Role of Hydrogen Sulfide and 3-Mercaptopyruvate Sulfurtransferase in the Regulation of the Endoplasmic Reticulum Stress Response in Hepatocytes


Abstract
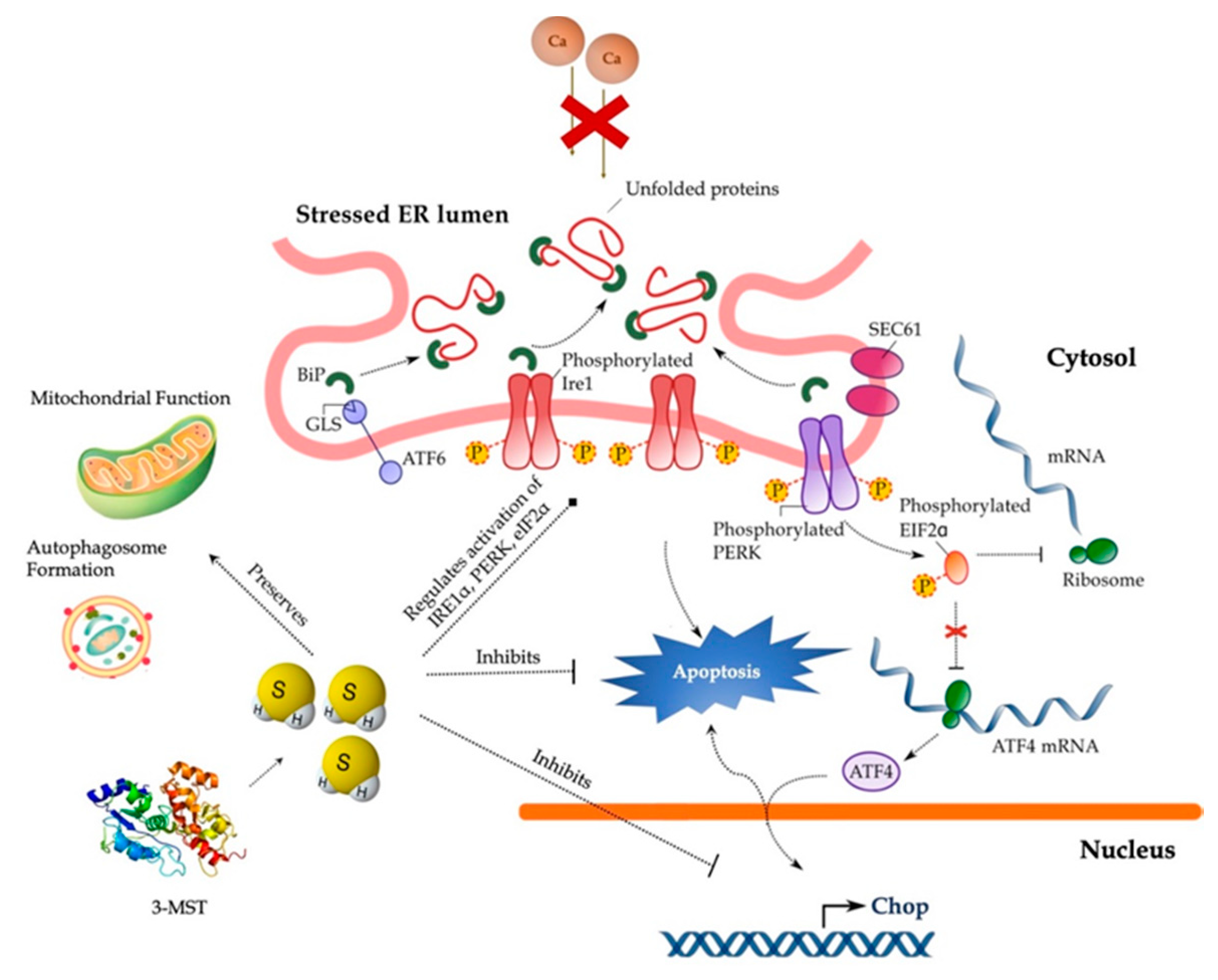
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Treatments
2.4. Cell Transfections
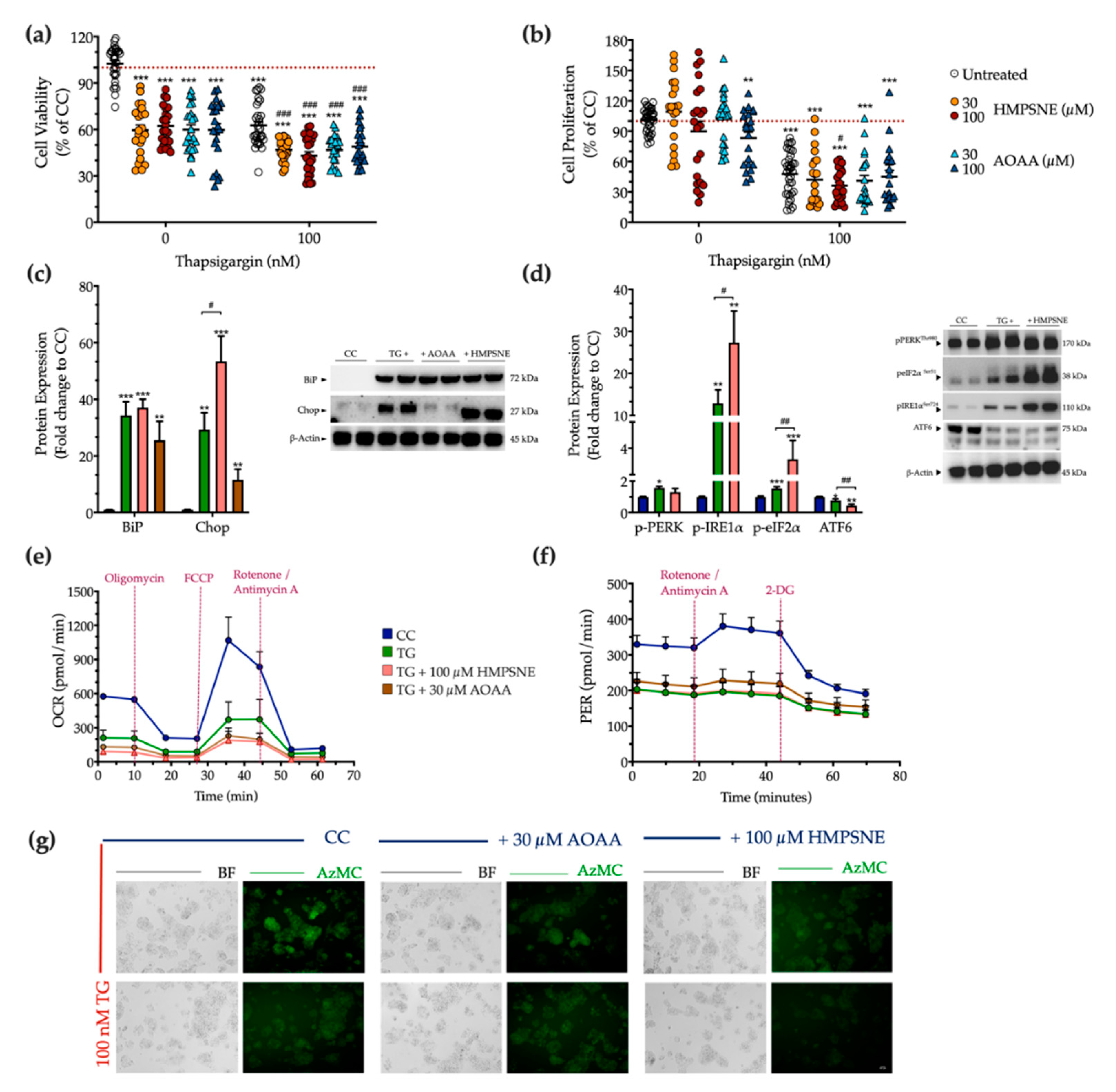
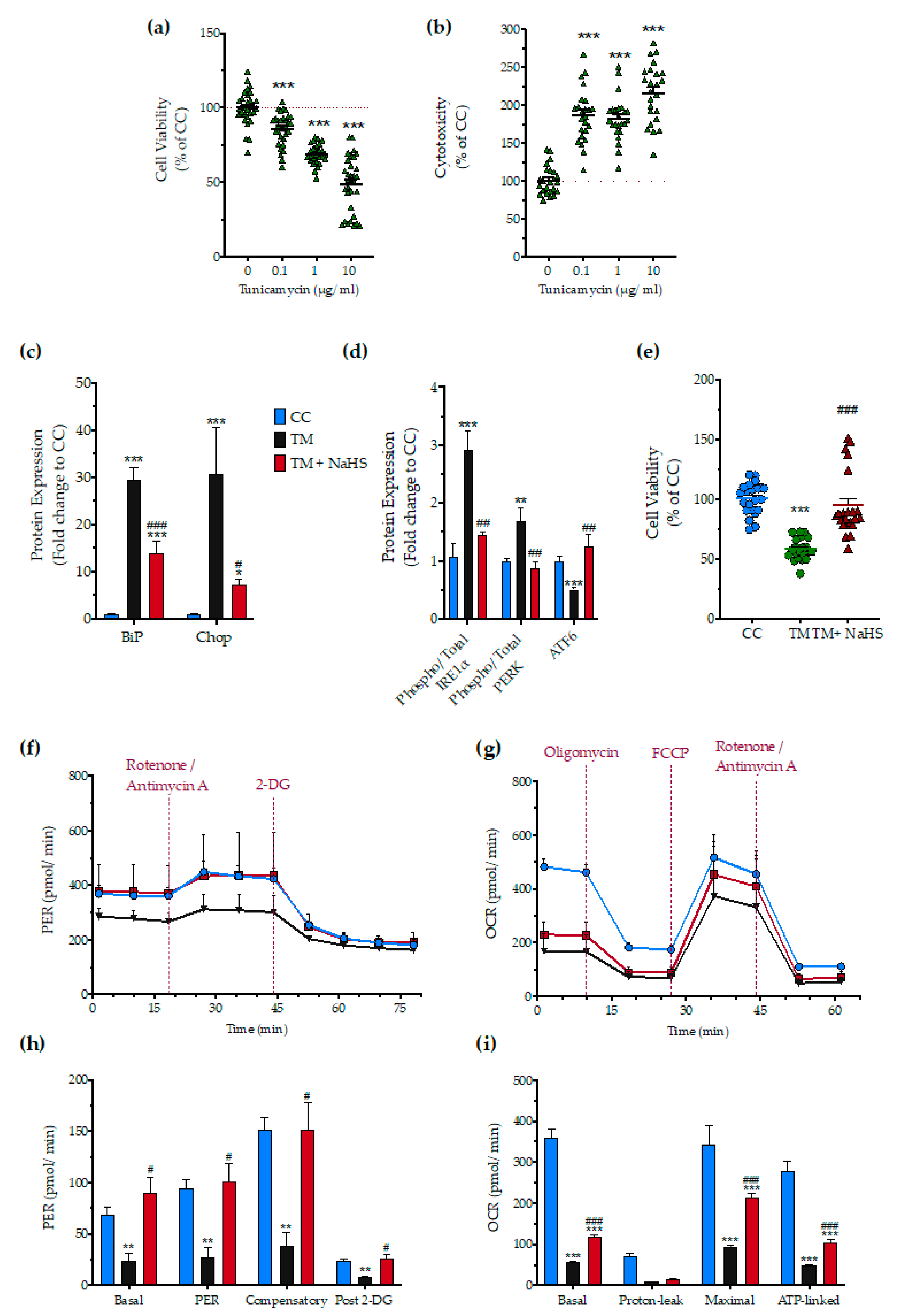
2.5. Assessment of Cell Viability and Proliferation
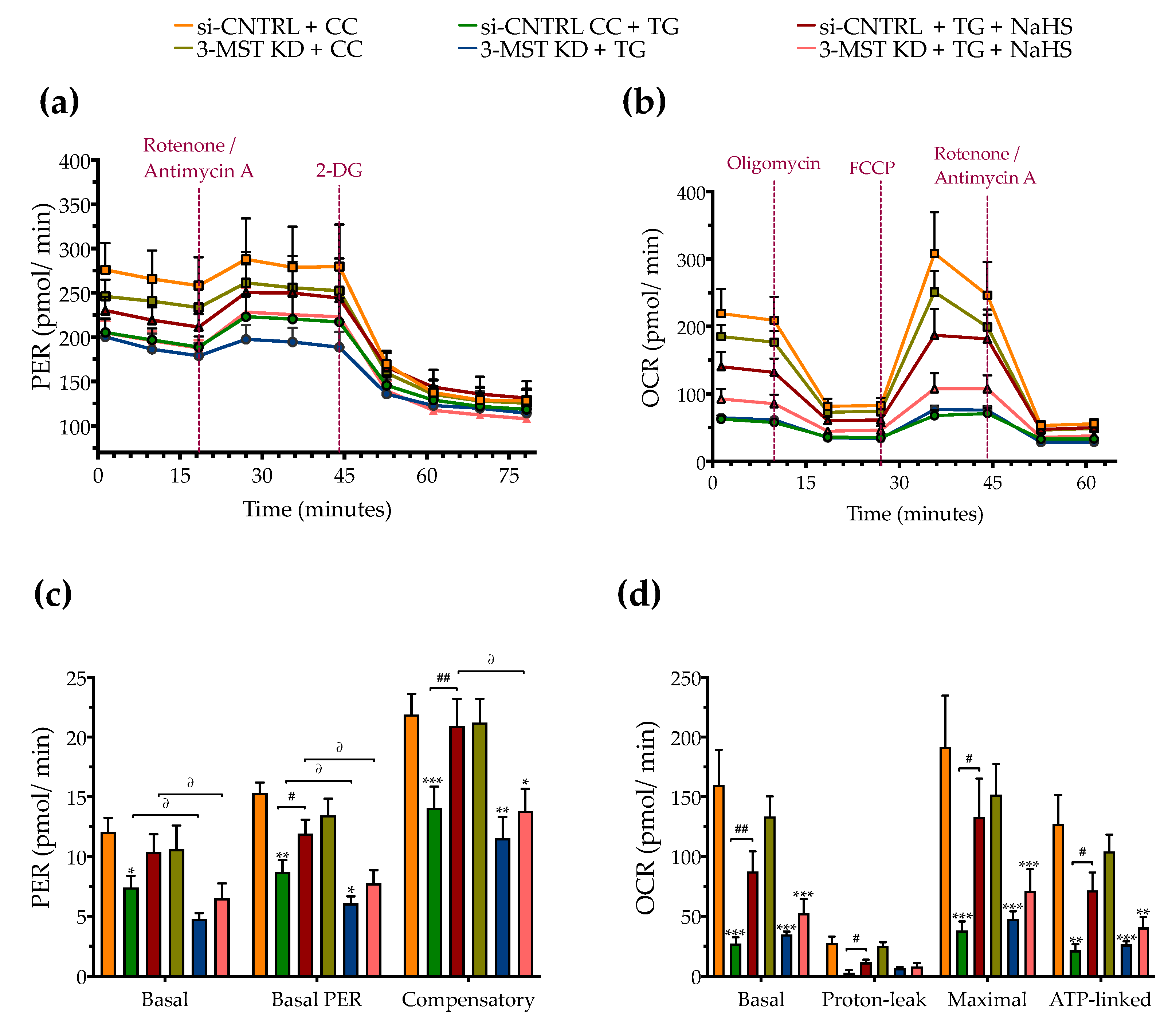
2.6. Assessment of Cellular Bioenergetics
2.7. Live-Cell Labelling for Autophagy and Endogenous H2S Quantification
2.8. Live-Cell Labeling for Mitochondrial Superoxide
2.9. Protein Extraction and Western Blotting
2.10. Statistics
3. Results
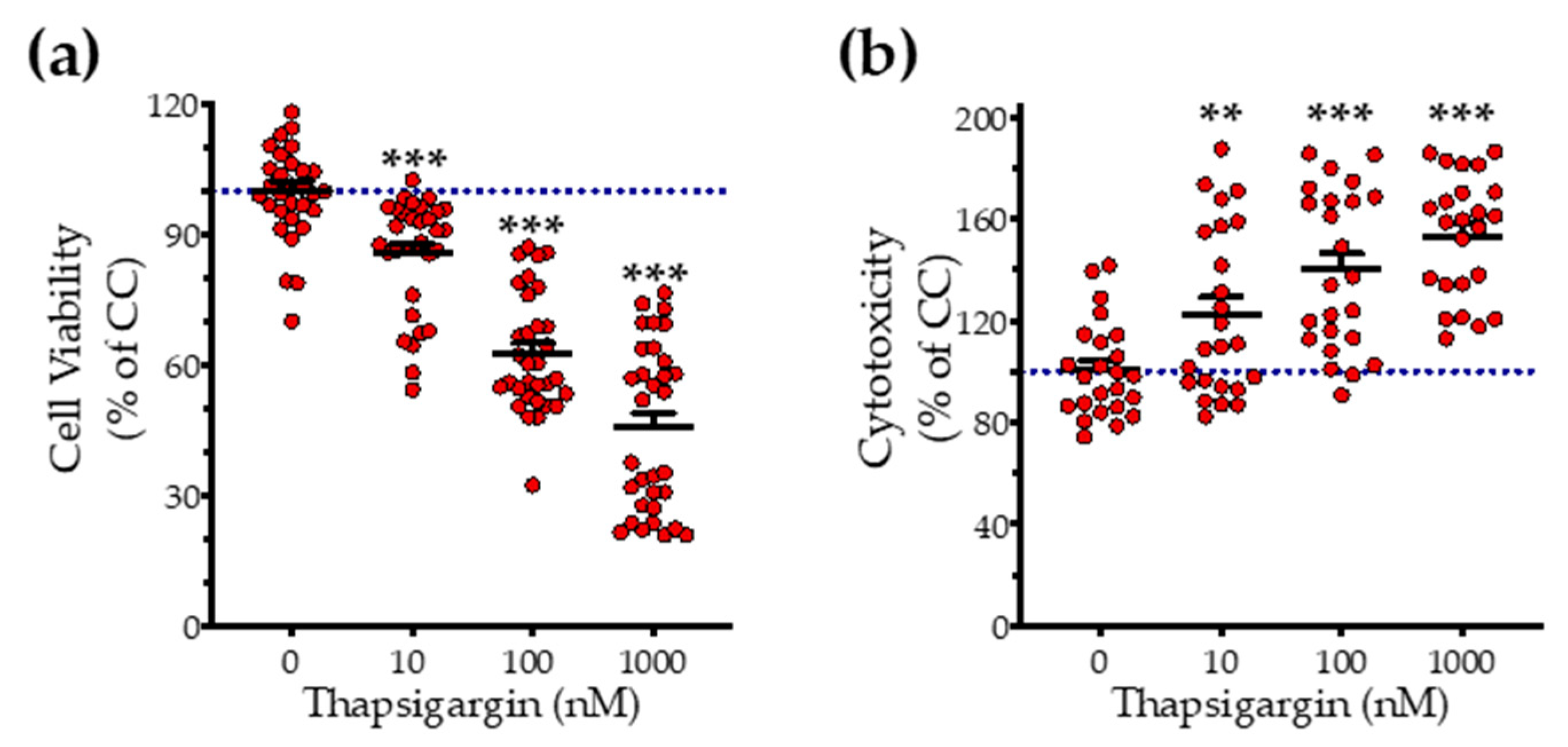
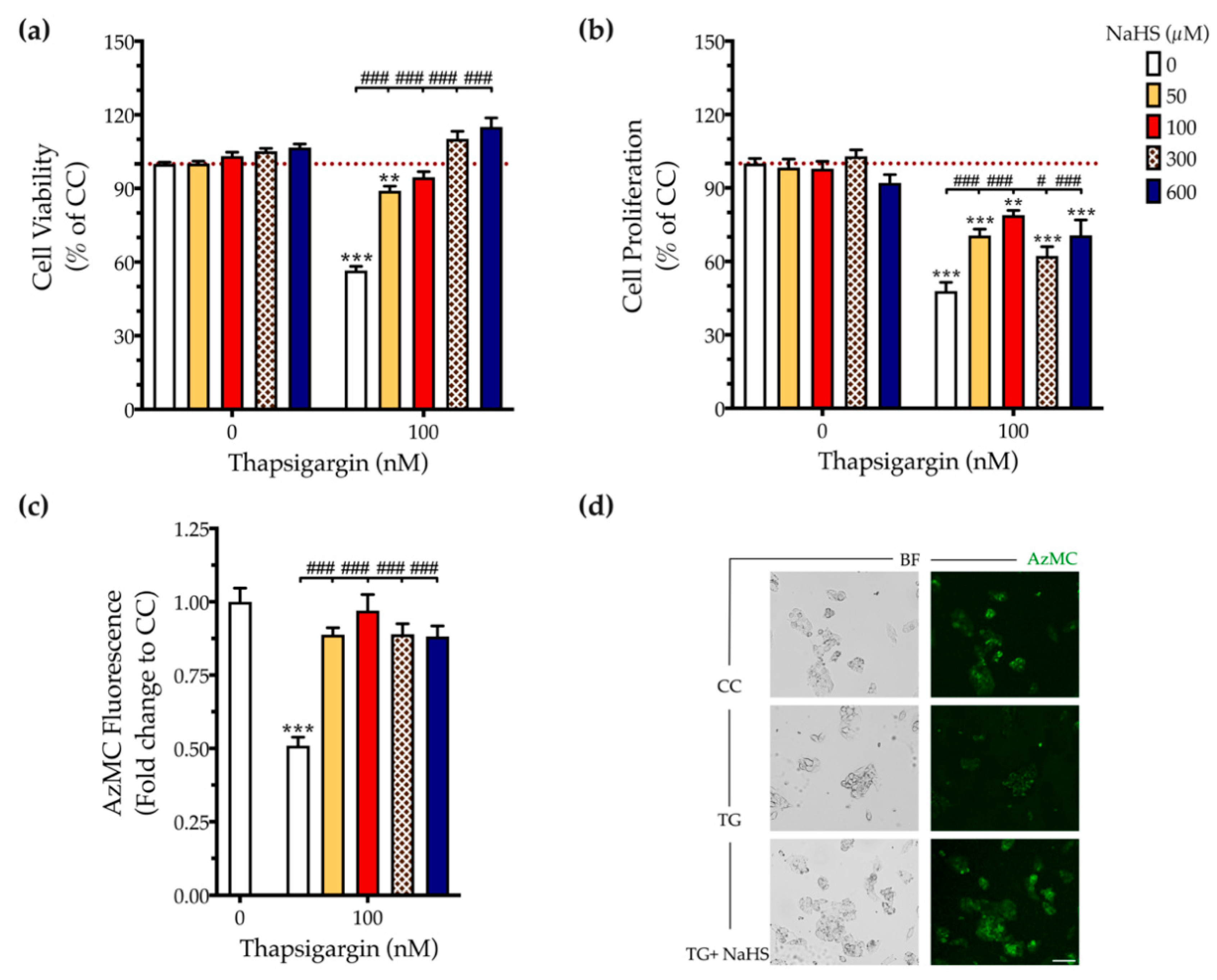
3.1. H2S Donation Rescues Thapsigargin-Induced Cell-Growth Arrest
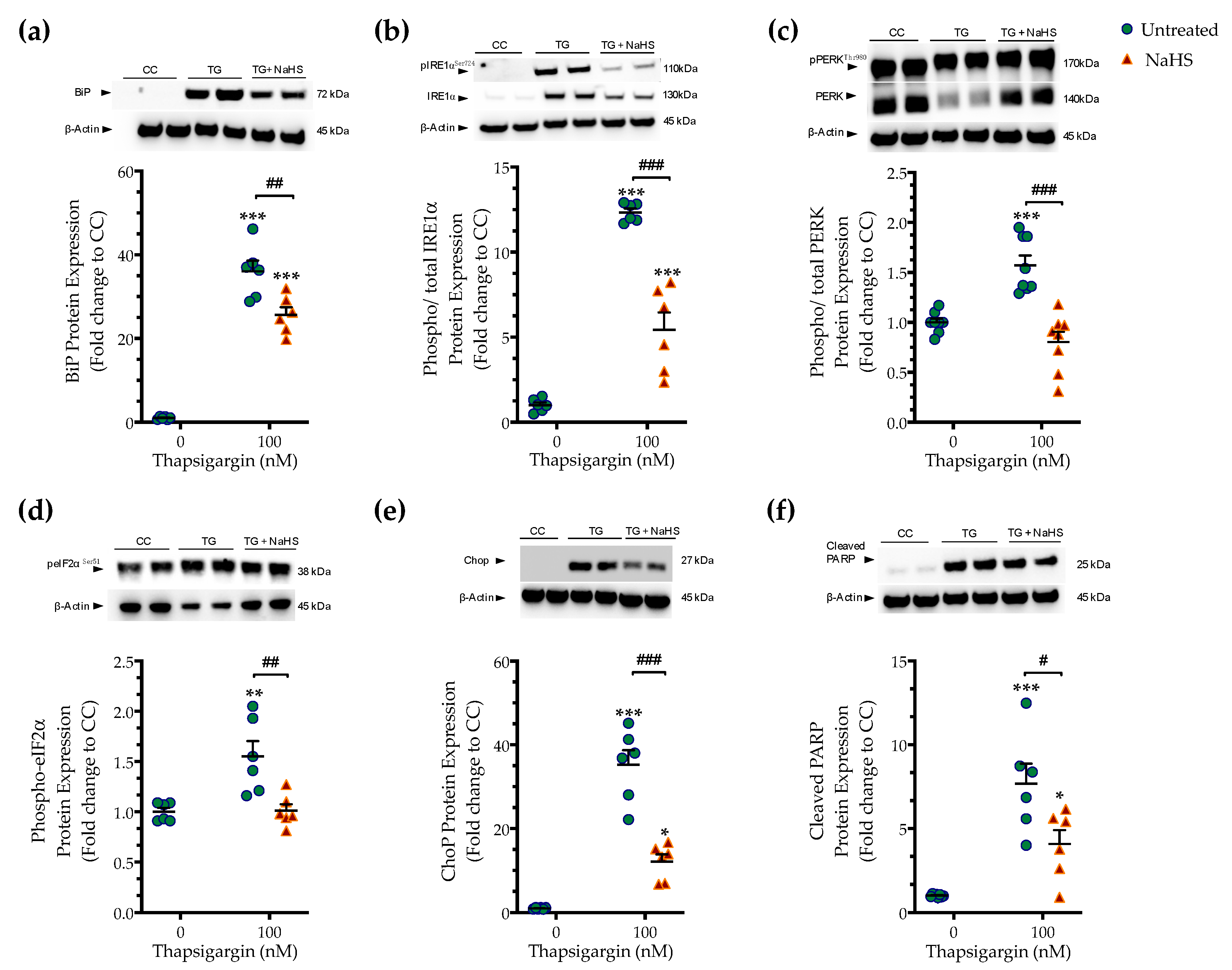
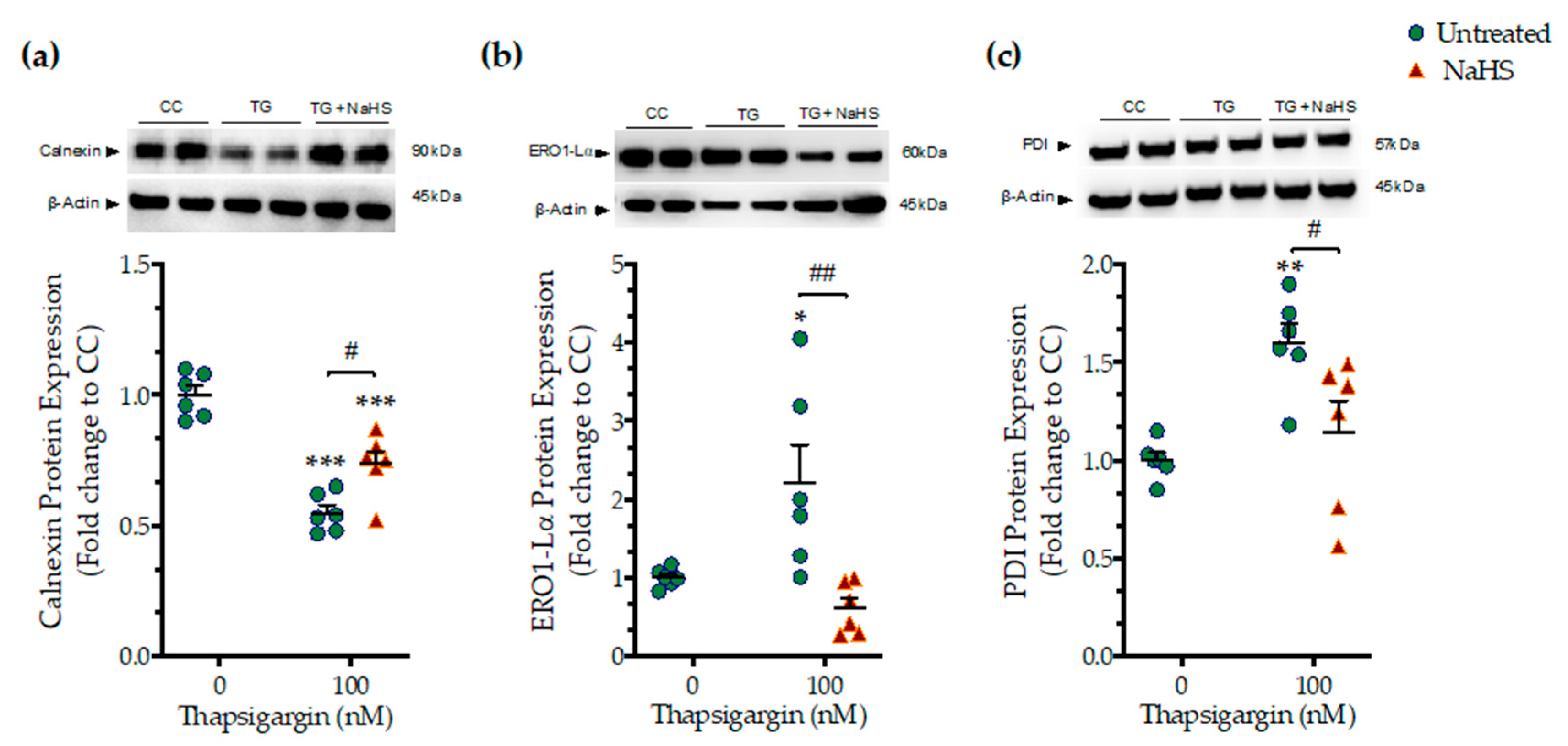
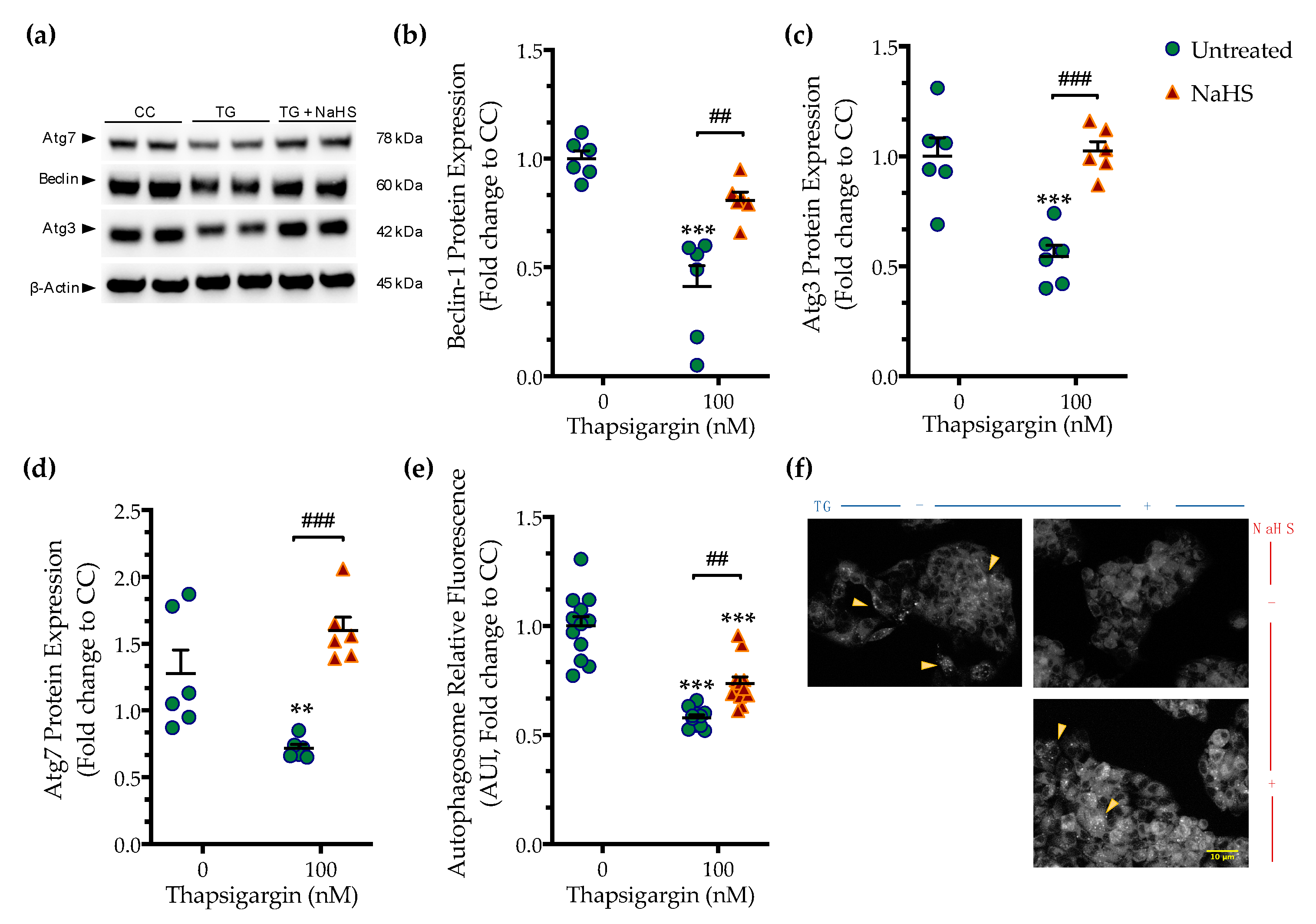
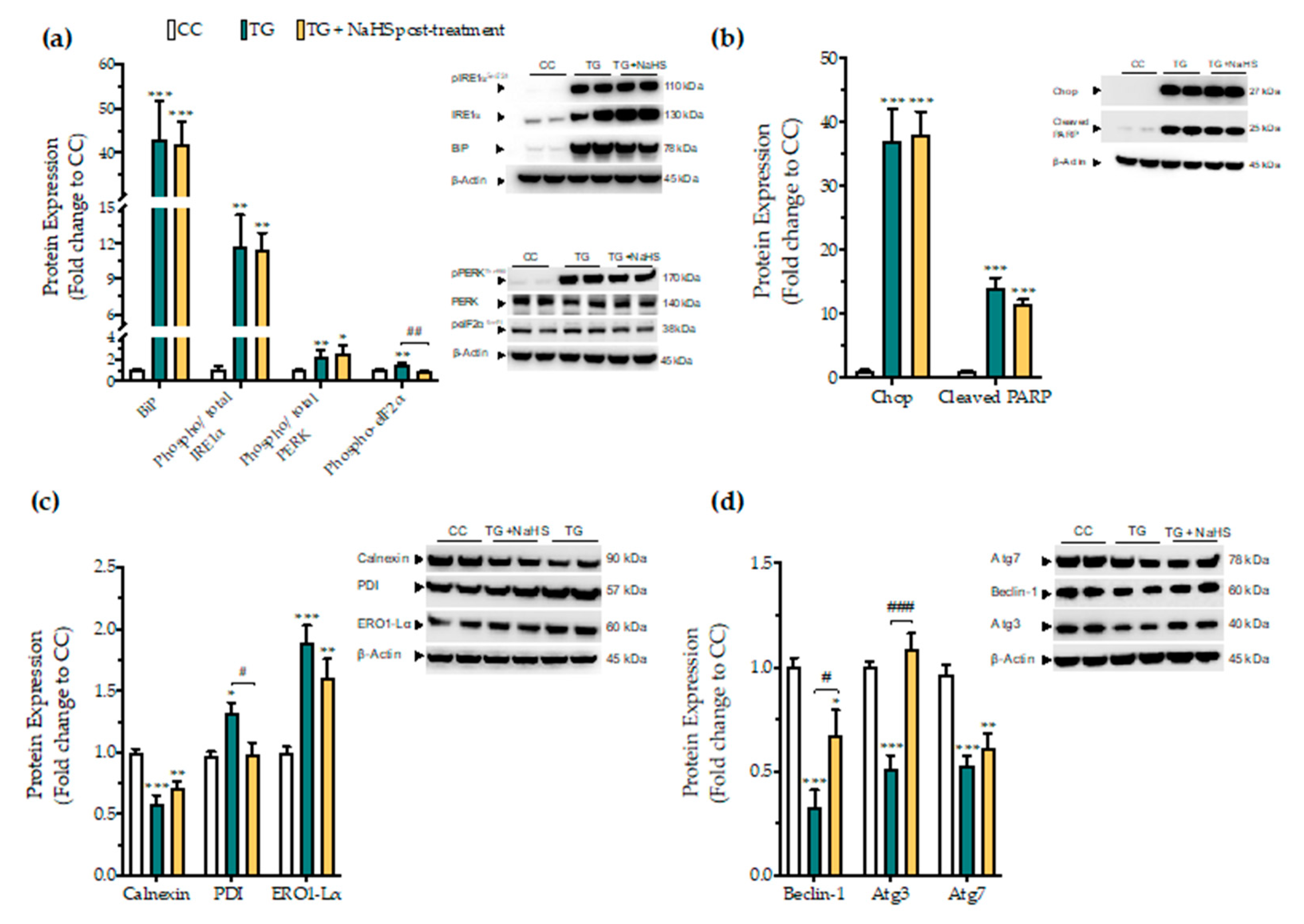
3.2. H2S Donation Ameliorates the Hepatic Unfolded Protein Response and Inhibits Autophagy Arrest
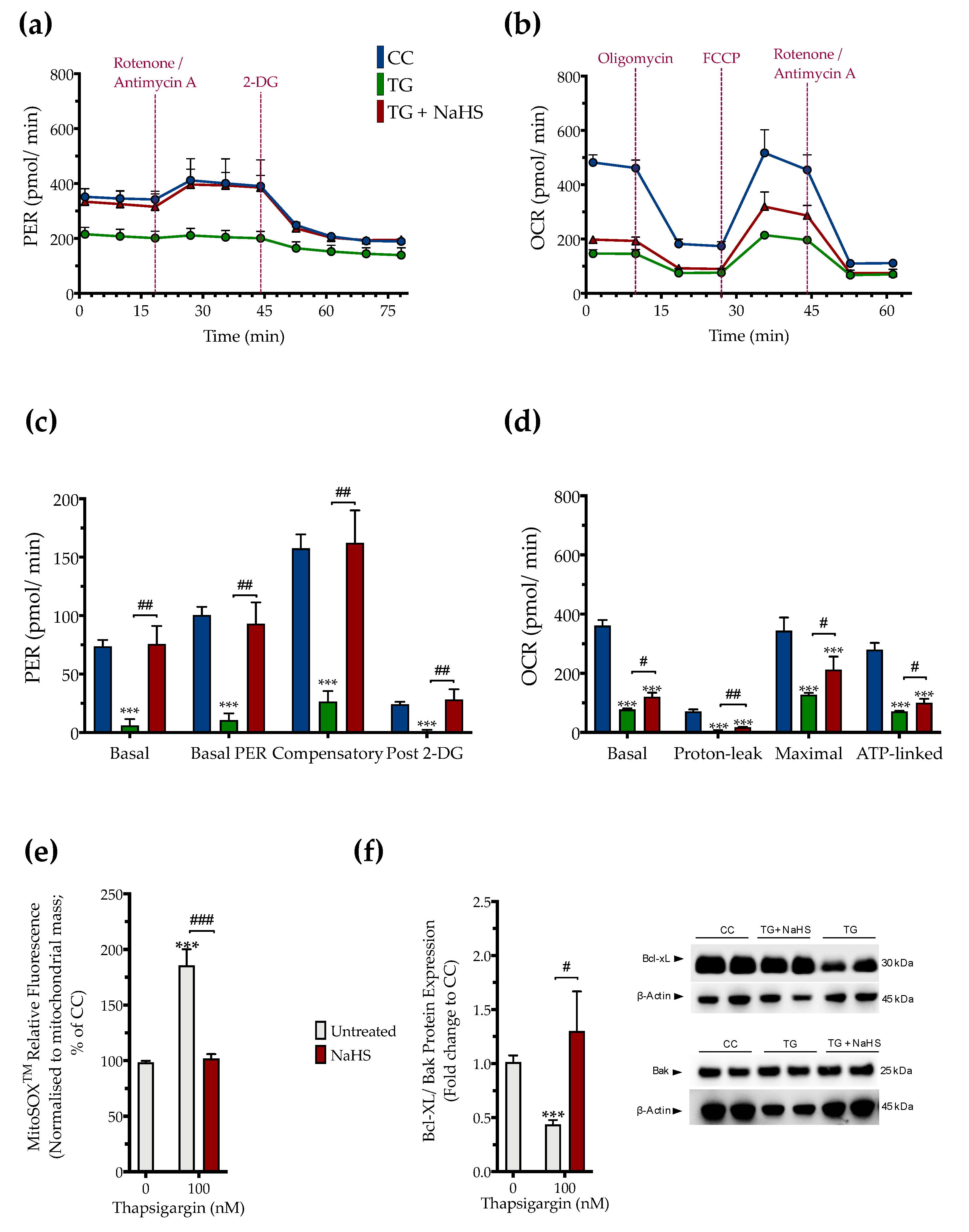
3.3. H2S Donation Restores Cellular Bioenergetics and Counteracts Mitochondrial Stress during ER Stress
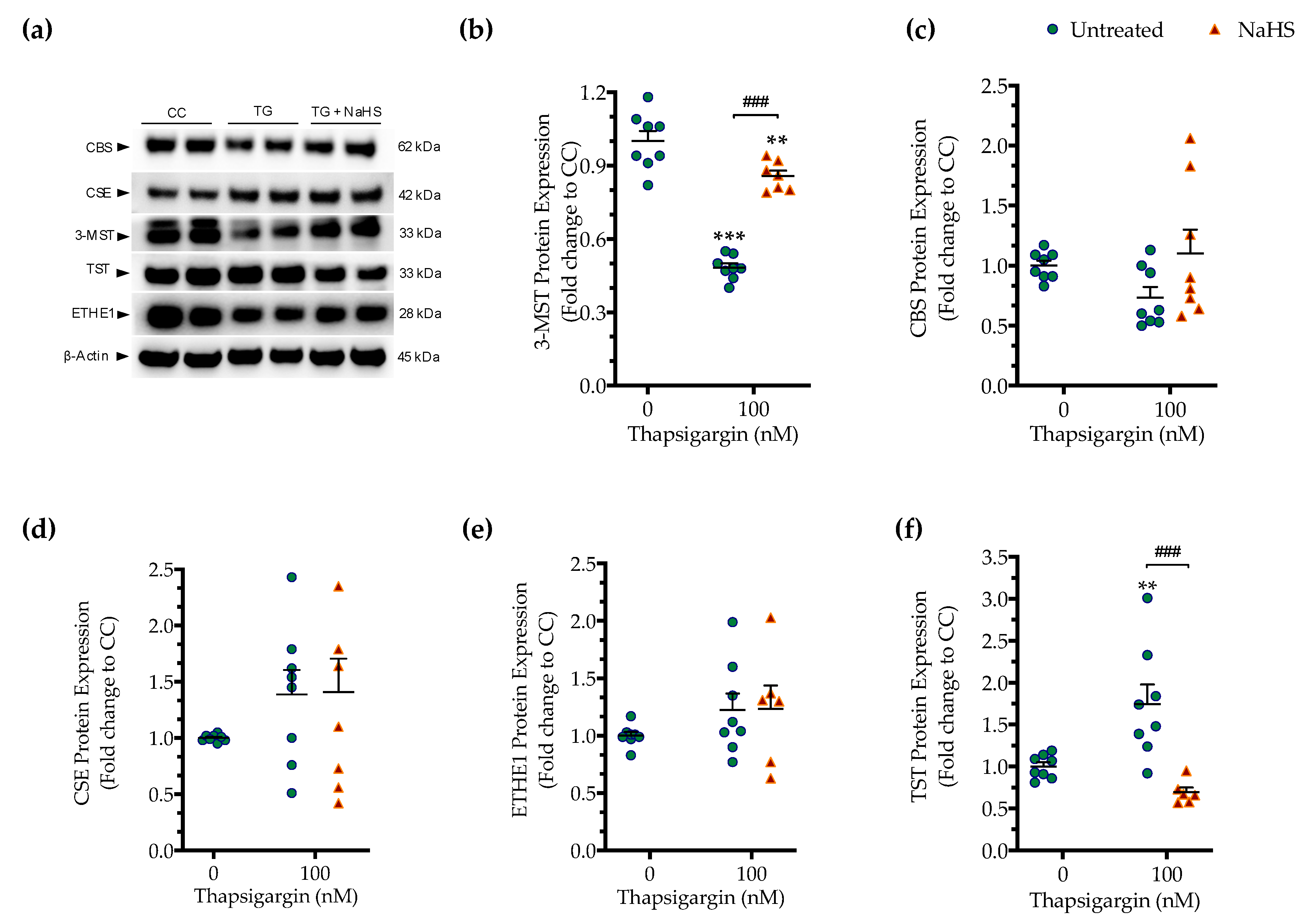
3.4. Effect of ER Stress on the Expression of Various H2S-Synthesizing and -Degrading Enzymes
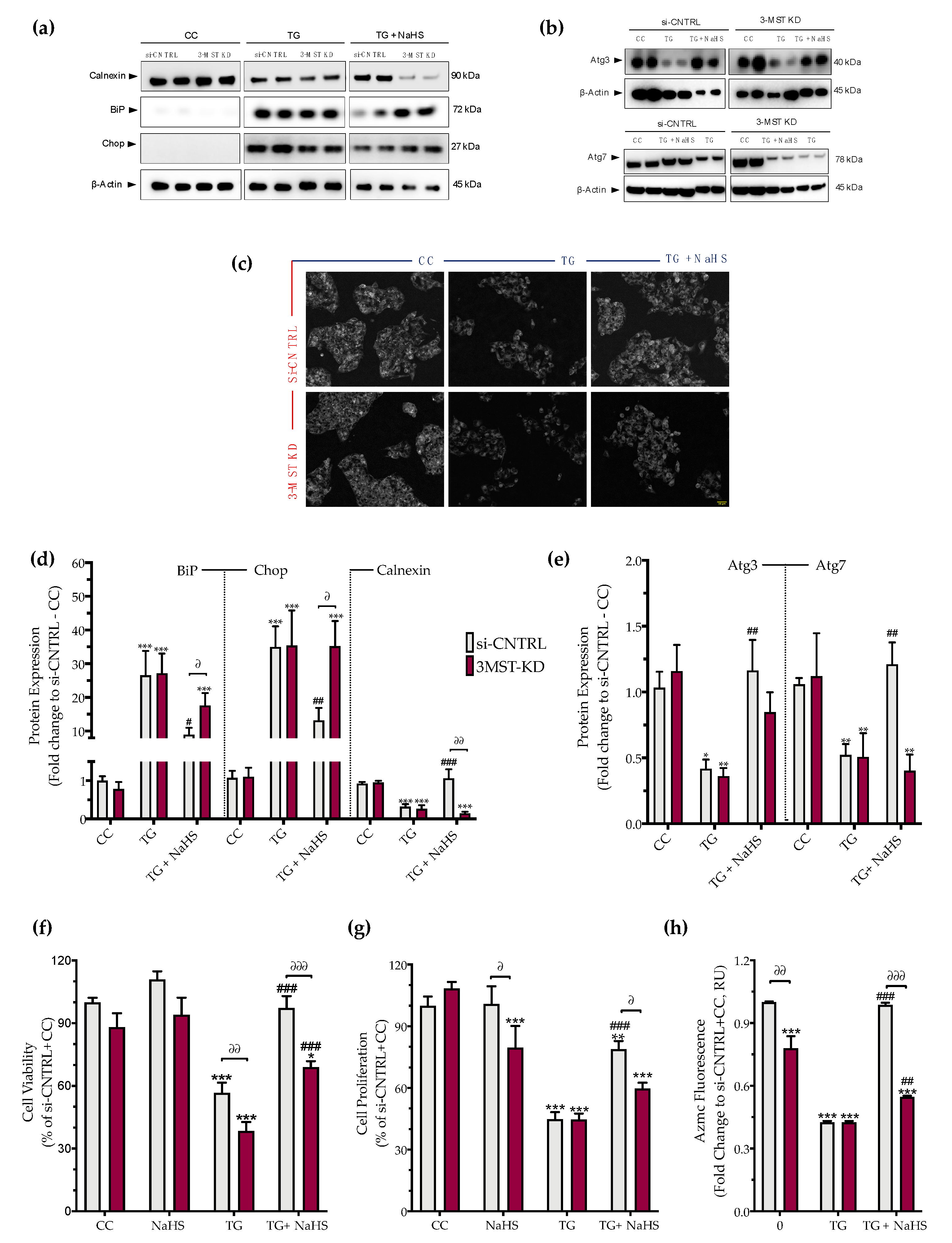
3.5. Effect of 3-MST Silencing on the Development of the ER Stress Response and on the Effect of H2S Donation
3.6. Effect of Pharmacological CBS and 3-MST Inhibition on the Development of ER Stress Response
3.7. Delayed Supplementation of H2S Exerts Partial Effects on the ER Stress Response
3.8. Effect of H2S on the Tunicamycin-Induced ER Stress Response
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, C.W.; Moore, P.K. H2S synthesizing enzymes: Biochemistry and molecular aspects. Handb. Exp. Pharmacol. 2015, 230, 3–25. [Google Scholar] [PubMed]
- Kimura, H. Physiological roles of hydrogen sulfide and polysulfides. Handb. Exp. Pharmacol. 2015, 230, 61–81. [Google Scholar] [PubMed]
- Szabo, C.; Papapetropoulos, A. International union of basic and clinical pharmacology. CII: Pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharmacol. 2018, 149, 5–19. [Google Scholar] [CrossRef]
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H2S and polysulfide production and possible SOx production. Br. J. Pharmacol. 2018, 175, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)-hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2020, 154, 104083. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Maiers, J.L.; Malhi, H. Endoplasmic reticulum stress in metabolic liver diseases and hepatic fibrosis. Semin. Liver Dis. 2019, 39, 235–248. [Google Scholar] [CrossRef]
- Oliva-Vilarnau, N.; Hankeova, S.; Vorrink, S.U.; Mkrtchian, S.; Andersson, E.R.; Lauschke, V.M. Calcium signaling in liver injury and regeneration. Front. Med. (Lausanne) 2018, 5, 192. [Google Scholar] [CrossRef]
- Hanaoka, K.; Sasakura, K.; Suwanai, Y.; Toma-Fukai, S.; Shimamoto, K.; Takano, Y.; Shibuya, N.; Terai, T.; Komatsu, T.; Ueno, T.; et al. Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteine persulfide. Sci. Rep. 2017, 7, 40227. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Randi, E.B.; Szabo, C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation and cellular bioenergetics in human Down syndrome fibroblasts. Biomolecules 2020, 10, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczesny, B.; Módis, K.; Yanagi, K.; Coletta, C.; Le Trionnaire, S.; Perry, A.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 2014, 41, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagaki, T.; Michael, M.; Hölscher, C. Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci. Rep. 2017, 7, 1615. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Szalai, P.; Parys, J.B.; Bultynck, G.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Nonlinear relationship between ER Ca2+ depletion versus induction of the unfolded protein response, autophagy inhibition, and cell death. Cell Calcium 2018, 76, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Toliver-Kinsky, T.; Cui, W.; Törö, G.; Lee, S.J.; Shatalin, K.; Nudler, E.; Szabo, C. H2S, a bacterial defense mechanism against the host immune response. Infect. Immun. 2018, 87, e00272-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augsburger, F.; Randi, E.B.; Jendly, M.; Ascencao, K.; Dilek, N.; Szabo, C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation, migration, and bioenergetics in murine colon cancer cells. Biomolecules 2020, 10, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollahi Govar, A.; Törő, G.; Szaniszlo, P.; Pavlidou, A.; Bibli, S.I.; Thanki, K.; Resto, V.A.; Chao, C.; Hellmich, M.R.; Szabo, C.; et al. 3-Mercaptopyruvate sulfurtransferase supports endothelial cell angiogenesis and bioenergetics. Br. J. Pharmacol. 2020, 177, 866–883. [Google Scholar] [CrossRef] [Green Version]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular regulation and pharmacological inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.; Zatarain, J.R.; Ding, Y.; Coletta, C.; Mrazek, A.A.; Druzhyna, N.; Johnson, P.; Chen, H.; Hellmich, J.L.; Asimakopoulou, A.; et al. Cystathionine-beta-synthase inhibition for colon cancer: Enhancement of the efficacy of aminooxyacetic acid via the prodrug approach. Mol. Med. 2016, 22, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, H.; Zhang, M.; Zhang, J.; Wei, X.; Ying, W. Malate-aspartate shuttle inhibitor aminooxyacetic acid leads to decreased intracellular ATP levels and altered cell cycle of C6 glioma cells by inhibiting glycolysis. Cancer Lett. 2016, 378, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, B.; Marcatti, M.; Zatarain, J.R.; Druzhyna, N.; Wiktorowicz, J.E.; Nagy, P.; Hellmich, M.R.; Szabo, C. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 2016, 6, 36125. [Google Scholar] [CrossRef] [Green Version]
- Untereiner, A.A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Randi, E.B.; Augsburger, F.; Szabo, C. Overproduction of H2S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 18769–18771. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Li, J.; Huang, B.; Kuang, L.; Xiao, F.; Zheng, D. Cystathionine β synthase/hydrogen sulfide signaling in multiple myeloma regulates cell proliferation and apoptosis. J. Environ. Pathol. Toxicol. Oncol. 2020, 39, 281–290. [Google Scholar] [CrossRef]
- Ye, F.; Li, X.; Sun, K.; Xu, W.; Shi, H.; Bian, J.; Lu, R.; Ye, Y. Inhibition of endogenous hydrogen sulfide biosynthesis enhances the anti-cancer effect of 3,3′-diindolylmethane in human gastric cancer cells. Life Sci. 2020, 261, 118348. [Google Scholar] [CrossRef]
- Yue, T.; Zuo, S.; Bu, D.; Zhu, J.; Chen, S.; Ma, Y.; Ma, J.; Guo, S.; Wen, L.; Zhang, X.; et al. Aminooxyacetic acid (AOAA) sensitizes colon cancer cells to oxaliplatin via exaggerating apoptosis induced by ROS. J. Cancer 2020, 11, 1828–1838. [Google Scholar] [CrossRef]
- Thastrup, O.; Cullen, P.J.; Drøbak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.W.; Wang, Z.M.; Sun, S.M.; Su, Y.; Li, Z.H.; Shao, J.J.; Tan, S.Z.; Chen, A.P.; Wang, S.J.; Zhang, Z.L.; et al. Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol. Res. 2020, 161, 105218. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bousette, N.; Abbasi, C.; Chis, R.; Gramolini, A.O. Calnexin silencing in mouse neonatal cardiomyocytes induces Ca2+ cycling defects, ER stress, and apoptosis. J. Cell Physiol. 2014, 229, 374–383. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1843, 2233–2239. [Google Scholar] [CrossRef] [Green Version]
- Jaronen, M.; Vehviläinen, P.; Malm, T.; Keksa-Goldsteine, V.; Pollari, E.; Valonen, P.; Koistinaho, J.; Goldsteins, G. Protein disulfide isomerase in ALS mouse glia links protein misfolding with NADPH oxidase-catalyzed superoxide production. Hum. Mol. Genet. 2013, 22, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.D.; Wang, D.Y.; Li, H.M.; Guo, J.C.; Duan, S.F.; Ji, X.Y. Hydrogen sulfide as a novel regulatory factor in liver health and disease. Oxid. Med. Cell Longev. 2019, 2019, 3831713. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Wang, X.-Y.; Bian, J.-S. Implications of hydrogen sulfide in liver pathophysiology: Mechanistic insights and therapeutic potential. J. Adv. Res. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Zhao, M.; Jiang, H.; Tan, G.; Pan, S.; Sun, X. Role of hydrogen sulfide in hepatic ischemia-reperfusion–induced injury in rats. Liver Transpl. 2009, 15, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Jiang, H.C.; Zhao, M.Y.; Sun, X.Y.; Pan, S.H. Protection of CSE/H2S system in hepatic ischemia reperfusion injury in rats. Zhonghua Wai Ke Za Zhi 2010, 48, 924–928. [Google Scholar]
- Morsy, M.A.; Ibrahim, S.A.; Abdelwahab, S.A.; Zedan, M.Z.; Elbitar, H.I. Curative effects of hydrogen sulfide against acetaminophen-induced hepatotoxicity in mice. Life Sci. 2010, 87, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Pan, S.; Li, J.; Dong, X.; Kang, K.; Zhao, M.; Jiang, X.; Kanwar, J.R.; Qiao, H.; Jiang, H.; et al. Hydrogen sulfide attenuates carbon tetrachloride-induced hepatotoxicity, liver cirrhosis and portal hypertension in rats. PLoS ONE 2011, 6, e25943. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zheng, N.; Qi, K.; Cheng, H.; Sun, Z.; Gao, B.; Zhang, Y.; Pang, W.; Huangfu, C.; Ji, S.; et al. Exogenous hydrogen sulfide mitigates the fatty liver in obese mice through improving lipid metabolism and antioxidant potential. Med. Gas Res. 2015, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Engedal, N.; Torgersen, M.L.; Guldvik, I.J.; Barfeld, S.J.; Bakula, D.; Sætre, F.; Hagen, L.K.; Patterson, J.B.; Proikas-Cezanne, T.; Seglen, P.O.; et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013, 9, 1475–1490. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Wong, P.-M.; Gammoh, N.; Jiang, X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Allaire, M.; Rautou, P.-E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hep. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Wang, H.; Teng, T.; Duan, S.; Ji, A.; Li, Y. Hydrogen sulfide and autophagy: A double edged sword. Pharmacol. Res. 2018, 131, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- B’chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- B’chir, W.; Chaveroux, C.; Carraro, V.; Averous, J.; Maurin, A.C.; Jousse, C.; Muranishi, Y.; Parry, L.; Fafournoux, P.; Bruhat, A. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. 2014, 26, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yang, W.; Wu, L.; Wang, R. H2S, endoplasmic reticulum stress, and apoptosis of insulin-secreting beta cells. J. Biol. Chem. 2007, 282, 16567–16576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullahi, A.; Stanojcic, M.; Parousis, A.; Patsouris, D.; Jeschke, M.G. Modeling acute ER stress in vivo and in vitro. Shock 2017, 47, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Zhang, R.; Jin, H.; Liu, D.; Tang, X.; Tang, C.; Du, J. Hydrogen sulfide attenuates hyperhomocysteinemia-induced cardiomyocytic endoplasmic reticulum stress in rats. Antioxid. Redox Signal. 2010, 12, 1079–1091. [Google Scholar] [CrossRef]
- Barr, L.A.; Shimizu, Y.; Lambert, J.P.; Nicholson, C.K.; Calvert, J.W. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide 2015, 46, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Luo, J.; Wu, Z.; Xiao, T.; Zeng, O.; Li, L.; Li, Y.; Yang, J. Hydrogen sulfide exhibits cardioprotective effects by decreasing endoplasmic reticulum stress in a diabetic cardiomyopathy rat model. Mol. Med. Rep. 2016, 14, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yu, X.; Li, T.; Wu, J.; Zhao, Y.; Liu, J.; Sun, A.; Dong, S.; Wu, J.; Zhong, X.; et al. Exogenous H2S regulates endoplasmic reticulum-mitochondria cross-talk to inhibit apoptotic pathways in STZ-induced type I diabetes. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E190–E203. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Yuan, J.; Tang, Z.J.; Wei, H.J.; Zhu, W.W.; Zhang, P.; Gu, H.F.; Wang, C.Y.; Tang, X.Q. Hydrogen sulfide ameliorates cognitive dysfunction in streptozotocin-induced diabetic rats: Involving suppression in hippocampal endoplasmic reticulum stress. Oncotarget 2017, 8, 64203–64216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, A.; Singh, M.; Behera, J.; Theilen, N.T.; George, A.K.; Tyagi, N.; Metreveli, N.; Tyagi, S.C. Hydrogen sulfide alleviates hyperhomocysteinemia-mediated skeletal muscle atrophy via mitigation of oxidative and endoplasmic reticulum stress injury. Am. J. Physiol. Cell Physiol. 2018, 315, C609–C622. [Google Scholar] [CrossRef]
- Wu, J.; Pan, W.; Wang, C.; Dong, H.; Xing, L.; Hou, J.; Fang, S.; Li, H.; Yang, F.; Yu, B. H2S attenuates endoplasmic reticulum stress in hypoxia-induced pulmonary artery hypertension. Biosci. Rep. 2019, 39, BSR20190304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.; Wu, N.; Wang, Q.; Yang, L.H. Endogenous hydrogen sulfide alleviates methotrexate-induced cognitive impairment by attenuating endoplasmic reticulum stress-induced apoptosis via CHOP and caspase-12. Fundam. Clin. Pharmacol. 2020, 34, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Ying, R.; Wang, X.Q.; Yang, Y.; Gu, Z.J.; Mai, J.T.; Qiu, Q.; Chen, Y.X.; Wang, J.F. Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. 2016, 144, 208–217. [Google Scholar] [CrossRef]
- Longchamp, A.; Mirabella, T.; Arduini, A.; MacArthur, M.R.; Das, A.; Treviño-Villarreal, J.H.; Hine, C.; Ben-Sahra, I.; Knudsen, N.H.; Brace, L.E.; et al. Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S Production. Cell 2018, 173, 117–129.e14. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell. 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [Green Version]
- Horsman, J.W.; Miller, D.L. Mitochondrial sulfide quinone oxidoreductase prevents activation of the unfolded protein response in hydrogen sulfide. J. Biol. Chem. 2016, 291, 5320–5325. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Wu, Z.; Jiang, J.; Liu, C.; Wu, B.; Li, X.; Li, T.; Mo, H.; He, S.; Li, S.; et al. New mechanism of lipotoxicity in diabetic cardiomyopathy: Deficiency of endogenous H2S production and ER stress. Mech. Ageing Dev. 2017, 162, 46–52. [Google Scholar] [CrossRef]
- George, A.K.; Homme, R.P.; Majumder, A.; Laha, A.; Metreveli, N.; Sandhu, H.S.; Tyagi, S.C.; Singh, M. Hydrogen sulfide intervention in cystathionine-β-synthase mutant mouse helps restore ocular homeostasis. Int. J. Ophthalmol. 2019, 12, 754–764. [Google Scholar]
- Rutkowski, D.T.; Wu, J.; Back, S.H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell. 2008, 15, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, F.; Gong, Q.; Cui, A.; Zhuo, S.; Hu, Z.; Han, Y.; Gao, J.; Sun, Y.; Liu, Z.; et al. Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosis in mice through peroxisome proliferator-activated receptor α. Diabetes 2016, 65, 1904–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.; Gao, X.H.; Willard, B.; Hatzoglou, M.; Banerjee, R.; Kabil, O. Hydrogen sulfide modulates eukaryotic translation initiation factor 2α (eIF2α) phosphorylation status in the integrated stress-response pathway. J. Biol. Chem. 2017, 292, 13143–13153. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Blocking Buffer | Dilution | Isotype | Manufacturer | Catalogue Number |
|---|---|---|---|---|---|
| β-Actin | 5% w/v SM | 1:2000 | Mouse IgG | Cell Signaling | 3700 |
| Beclin-1 | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 3495 |
| Atg3 | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 3415 |
| Atg7 | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 8558 |
| ATF6 | 5% w/v SM | 1:1000 | Mouse IgG | Abcam | ab122897 |
| BiP | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 9956 |
| Calnexin | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 9956 |
| PDI | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 9956 |
| Ero1-Lα | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 9956 |
| Chop | 5% w/v SM | 1:1000 | Mouse IgG | Cell Signaling | 9956 |
| IRE1α | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 9956 |
| Phospho-IRE1 (Ser724) | 5% w/v SM | 1:1000 | Rabbit IgG | Abcam | ab48187 |
| PERK | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 3192 |
| Phospho-PERK (Thr980) | 5% w/v BSA | 1:1000 | Rabbit IgG | Thermo Fisher | 15033 |
| Phospho-eIF2α (Ser51) | 5% w/v BSA | 1:1000 | Rabbit IgG | Cell Signaling | 9721 |
| Bak | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 12105 |
| Bcl-xL | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 2764 |
| Cleaved PARP | 5% w/v SM | 1:500 | Rabbit IgG | Abcam | ab32064 |
| CBS | 5% w/v SM | 1:1000 | Rabbit IgG | Cell Signaling | 14782 |
| 3-MST | 5% w/v SM | 1:1000 | Rabbit IgG | Abcam | ab154514 |
| CSE | 5% w/v SM | 1:1000 | Rabbit IgG | Abcam | ab151769 |
| TST | 5% w/v SM | 1:1000 | Rabbit IgG | Abcam | ab166625 |
| ETHE1 | 5% w/v SM | 1:1000 | Rabbit IgG | Abcam | ab174302 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panagaki, T.; Randi, E.B.; Szabo, C. Role of Hydrogen Sulfide and 3-Mercaptopyruvate Sulfurtransferase in the Regulation of the Endoplasmic Reticulum Stress Response in Hepatocytes. Biomolecules 2020, 10, 1692. https://doi.org/10.3390/biom10121692
Panagaki T, Randi EB, Szabo C. Role of Hydrogen Sulfide and 3-Mercaptopyruvate Sulfurtransferase in the Regulation of the Endoplasmic Reticulum Stress Response in Hepatocytes. Biomolecules. 2020; 10(12):1692. https://doi.org/10.3390/biom10121692
Chicago/Turabian StylePanagaki, Theodora, Elisa B. Randi, and Csaba Szabo. 2020. "Role of Hydrogen Sulfide and 3-Mercaptopyruvate Sulfurtransferase in the Regulation of the Endoplasmic Reticulum Stress Response in Hepatocytes" Biomolecules 10, no. 12: 1692. https://doi.org/10.3390/biom10121692