 1977, 37, 1692–1696. [Google Scholar] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef]
- Dose-finding and Pharmacokinetic Study of DpC, Administered Orally to Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02688101 (accessed on 26 July 2020).
- Study of COTI-2 as Monotherapy or Combination Therapy for the Treatment of Malignancies (COTI2-101). Available online: https://clinicaltrials.gov/ct2/show/NCT02433626 (accessed on 26 July 2020).
- Aye, Y.; Long, M.J.C.; Stubbe, J.J. Mechanistic Studies of Semicarbazone Triapine Targeting Human Ribonucleotide Reductase in Vitro and in Mammalian Cells: Tyrosyl Radical Quenching Not Involving Reactive Oxygen Species. Biol. Chem. 2012, 287, 35768–35778. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Thiosemicarbazones from the Old to New: Iron Chelators That Are More Than Just Ribonucleotide Reductase Inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef]
- Guo, Z.-L.; Richardson, D.R.; Kalinowski, D.S.; Kovacevic, Z.; Tan-Un, K.C.; Chan, G.C.-F. The novel thiosemicarbazone, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC), inhibits neuroblastoma growth in vitro and in vivo via multiple mechanisms. J. Hematol. Oncol. 2016, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Zeglis, B.M.; Divilov, V.; Lewis, J.S. Role of Metalation in the Topoisomerase IIα Inhibition and Antiproliferation Activity of a Series of α-Heterocyclic-N4-Substituted Thiosemicarbazones and Their Cu(II) Complexes. J. Med. Chem. 2011, 54, 2391–2398. [Google Scholar] [CrossRef]
- Heffeter, P.; Pape, V.F.S.; Enyedy, É.A.; Keppler, B.K.; Szakacs, G.; Kowol, C.R. Anticancer Thiosemicarbazones: Chemical Properties, Interaction with Iron Metabolism, and Resistance Development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef]
- Paterson, B.M.; Donnelly, P.S. Copper complexes of bis(thiosemicarbazones): From chemotherapeutics to diagnostic and therapeutic radiopharmaceuticals. Chem. Soc. Rev. 2011, 40, 3005–3018. [Google Scholar] [CrossRef]
- De Siqueira, L.R.P.; de Moraes Gomes, P.A.T.; de Lima Ferreira, L.P.; de Melo Rêgo, M.J.B.; Leite, A.C.L. Multi-target compounds acting in cancer progression: Focus on thiosemicarbazone, thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019, 170, 237–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, D.; Xie, C.; Zheng, T.; Liang, Y.; Hong, X.; Lu, Z.; Song, X.; Song, R.; Yang, H.; et al. The Iron Chelator Dp44mT Inhibits Hepatocellular carcinoma Metastasis via N-Myc Downstream-regulated Gene 2 (NDRG2)/gp130/STAT3 Pathway. Oncotarget 2014, 5, 8478–8491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J. Novel Di-2-pyridyl-derived Iron Chelators with Marked and Selective Antitumor Activity: In vitro and in vivo Assessment. Blood 2004, 104, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Novel chelators for cancer treatment: Where are we now? Antioxid. Redox Signal. 2013, 18, 973–1006. [Google Scholar] [CrossRef] [PubMed]
- Serda, M.; Kalinowski, D.S.; Rasko, N.; Potůčková, E.; Mrozek-Wilczkiewicz, A.; Musiol, R.; Małecki, J.G.; Sajewicz, M.; Ratuszna, A.; Muchowicz, A.; et al. Exploring the Anti-Cancer Activity of Novel Thiosemicarbazones Generated through the Combination of Retro-Fragments: Dissection of Critical Structure-Activity Relationships. PLoS ONE 2014, 9, e110291. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.R.; Sharpe, P.C.; Lovejoy, D.B.; Senaratne, D.; Kalinowski, D.S.; Islam, M.; Bernhardt, P.V. Dipyridyl Thiosemicarbazone Chelators with Potent and Selective Antitumor Activity Form Iron Complexes with Redox Activity. J. Med. Chem. 2006, 49, 6510–6521. [Google Scholar] [CrossRef]
- Pelosi, G. Thiosemicarbazone Metal Complexes: From Structure to Activity. Open Crystallogr. J. 2010, 3, 16–28. [Google Scholar] [CrossRef]
- Bisceglie, F.; Tavone, M.; Mussi, F.; Azzoni, S.; Montalbano, S.; Franzoni, S.; Tarasconi, P.; Buschini, A.; Pelosi, G.J. Effects of polar substituents on the biological activity of thiosemicarbazone metal complexes. Inorg. Biochem. 2018, 179, 60–70. [Google Scholar] [CrossRef]
- Milunovic, M.N.M.; Enyedy, É.A.; Nagy, N.V.; Kiss, T.; Trondl, R.; Jakupec, M.A.; Keppler, B.K.; Krachler, R.; Novitchi, G.; Arion, V.B. L- and D-Proline Thiosemicarbazone Conjugates: Coordination Behavior in Solution and the Effect of Copper(II) Coordination on Their Antiproliferative Activity. Inorg. Chem. 2012, 51, 9309–9321. [Google Scholar] [CrossRef] [Green Version]
- Dobrova, A.; Platzer, S.; Bacher, F.; Milunovic, M.N.M.; Dobrov, A.; Spengler, G.; Enyedy, É.A.; Novitchi, G.; Arion, V.B. Structure–antiproliferative Activity Studies on L-proline- and Homoproline-4-N-pyrrolidine-3-thiosemicarbazon Hybrids and Their Nickel(ii), Palladium(ii) and Copper(ii) Complexes. Dalton Trans. 2016, 45, 13427–13439. [Google Scholar] [CrossRef] [Green Version]
- Zaltariov, M.F.; Hammerstad, M.; Arabshahi, H.J.; Jovanović, K.; Richter, K.W.; Cazacu, M.; Shova, S.; Balan, M.; Andersen, N.H.; Radulović, S.; et al. New Iminodiacetate–Thiosemicarbazone Hybrids and Their Copper(II) Complexes Are Potential Ribonucleotide Reductase R2 Inhibitors with High Antiproliferative Activity. Inorg. Chem. 2017, 56, 3532–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohui, K.; Afanasenko, E.; Bacher, F.; Ting, R.L.X.; Zafar, A.; Blanco-Cabra, N.; Torrents, E.; Dömötör, O.; May, N.V.; Darvasiova, D.; et al. New Water-Soluble Copper(II) Complexes with Morpholine–Thiosemicarbazone Hybrids: Insights into the Anticancer and Antibacterial Mode of Action. J. Med. Chem. 2019, 62, 512–530. [Google Scholar] [PubMed] [Green Version]
- Đilović, I.; Rubčić, M.; Vrdoljak, V.; Pavelić, S.K.; Kralj, M.; Piantanida, I.; Cindrić, M. Novel thiosemicarbazone derivatives as potential antitumor agents: Synthesis, physicochemical and structural properties, DNA interactions and antiproliferative activity. Bioorg. Med. Chem. 2008, 16, 5189–5198. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharm. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Keogh, J.; Hagenbuch, B.; Rynn, C.; Stieger, B.; Nicholls, G. Membrane Transporters: Fundamentals, Function and Their Role in ADME; Royal Society of Chemistry: London, UK, 2016; pp. 1–56. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, T.; Wu, F.; Li, N.; Wang, R.; Zhao, Q.; Ma, Y.-M.; Zhang, J.-Q.; Ma, B.-L. Subcellular drug distribution: Mechanisms and roles in drug efficacy, toxicity, resistance, and targeted delivery. Drug Metab. Rev. 2018, 50, 430–447. [Google Scholar] [CrossRef]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef] [Green Version]
- Taubert, D.; Grimberg, G.; Stenzel, W.; Schömig, E. Identification of the Endogenous Key Substrates of the Human Organic Cation Transporter OCT2 and Their Implication in Function of Dopaminergic Neurons. PLoS ONE 2007, 2, e385. [Google Scholar] [CrossRef] [Green Version]
- Hayer-Zillgen, M.; Brüss, M.; Bönisch, H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br. J. Pharm. 2002, 136, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Jonker, J.W.; Schinkel, A.H. Pharmacological and Physiological Functions of the Polyspecific Organic Cation Transporters: OCT1, 2, and 3 (SLC22A1-3). J. Pharm. Exp. 2004, 308, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; Barilli, A.; Riccardi, B.; Puccini, P.; Dall’Asta, V. Functional characterization of the organic cation transporters (OCTs) in human airway pulmonary epithelial cells. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1563–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrach, S.; Ciarimboli, G. Role of transporters in the distribution of platinum-based drugs. Front. Pharm. 2015, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarimboli, G. Role of organic cation transporters in drug-induced toxicity. Expert. Opin. Drug Metab. Toxicol. 2011, 7, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Pantarat, N.; Lovejoy, D.B.; Kalinowski, D.S.; Richardson, D.R. Membrane Transport and Intracellular Sequestration of Novel Thiosemicarbazone Chelators for the Treatment of Cancer. Mol. Pharm. 2010, 78, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Price, K.A.; Crouch, P.J.; Volitakis, I.; Paterson, B.M.; Lim, S.; Donnelly, P.S.; White, A.R. Mechanisms Controlling the Cellular Accumulation of Copper Bis(thiosemicarbazonato) Complexes. Inorg. Chem. 2011, 50, 9594–9605. [Google Scholar] [CrossRef]
- Fung, K.L.; Tepede, A.K.; Pluchino, K.M.; Pouliot, L.M.; Pixley, J.N.; Hall, M.D.; Gottesman, M.M. Uptake of Compounds That Selectively Kill Multidrug-Resistant Cells: The Copper Transporter SLC31A1 (CTR1) Increases Cellular Accumulation of the Thiosemicarbazone NSC73306. Mol. Pharm. 2014, 11, 2692–2702. [Google Scholar] [CrossRef] [Green Version]
- Jansson, P.J.; Yamagishi, T.; Arvind, A.; Seebacher, N.; Gutierrez, E.; Stacy, A.; Maleki, S.; Sharp, D.; Sahni, S.; Richardson, D.R. Di-2-pyridylketone 4,4-Dimethyl-3-thiosemicarbazone (Dp44mT) Overcomes Multidrug Resistance by a Novel Mechanism Involving the Hijacking of Lysosomal P-Glycoprotein (Pgp). J. Biol. Chem. 2015, 290, 9588–9603. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, V.; Fogagnolo, M.; Marchi, A.; Marvelli, L.; Sforza, F.; Bergamini, P. Acetylcholine-like and Trimethylglycine-like PTA (1,3,5-Triaza-7-phosphaadamantane) Derivatives for the Development of Innovative Ru- and Pt-Based Therapeutic Agents. Inorg. Chem. 2014, 53, 4881–4890. [Google Scholar] [CrossRef]
- Sala-Rabanal, M.; Li, D.C.; Dake, G.R.; Kurata, H.T.; Inyushin, M.; Skatchkov, S.N.; Nichols, C.G. Polyamine Transport by the Polyspecific Organic Cation Transporters OCT1, OCT2, and OCT3. Mol. Pharm. 2013, 10, 1450–1458. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Tsurutani, K.; Komatsu, A.; Ito, T.; Iida, K.; Fujii, Y.; Nakano, Y.; Usui, Y.; Fukuda, Y.; Chikira, M. Synthesis of New Cationic Shiff Base Complexes of Copper(II) and Their Selective Binding with DNA. Bull. Chem. Soc. Jpn. 1997, 70, 615–629. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. Computational Methods for the Determination of Stability Constants; Leggett, D.J., Ed.; Plenum Press: New York, NY, USA, 1985; pp. 291–353. [Google Scholar]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A Study of Some Problems in Determining the Stoicheiometric Proton Dissociation Constants of Complexes by Potentiometric Titrations Using a Glass Electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Primik, M.F.; Kowol, C.R.; Arion, V.B.; Kiss, T.; Keppler, B.K. Interaction of Triapine and Related Thiosemicarbazones with Iron(III)/(II) and Gallium(III): A Comparative Solution Equilibrium Study. Dalton Trans. 2011, 40, 5895–5905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowol, C.R.; Nagy, N.V.; Jakusch, T.; Roller, A.; Heffeter, P.; Keppler, B.K.; Enyedy, É.A. Vanadium(IV/V) Complexes of Triapine and Related Thiosemicarbazones: Synthesis, Solution Equilibrium and Bioactivity. J. Inorg. Biochem. 2015, 152, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CrysAlis RED; Version 1.171.40.53; Oxford Diffraction Ltd.: Oxford, UK, 2003.
- Kabsch, W. XDS. Acta Cryst. D Biol. Cryst. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- GraphPad Software I, Version 5.0; GraphPad Prism; GraphPad Software Inc.: New York, NY, USA, 2007.
- Fraser-Spears, R.; Krause-Heuer, A.M.; Basiouny, M.; Mayer, F.P.; Manishimwe, R.; Wyatt, N.A.; Dobrowolski, J.C.; Roberts, M.P.; Greguric, I.; Kumar, N.; et al. Comparative analysis of novel decynium-22 analogs to inhibit transport by the low-affinity, high-capacity monoamine transporters, organic cation transporters 2 and 3, and plasma membrane monoamine transporter. Eur. J. Pharm. 2019, 842, 351–364. [Google Scholar] [CrossRef]
- Kalaba, P.; Ilić, M.; Aher, N.Y.; Dragačević, V.; Wieder, M.; Zehl, M.; Wackerlig, J.; Beyl, S.; Sartori, S.B.; Ebner, K.; et al. Structure–Activity Relationships of Novel Thiazole-Based Modafinil Analogues Acting at Monoamine Transporters. Med. Chem. 2020, 63, 391–417. [Google Scholar] [CrossRef]
- Sîrbu, A.; Palamarciuc, O.; Babak, M.V.; Lim, J.M.; Ohui, K.; Enyedy, E.A.; Shova, S.; Darvasiová, D.; Rapta, P.; Ang, W.H.; et al. Copper(II) thiosemicarbazone complexes induce marked ROS accumulation and promote nrf2-mediated antioxidant response in highly resistant breast cancer cells. Dalton Trans. 2017, 46, 3833–3847. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Gou, S.; Zhu, H.; Huang, W. Cleavage of C–S Bonds with the Formation of a Tetranuclear Cu(I) Cluster. Inorg. Chem. 2007, 46, 5537–5543. [Google Scholar] [CrossRef]
- Yang, Y.; Guan, J.; Qiu, P.; Kan, Q. Synthesis, characterization and catalytic properties of heterogeneous iron(III) tetradentate Schiff base complexes for the aerobic epoxidation of styrene. Transit. Met. Chem. 2010, 35, 263–270. [Google Scholar] [CrossRef]
- Kowol, C.R.; Miklos, W.; Pfaff, S.; Hager, S.; Kallus, S.; Pelivan, K.; Kubanik, M.; Enyedy, É.A.; Berger, W.; Heffeter, P.; et al. Impact of Stepwise NH2-Methylation of Triapine on the Physicochemical Properties, Anticancer Activity, and Resistance Circumvention. J. Med. Chem. 2016, 59, 6739–6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enyedy, É.A.; Zsigó, É.; Nagy, N.V.; Kowol, C.R.; Roller, A.; Keppler, B.K.; Kiss, T. Complex-Formation Ability of Salicylaldehyde Thiosemicarbazone towards ZnII, CuII, FeII, FeIII and GaIII Ions. Eur. J. Inorg. Chem. 2012, 2012, 4036–4047. [Google Scholar] [CrossRef] [Green Version]
- West, D.W.; Ives, J.S.; Krejci, J.; Salberg, M.M.; Zumbahlen, T.L.; Bain, G.A.; Liberta, A.E.; Valdes-Martinez, J.; Hernandez-Ortiz, S.; Toscano, R.A. Copper(II) Complexes of 2-Benzoylpyridine 4N-Substituted Thiosemicarbazones. Polyhedrone 1995, 14, 2189–2200. [Google Scholar] [CrossRef]
- Bernhardt, P.V.; Martínez, M.; Rodríguez, C.; Vazquez, M. Biologically Active Thiosemicarbazone Fe Chelators and Their Reactions with Ferrioxamine B and Ferric EDTA; a Kinetic Study. Dalton Trans. 2012, 41, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- Kowol, C.R.; Eichinger, R.; Jakupec, M.A.; Galanski, M.; Arion, V.B.; Keppler, B.K. Effect of Metal Ion Complexation and Chalcogen Donor Identity on the Antiproliferative Activity of 2-Acetylpyridine N,N-Dimethyl(chalcogen)semicarbazones. J. Inorg. Biochem. 2007, 101, 1946–1957. [Google Scholar] [CrossRef]
- Dömötör, O.; May, N.V.; Pelivan, K.; Kiss, T.; Keppler, B.K.; Kowol, C.R.; Enyedy, É.A. A Comparative Study of α-N-pyridyl Thiosemicarbazones: Spectroscopic Properties, Solution Stability and Copper(II) Complexation. Inorg. Chim. Acta 2018, 472, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Hager, S.; Pape, V.F.S.; Pósa, V.; Montsch, B.; Uhlik, L.; Szakács, G.; Tóth, S.; Jabronka, N.; Keppler, B.K.; Kowol, C.R.; et al. High Copper Complex Stability and Slow Reduction Kinetics as Key Parameters for Improved Activity, Paraptosis Induction and Impact on Drug-resistant Cells of Anticancer Thiosemicarbazones. Antioxid. Redox Signal. 2020, 33, 395–414. [Google Scholar] [CrossRef]
- Park, K.C.; Fouani, L.; Jansson, P.J.; Wooi, D.; Sahni, S.; Lane, D.J.R.; Palanimuthu, D.; Lok, H.C.; Kovačević, Z.; Huang, M.L.H.; et al. Copper and Conquer: Copper Complexes of Di-2-pyridylketone Thiosemicarbazones as Novel Anti-cancer Therapeutics. Metallomics 2016, 8, 874–886. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Vileno, B.; Palacios, Ò.; Peris-Díaz, M.D.; Riegel, G.; Gaiddon, C.; Krężel, A.; Faller, P. Reactivity of Cu(II)–, Zn(II)– and Fe(II)–thiosemicarbazone Complexes with Glutathione and Metallothionein: From Stability to Dissociation to Transmetallation. Metallomics 2019, 11, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.R.; Dilworth, J.R.; Donnelly, P.S.; Labisbal, E.; Sousa, A.J. An Unusual Dimeric Structure of a Cu(I) Bis(thiosemicarbazone) Complex: Implications for the Mechanism of Hypoxic Selectivity of the Cu(II) Derivatives. Am. Chem. Soc. 2002, 124, 5270–5271. [Google Scholar] [CrossRef] [PubMed]
- Lobana, T.S.; Sharma, R.; Hundal, G.; Castineiras, A.; Butcher, R.J. The Influence of Substituents (R) at N1 atom of Furan-2-carbaldehyde Thiosemicarbazones {(C4H3O)HC2N3–N(H)–C1(S)N1HR} on Bonding, Nuclearity, H-bonded Networks of Copper(I) Complexes. Polyhedron 2012, 47, 134–142. [Google Scholar] [CrossRef]
- Srinivas, K.; Prabusankar, G. Role of C, S, Se and P donor ligands in copper(I) mediated C–N and C–Si bond formation reactions. RSC Adv. 2018, 8, 32269–32282. [Google Scholar] [CrossRef] [Green Version]
- Ashfield, L.J.; Cowley, A.R.; Dilworth, J.R.; Donnelly, P.S. Functionalized Thiosemicarbazone Clusters of Copper(I) and Silver(I). Inorg. Chem. 2004, 43, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Mahendiran, D.; Pravin, N.; Bhuvanesh, N.S.P.; Kumar, R.S.; Viswanathan, V.; Velmurugan, D.; Rahiman, A.K. Bis(thiosemicarbazone)copper(I) Complexes as Prospective Therapeutic Agents: Interaction with DNA/BSA Molecules, and In Vitro and In Vivo Anti-Proliferative Activities. ChemistrySelect 2018, 3, 7100–7111. [Google Scholar] [CrossRef]
- Jansson, P.J.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Novel Thiosemicarbazones of the ApT and DpT Series and Their Copper Complexes: Identification of Pronounced Redox Activity and Characterization of Their Antitumor Activity. J. Med. Chem. 2010, 53, 5759–5769. [Google Scholar] [CrossRef]
- Casella, L.; Gullotti, M.; Pintar, A.; Pinciroli, F.; Viganò, R.; Zanello, P.J. Blue copper models. Spectroscopic and electrochemical studies of copper(II) complexes with new ligand systems containing sulphur and nitrogen donor atoms. Chem. Soc. Dalton Trans. 1989, 1161–1169. [Google Scholar] [CrossRef]
- Yu, Q.; Salhi, C.A.; Ambundo, E.A.; Heeg, M.J.; Ochrymowycz, L.A.; Rorabacher, D.B. Direct Evidence for a Geometrically Constrained “Entatic State” Effect on Copper(II/I) Electron-Transfer Kinetics As Manifested in Metastable Intermediates. J. Am. Chem. Soc. 2001, 123, 5720–5729. [Google Scholar] [CrossRef]
- Xie, B.; Wilson, L.J.; Stanbury, D.M. Cross-Electron-Transfer Reactions of the [CuII/I(bite)]2+/+ Redox Couple. Inorg. Chem. 2001, 40, 3606–3614. [Google Scholar] [CrossRef]
- Rapta, P.; Kožíšek, J.; Breza, M.; Gembický, M.; Dunsch, L.J. ESR/UV–Vis–NIR cyclovoltammetry of macrocyclic complex [CuI(bite)]BF4 at different temperatures. Electroanal. Chem. 2004, 566, 123–129. [Google Scholar] [CrossRef]
- Santoro, A.; Calvo, J.S.; Peris-Díaz, M.D.; Krężel, A.; Meloni, G.; Faller, P. The Glutathione/Metallothionein System Challenges the Design of Efficient O2-Activating Copper Complexes. Angew. Chem. Int. Ed. 2020, 59, 7830–7835. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Sun, D.L.; Poddar, S.; Pan, R.D.; Rosser, E.W.; Abt, E.R.; Van Valkenburgh, J.; Le, T.M.; Lok, V.; Hernandez, S.P.; Song, J.; et al. Isoquinoline thiosemicarbazone displays potent anticancer activity with in vivo efficacy against aggressive leukemias. RSC Med. Chem. 2020, 11, 392–410. [Google Scholar] [CrossRef]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a Clinically Used Anti-Alcoholism Drug and Copper-Binding Agent, Induces Apoptotic Cell Death in Breast Cancer Cultures and Xenografts via Inhibition of the Proteasome Activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef] [Green Version]
- Brewer, G.J.; Dick, R.D.; Grover, D.K.; LeClaire, V.; Tseng, M.; Wicha, M.; Pienta, K.; Redman, B.G.; Jahan, T.; Sondak, V.K.; et al. Treatment of Metastatic Cancer with Tetrathiomolybdate, an Anticopper, Antiangiogenic Agent: Phase I Study. Clin. Cancer Res. 2000, 6, 1–10. [Google Scholar] [PubMed]
- Whitnall, M.; Howard, J.; Ponka, P.; Richardson, D.R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. USA 2006, 103, 14901–14906. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Reznik, E.; Stokes, M.E.; Krishnamoorthy, L.; Bos, P.H.; Song, Y.; Quartararo, C.E.; Pagano, N.C.; Carpizo, D.R.; de Carvalho, A.C.; et al. Copper-Binding Small Molecule Induces Oxidative Stress and Cell-Cycle Arrest in Glioblastoma-Patient-Derived Cells. Cell Chem. Biol. 2018, 25, 585–594. [Google Scholar] [CrossRef]
- Neuhoff, S.; Ungell, A.-L.; Zamora, I.; Artursson, P. pH-Dependent Bidirectional Transport of Weakly Basic Drugs Across Caco-2 Monolayers: Implications for Drug–Drug Interactions. Pharm. Res. 2003, 20, 1141–1148. [Google Scholar] [CrossRef]
- Wagner, D.J.; Hu, T.; Wang, J. Polyspecific organic cation transporters and their impact on drug intracellular levels and pharmacodynamics. Pharm. Res. 2016, 111, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Engel, K.; Wang, J. Interaction of Organic Cations with a Newly Identified Plasma Membrane Monoamine Transporter. Mol. Pharm. 2005, 68, 1397–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, F.P.; Schmid, D.; Owens, W.A.; Gould, G.G.; Apuschkin, M.; Kudlacek, O.; Salzer, I.; Boehm, S.; Chiba, P.; Williams, P.H.; et al. An unsuspected role for organic cation transporter 3 in the actions of amphetamine. Neuropsychopharmacology 2018, 43, 2408–2417. [Google Scholar] [CrossRef] [PubMed]









 ), [H2LPh]Cl (
), [H2LPh]Cl (  ), 1 (●), 2 (■), 3 (▲), 4 (▼).
), [H2LPh]Cl ( ), 1 (●), 2 (■), 3 (▲), 4 (▼).
), 1 (●), 2 (■), 3 (▲), 4 (▼).
), [H2LPh]Cl ( ), 1 (●), 2 (■), 3 (▲), 4 (▼).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 2 | 3 | 4 |
|---|---|---|---|
| Empirical formula | C26H49.81Cl4Cu2N8O6.91S2 | C14H28.45ClCuN4O4.22S | C18H32Cl2CuN4O6S |
| Formula weight | 918.03 | 451.51 | 566.97 |
| Temperature/K | 293 | 100 | 293 |
| Crystal system | triclinic | monoclinic | monoclinic |
| Space group | P-1 | C2/c | P21/c |
| a/Å | 11.8147(4) | 16.951(3) | 8.7395(8) |
| b/Å | 13.5950(5) | 25.050(5) | 34.147(2) |
| c/Å | 13.7575(5) | 9.7040(19) | 8.8692(6) |
| α/° | 89.099(3) | ||
| β/° | 65.774(4) | 106.39(3) | 108.701(9) |
| γ/° | 86.133(3) | ||
| V/Å3 | 2010.38(14) | 3952.9(15) | 2507.1(4) |
| Z | 2 | 8 | 4 |
| Dcalc/mg/mm3 | 1.517 | 1.517 | 1.502 |
| μ/mm−1 | 5.143 | 1.320 | 1.208 |
| Crystal size/mm3 | 0.30 × 0.10 × 0.02 | 0.05 × 0.04 × 0.015 | 0.01 × 0.01 × 0.01 |
| θmin,θmax(°) | 6.516 to 133.18 | 2.94 to 48.586 | 6.028 to 50.05 |
| Reflections collected | 12,762 | 20,627 | 16,513 |
| Independent reflections | 70,383 [Rint = 0.0327] | 3294 [Rint = 0.0780] | 4421 [Rint = 0.0718] |
| Data/restraints/parameters | 7038/0/467 | 3294/0/253 | 4421/0/292 |
| GOF c | 0.984 | 1.04 | 1.043 |
| R1a (I > 2σ(I)) | 0.0482 | 0.0826 | 0.0584 |
| wR2b (all data) | 0.1395 | 0.2586 | 0.1035 |
| Largest diff. peak/hole/e Å−3 | 0.76/−0.84 | 1.08/−0 | 0.34/−0.38 |
| [H2LH]Cl | [H2LMe]Cl | [H2LEt]Cl | [H2LPh]Cl | |
|---|---|---|---|---|
| pKa (H2L+) | 7.46 ± 0.01 | 7.54 ± 0.01 | 7.31 ± 0.01 | 7.03 ± 0.01 |
| % HL at pH 7.4 | 47% | 42% | 55% | 70% |
| logD7.4 (proligand) | −0.84 ± 0.03 | −0.39 ± 0.01 | 0.06 ± 0.04 | 0.68 ± 0.04 |
| logβ [Cu(HL)]2+ [a] | 12.00 ± 0.01 | 11.73 ± 0.02 | 11.66 ± 0.03 | 11.26 ± 0.02 |
| logβ [CuL]+ [b] | 8.14 ± 0.01 | 7.76 ± 0.02 | 7.67 ± 0.03 | 8.55 ± 0.02 |
| logβ [CuLH−1] [c] | −1.66 ± 0.02 | −1.48 ± 0.08 | −1.65 ± 0.09 | −1.31 ± 0.05 |
| pKa [Cu(HL)]2+ | 3.86 | 3.97 | 3.99 | 2.71 |
| pKa [CuL]+ | 9.80 | 9.24 | 9.32 | 9.86 |
| logKderived [CuLH−1]+ [d] | 0.68 | 0.22 | 0.36 | 1.52 |
| pCu [e] | 12.21 | 11.79 | 11.82 | 12.80 |
| logD7.4 (complex) | −1.00 ± 0.01 | −0.79 ± 0.01 | −0.40 ± 0.01 | −0.17 ± 0.01 |
| kobs (min−1) | 0.080 ± 0.008 | 0.073 ± 0.002 | 0.058 ± 0.001 | 0.025 ± 0.005 |
| Compound | Colo205 (IC50, μM) [a] | Colo320 (IC50, μM) | SH-SY5Y (IC50, μM) | MRC-5 (IC50, μM) | ||||
|---|---|---|---|---|---|---|---|---|
| 24 h [b] | 72 h [b] | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |
| [H2LH]Cl | >100 [c] | >100 | >100 | 88.77 ± 5.32 | >100 | >100 | >100 | >100 |
| [H2LMe]Cl | >100 | >100 | >100 | 74.43 ± 3.85 | >100 | >100 | >100 | >100 |
| [H2LEt]Cl | >100 | >100 | 63.83 ± 3.92 | 53.30 ± 2.44 | 88.13 ± 9.19 | 49.86 ± 3.97 | >100 | >100 |
| [H2LPh]Cl | >100 | >100 | >100 | 95.81 ± 1.58 | >100 | 31.46 ± 2.66 | >100 | >100 |
| 1 | 88.79 ± 4.40 | >100 | >100 | 65.38 ± 3.71 | 45.44 ± 9.1 | 38.15 ± 1.15 | >100 | >100 |
| 2 | 73.4 ± 2.24 | 53.34 ± 3.31 | >100 | 80.64 ± 1.57 | 23.35 ± 3.57 | 15.35 ± 1.74 | >100 | 51.53 ± 4.67 |
| 3 | 27.51 ± 1.68 | 28.59 ± 2.02 | 30.47 ± 3.27 | 35.41 ± 1.87 | 10.34 ± 2.65 | 8.93 ± 0.5 | 57.79 ± 2.65 | 22.6 ± 1.81 |
| 4 | 36.99 ± 3.06 | 42.81 ± 4.23 | 45.02 ± 2.49 | 65.14 ± 2.93 | 38.07 ± 5.18 | 58.64 ± 7.18 | 52.71 ± 6.49 | 25.64 ± 2.57 |
| Cisplatin | 68.82 ± 5.08 | 8.14 ± 1.59 | 12.69 ± 0.79 | 2.12 ± 0.14 | 26.03 ± 2.38 | 0.65 ± 0.08 | 55.67 ± 4.06 | 1.73 ± 0.28 |
| Compound | OCT1 (IC50, μM) [a] | OCT2 (IC50, μM) [a] | OCT3 (IC50, μM) [a] |
|---|---|---|---|
| [H2L1]Cl | 740.2 ± 194.8 | >4000 | 105.1 ± 32.8 |
| [H2L2]Cl | 390.8 ± 40.0 | >4000 | 102.8 ± 23.4 |
| [H2L3]Cl | 168.6 ± 26.2 | 236.2 ± 97.1 | 63.1 ± 8.9 |
| [H2L4]Cl | 118.9 ± 50.0 | 370.8 ± 123.2 | 27.8 ± 5.0 |
| 1 | 268.6 ± 31.5 | 8.5 ± 0.9 | 6.6 ± 1.9 |
| 2 | 113.4 ± 21.8 | 7.6 ± 0.2 | 2.4 ± 0.4 |
| 3 | 32.2 ± 2.9 | 4.0 ± 0.3 | 1.3 ± 0.5 |
| 4 | 0.25 ± 0.08 | 0.20 ± 0.02 | 0.62 ± 0.27 |
| D-22 [b] | 0.98 ± 0.31 | 1.13 ± 0.19 | 0.09 ± 0.01 |
| Prazosin [b] | 1.84 ± 0.48 | >100 | 12.6 ± 2.93 |
| Corticosterone [b] | 21.7 ± 2.44 | 34.2 ± 6.47 | 0.29 ± 0.04 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milunović, M.N.M.; Palamarciuc, O.; Sirbu, A.; Shova, S.; Dumitrescu, D.; Dvoranová, D.; Rapta, P.; Petrasheuskaya, T.V.; Enyedy, E.A.; Spengler, G.; et al. Insight into the Anticancer Activity of Copper(II) 5-Methylenetrimethylammonium-Thiosemicarbazonates and Their Interaction with Organic Cation Transporters. Biomolecules 2020, 10, 1213. https://doi.org/10.3390/biom10091213
Milunović MNM, Palamarciuc O, Sirbu A, Shova S, Dumitrescu D, Dvoranová D, Rapta P, Petrasheuskaya TV, Enyedy EA, Spengler G, et al. Insight into the Anticancer Activity of Copper(II) 5-Methylenetrimethylammonium-Thiosemicarbazonates and Their Interaction with Organic Cation Transporters. Biomolecules. 2020; 10(9):1213. https://doi.org/10.3390/biom10091213
Chicago/Turabian StyleMilunović, Miljan N. M., Oleg Palamarciuc, Angela Sirbu, Sergiu Shova, Dan Dumitrescu, Dana Dvoranová, Peter Rapta, Tatsiana V. Petrasheuskaya, Eva A. Enyedy, Gabriella Spengler, and et al. 2020. "Insight into the Anticancer Activity of Copper(II) 5-Methylenetrimethylammonium-Thiosemicarbazonates and Their Interaction with Organic Cation Transporters" Biomolecules 10, no. 9: 1213. https://doi.org/10.3390/biom10091213