The Non-Fibrillating N-Terminal of α-Synuclein Binds and Co-Fibrillates with Heparin

 , , ,
, , ,  , , and
, , and
Abstract
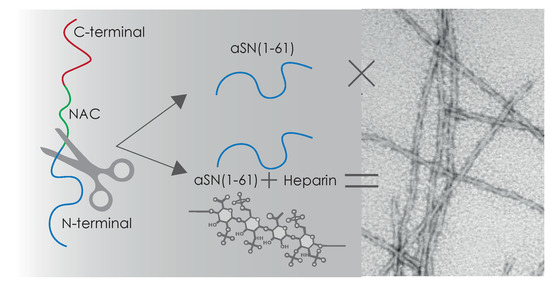
:
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Expression and Purification of Recombinant aSN1–61
2.3. Purification of Full-Length Recombinant aSN
2.4. Expression and Purification of Recombinant His-ULP1
2.5. Protein Fibrillation Assay
2.6. Seeding Experiments
2.7. Negative Stain Transmission Electron Microscopy
2.8. Circular Dichroism (CD)
2.9. Small Angle X-ray Scattering (SAXS)
2.10. NMR Sample Preparation
2.11. NMR Data Recording and Processing
2.12. Wide-Field Fluorescence Microscopy
3. Results
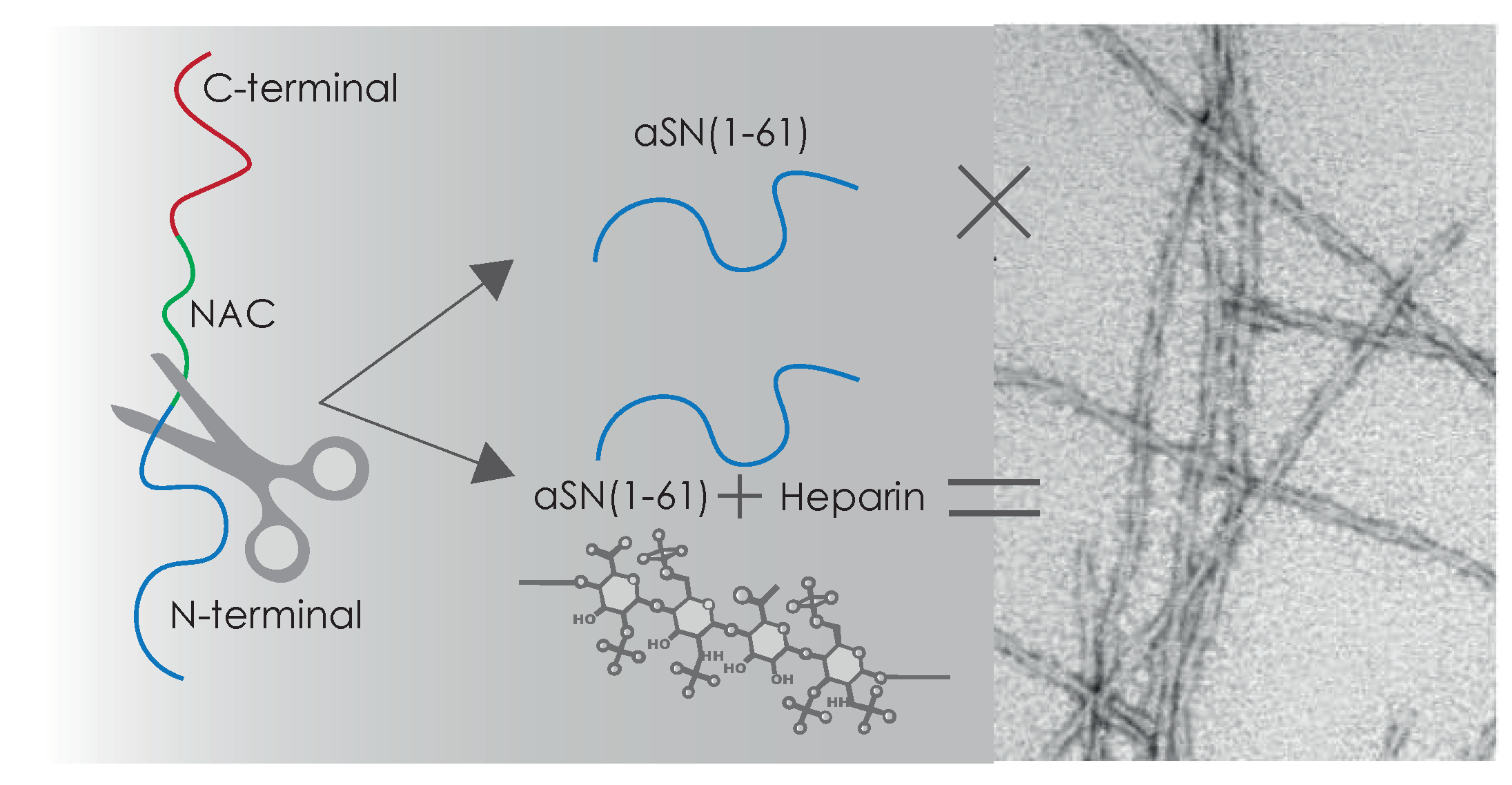
3.1. This aSN1–61 Maintains Disorder as in Full-Length aSN
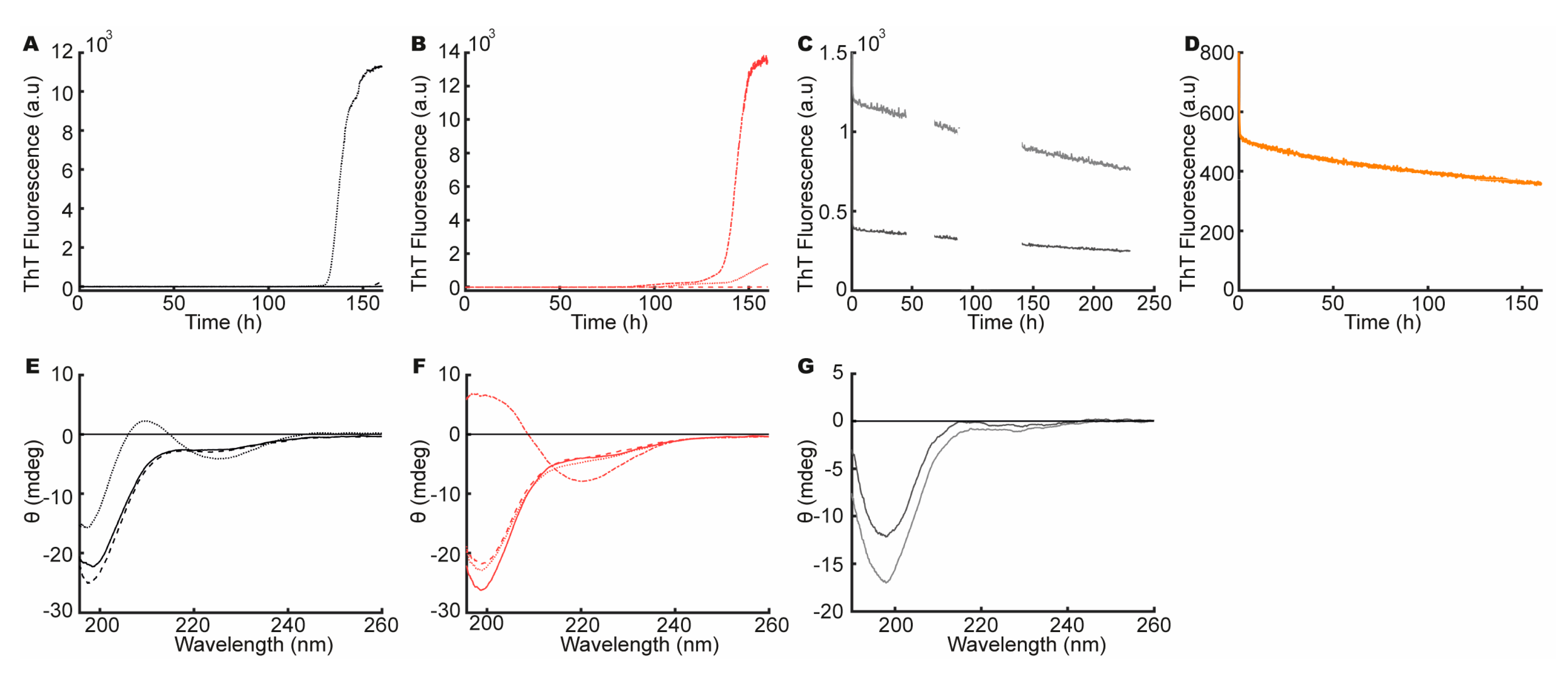
3.2. aSN1–61 Lacks Intrinsic Fibrillation Propensity
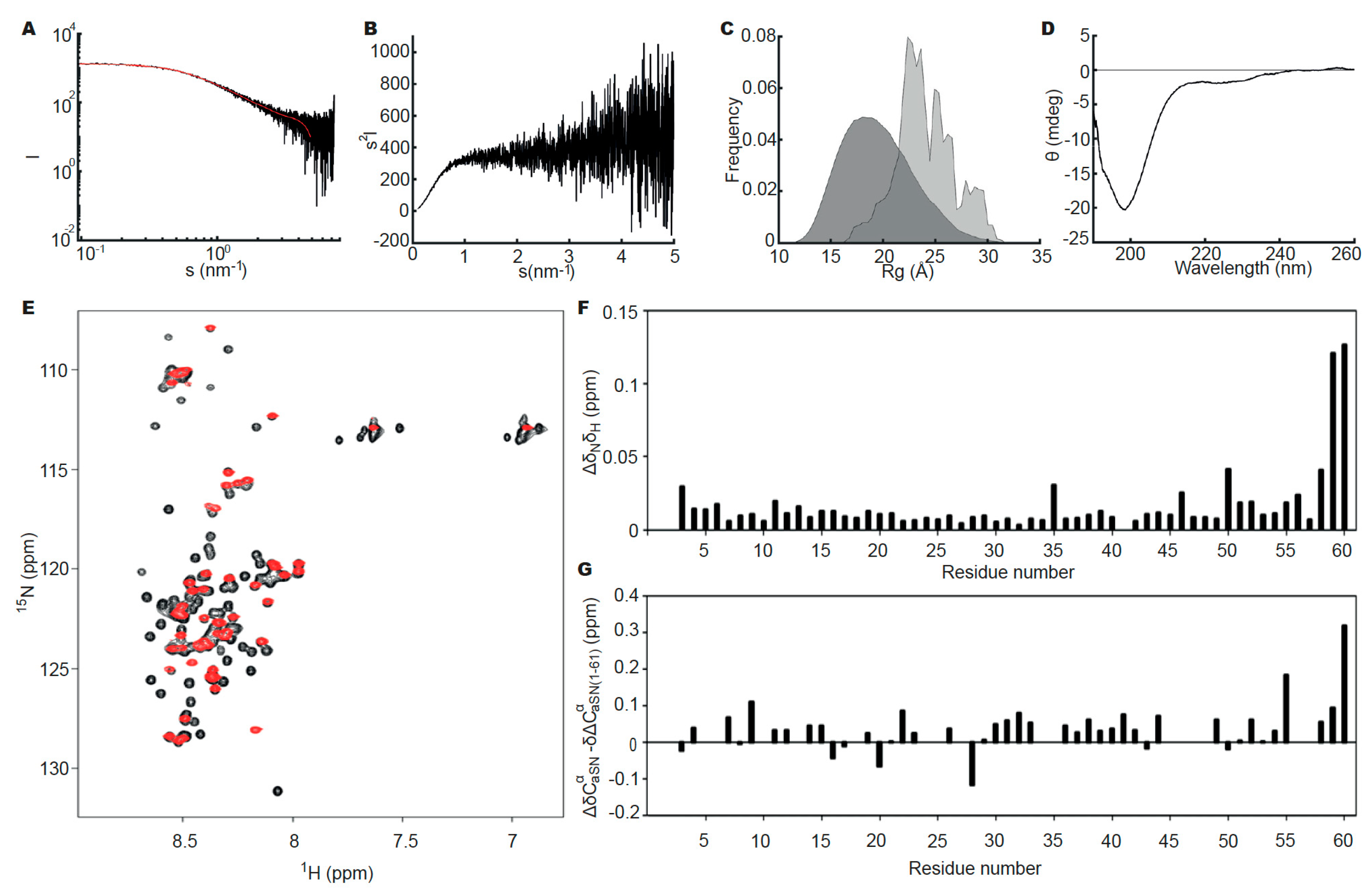
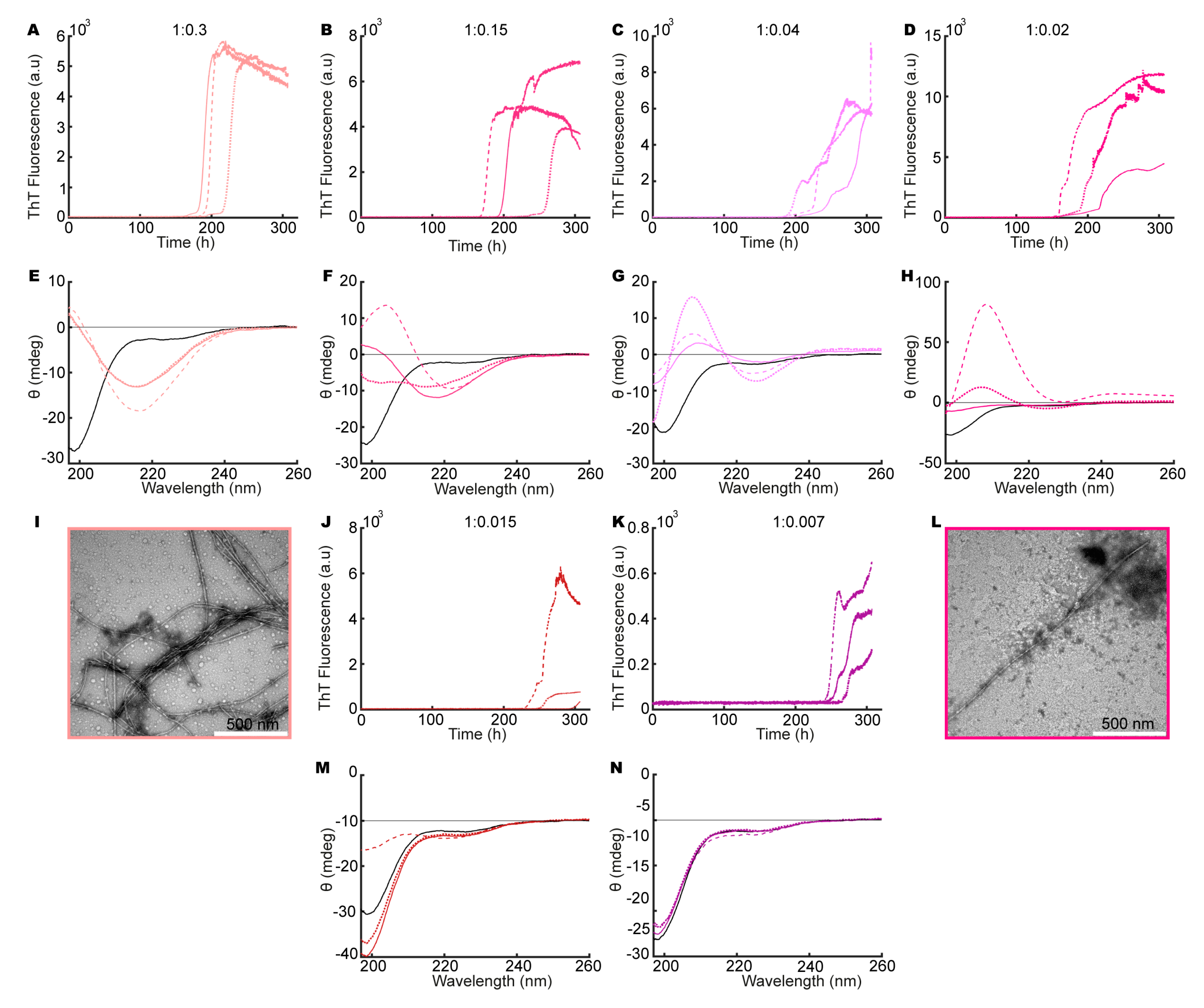
3.3. Heparin Induces Fibril Formation of aSN1–61
3.4. Heparin Has Its Own Concentration-Dependent Structure
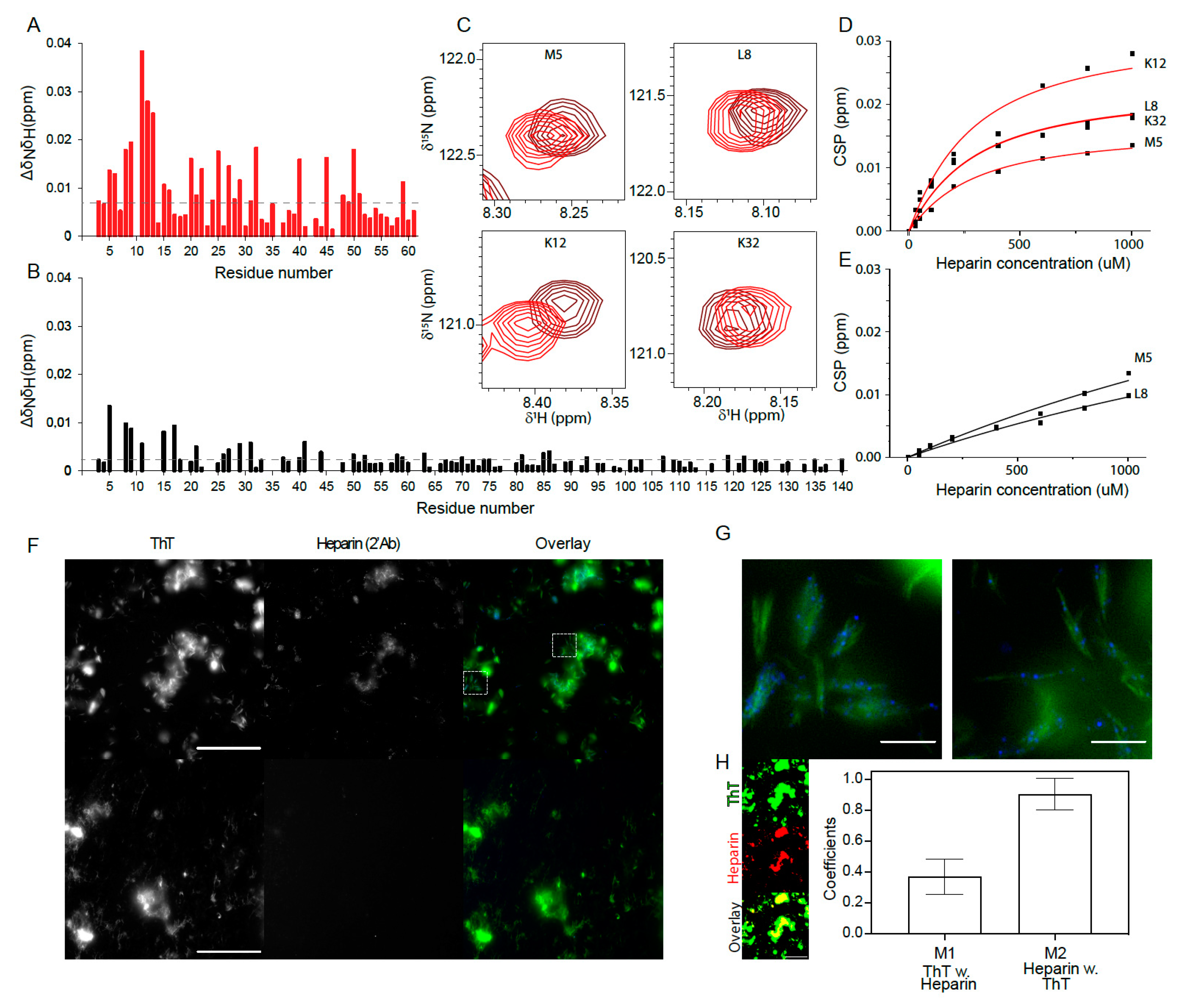
3.5. Heparin Interaction with aSN1–61 Is Specific and Mainly Driven by Electrostatics
3.6. Heparin Binds Weakly But Preferably to aSN1–61
3.7. Heparin and aSN1–61 Co-Exist in the Fibrils
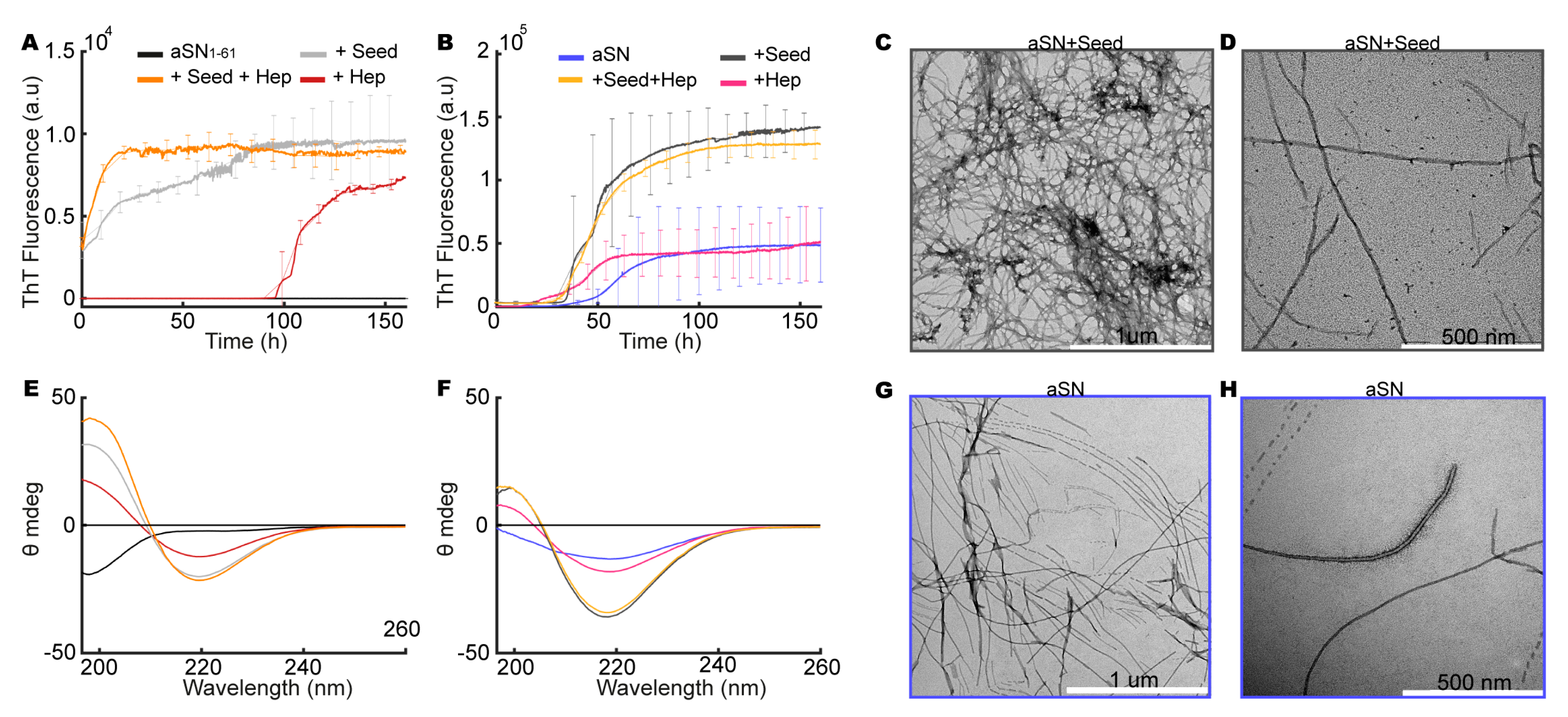
3.8. aSN1–61/Heparin Co-Fibrils Seed Fibrillation of Full-Length aSN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, R.; Meisl, G.; Buell, A.K.; Young, L.; Kaminski, C.F.; Knowles, T.P.J.; Sparr, E.; Linse, S. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q. Rev. Biophys. 2017, 50, e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.J.; Roy, S. α-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol. 2014, 24, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Scialò, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and Prion-Like Protein Strains: Deciphering the Molecular Basis of Heterogeneity in Neurodegeneration. Viruses 2019, 11, E261. [Google Scholar] [CrossRef] [Green Version]
- Deleersnijder, A.; Gerard, M.; Debyser, Z.; Baekelandt, V. The remarkable conformational plasticity of alpha-synuclein: Blessing or curse. Trends Mol. Med. 2013, 19, 368–377. [Google Scholar] [CrossRef]
- Bodner, C.R.; Maltsev, A.S.; Dobson, C.M.; Bax, A. Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 2010, 49, 862–871. [Google Scholar]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [Green Version]
- Crowther, R.A.; Daniel, S.E.; Goedert, M. Characterisation of isolated alpha-synuclein filaments from substantia nigra of Parkinson’s disease brain. Neurosci. Lett. 2000, 292, 128–130. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. eLife 2018, 7, e36402. [Google Scholar] [CrossRef] [PubMed]
- Heise, H.; Hoyer, W.; Becker, S.; Andronesi, O.C.; Riedel, D.; Baldus, M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA 2005, 102, 15871–15876. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Zhao, X.; Liu, F.; Yan, S.; Wang, Y.; Du, C.; Cui, X.; Li, R.; Zhang, C.X. Pathogenic Mutations Differentially Regulate Cell-to-Cell Transmission of α-Synuclein. Front. Cell. Neurosci. 2020, 14, 159. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.C.; Rochet, J.C.; Lansbury, P.T. The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry 2003, 42, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Song, G.; Yang, H.; Lin, X.; Brown, B.; Hong, Y.; Cai, J.; Cao, C. Identifying the Pathological Domain of Alpha- Synuclein as a Therapeutic for Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, E2338. [Google Scholar] [CrossRef] [Green Version]
- Perry, G.; Richey, P.; Siedlak, S.L.; Galloway, P.; Kawai, M.; Cras, P. Basic fibroblast growth factor binds to filamentous inclusions of neurodegenerative diseases. Brain Res. 1992, 579, 350–352. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Uchimura, K. Sulfated glycosaminoglycans in protein aggregation diseases. Glycoconj. J. 2017, 34, 453–466. [Google Scholar] [CrossRef]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 1998, 20, 156–167. [Google Scholar] [CrossRef]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieri, G.; Gitler, A.D.; Brahic, M. Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis. 2018, 109, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Ihse, E.; Yamakado, H.; van Wijk, X.M.; Lawrence, R.; Esko, J.D.; Masliah, E. Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 2017, 7, 9008. [Google Scholar] [CrossRef]
- Feyzi, E.; Saldeen, T.; Larsson, E.; Lindahl, U.; Salmivirta, M. Age-dependent modulation of heparan sulfate structure and function. J. Biol. Chem. 1998, 273, 13395–13398. [Google Scholar] [CrossRef] [Green Version]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Torrent, M.; Nogués, M.V.; Andreu, D.; Boix, E. The “CPC clip motif”: A conserved structural signature for heparin-binding proteins. PLoS ONE 2012, 7, e42692. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Ghosh, D.; Kumar, R.; Mondal, M.; Gadhe, L.G.; Das, S.; Anoop, A.; Jha, N.N.; Jacob, R.S.; Chatterjee, D.; et al. Glycosaminoglycans have variable effects on α-synuclein aggregation and differentially affect the activities of the resulting amyloid fibrils. J. Biol. Chem. 2018, 293, 12975–12991. [Google Scholar] [CrossRef] [Green Version]
- Cohlberg, J.A.; Li, J.; Uversky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef]
- Trexler, A.J.; Rhoades, E. Single molecule characterization of α-synuclein in aggregation-prone states. Biophys. J. 2010, 99, 3048–3055. [Google Scholar] [CrossRef] [Green Version]
- Madine, J.; Clayton, J.C.; Yates, E.A.; Middleton, D.A. Exploiting a (13)C-labelled heparin analogue for in situ solid-state NMR investigations of peptide-glycan interactions within amyloid fibrils. Org. Biomol. Chem. 2009, 7, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Vilasi, S.; Sarcina, R.; Maritato, R.; De Simone, A.; Irace, G.; Sirangelo, I. Heparin induces harmless fibril formation in amyloidogenic W7FW14F apomyoglobin and amyloid aggregation in wild-type protein in vitro. PLoS ONE 2011, 6, e22076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madine, J.; Davies, H.A.; Hughes, E.; Middleton, D.A. Heparin promotes the rapid fibrillization of a peptide with low intrinsic amyloidogenicity. Biochemistry 2013, 52, 8984–8992. [Google Scholar] [PubMed]
- Van Maarschalkerweerd, A.; Vetri, V.; Langkilde, A.E.; Foderà, V.; Vestergaard, B. Protein/lipid coaggregates are formed during α-synuclein-induced disruption of lipid bilayers. Biomacromolecules 2014, 15, 3643–3654. [Google Scholar] [CrossRef] [PubMed]
- Prestel, A.; Wichmann, N.; Martins, J.M.; Marabini, R.; Kassem, N.; Broendum, S.S.; Otterlei, M.; Nielsen, O.; Willemoës, M.; Ploug, M.; et al. The PCNA interaction motifs revisited: Thinking outside the PIP-box. Cell. Mol. Life Sci. 2019, 76, 4923–4943. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, C.E.; Spilotros, A.; Schwemmer, F.; Graewert, M.A.; Kikhney, A.; Jeffries, C.M.; Franke, D.; Mark, D.; Zengerle, R.; Cipriani, F.; et al. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Cryst. 2015, 48, 431–443. [Google Scholar] [CrossRef]
- Franke, D.; Kikhney, A.G.; Svergun, D.I. Automated acquisition and analysis of small angle X-ray scattering data. Nucl. Instrum. Methods Phys. Res. A 2012, 689, 52–59. [Google Scholar] [CrossRef]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Cholak, E.; Bucciarelli, S.; Bugge, K.; Johansen, N.T.; Vestergaard, B.; Arleth, L.; Kragelund, B.B.; Langkilde, A.E. Distinct α-Synuclein:Lipid Co-Structure Complexes Affect Amyloid Nucleation through Fibril Mimetic Behavior. Biochemistry 2019, 58, 5052–5065. [Google Scholar]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, V.Y.; Jaravine, V.A. Analysis of non-uniformly sampled spectra with multi-dimensional decomposition. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Stilbs, P. Molecular self-diffusion coefficients in Fourier transform nuclear magnetic resonance spectrometric analysis of complex mixtures. Anal. Chem. 1981, 53, 2135–2137. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Kunze, M.B.; Erlendsson, S.; Kragelund, B.B. (S)Pinning down protein interactions by NMR. Protein Sci. 2017, 26, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Schwarzinger, S.; Kroon, G.J.; Foss, T.R.; Chung, J.; Wright, P.E.; Dyson, H.J. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001, 123, 2970–2978. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Poulsen, F.M. Sequence correction of random coil chemical shifts: Correlation between neighbor correction factors and changes in the Ramachandran distribution. J. Biomol. NMR 2011, 50, 157–165. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Brander, S.; Poulsen, F.M. Random coil chemical shift for intrinsically disordered proteins: Effects of temperature and pH. J. Biomol. NMR 2011, 49, 139–149. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D.; Nespovitaya, N.; Zacharopoulou, M.; Kaminski, C.F.; Phillips, J.J.; Kaminski Schierle, G.S. Different Structural Conformers of Monomeric α-Synuclein Identified after Lyophilizing and Freezing. Anal. Chem. 2018, 90, 6975–6983. [Google Scholar] [CrossRef] [PubMed]
- Paslawski, W.; Lorenzen, N.; Otzen, D.E. Formation and Characterization of α-Synuclein Oligomers. Methods Mol. Biol. 2016, 1345, 133–150. [Google Scholar]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, M.; Kragelund, B.B.; Papaleo, E.; Lindorff-Larsen, K. An Efficient Method for Estimating the Hydrodynamic Radius of Disordered Protein Conformations. Biophys. J. 2017, 113, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Skolnick, J.; Kolinski, A.; Ortiz, A.R. MONSSTER: A method for folding globular proteins with a small number of distance restraints. J. Mol. Biol. 1997, 265, 217–241. [Google Scholar] [CrossRef] [Green Version]
- Kohn, J.E.; Millett, I.S.; Jacob, J.; Zagrovic, B.; Dillon, T.M.; Cingel, N.; Dothager, R.S.; Seifert, S.; Thiyagarajan, P.; Sosnick, T.R.; et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12491–12496. [Google Scholar] [CrossRef] [Green Version]
- Van Maarschalkerweerd, A.; Pedersen, M.N.; Peterson, H.; Nilsson, M.; Nguyen, T.; Skamris, T.; Rand, K.; Vetri, V.; Langkilde, A.E.; Vestergaard, B. Formation of covalent di-tyrosine dimers in recombinant α-synuclein. Intrinsically Disord. Proteins 2015, 3, e1071302. [Google Scholar] [CrossRef]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef] [Green Version]
- Iwai, A.; Yoshimoto, M.; Masliah, E.; Saitoh, T. Non-Aβ Component of Alzheimer’s Disease Amyloid (NAC) Is Amyloidogenic. Biochemistry 1995, 34, 10139–10145. [Google Scholar] [CrossRef] [PubMed]
- Perczel, A.; Hollósi, M.; Foxman, B.M.; Fasman, G.D. Conformational Analysis of Pseudocyclic Hexapeptides Based on Quantitative Circular Dichroism (CD), NOE, and X-ray Data. The Pure CD Spectra of Type I and Type II β-Turns. J. Am. Chem. Soc. 1991, 113, 9772–9784. [Google Scholar] [CrossRef]
- Perlin, A.S.; Casu, B.; Sanderson, G.R. 200 MHz spectra of heparin, chondroitins, and other muscopolysaccharides. Can. J. Chem. 1970, 48, 2260–2268. [Google Scholar] [CrossRef]
- Thompson, M.P.; Gordon, W.G.; Pepper, L.; Greenberg, R. Amino acid composition of beta-caseins from the milks of Bos indicus and Bos taurus cows: A comparative study. Comp. Biochem. Physiol. 1969, 30, 91–98. [Google Scholar] [CrossRef]
- Pornsuwan, S.; Giller, K.; Riedel, D.; Becker, S.; Griesinger, C.; Bennati, M. Long-range distances in amyloid fibrils of α-synuclein from PELDOR spectroscopy. Angew. Chem. Int. Ed. Engl. 2013, 125, 10480–10484. [Google Scholar] [CrossRef]
- Ramachandran, G.; Udgaonkar, J.B. Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by tau protein. J. Biol. Chem. 2011, 286, 38948–38959. [Google Scholar] [CrossRef] [Green Version]
- Noborn, F.; O’Callaghan, P.; Hermansson, E.; Zhang, X.; Ancsin, J.B.; Damas, A.M.; Dacklin, I.; Presto, J.; Johansson, J.; Saraiva, M.J.; et al. Heparan sulfate/heparin promotes transthyretin fibrillization through selective binding to a basic motif in the protein. Proc. Natl. Acad. Sci. USA 2011, 108, 5584–5589. [Google Scholar] [CrossRef] [Green Version]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 2015, 20, 2510–2528. [Google Scholar] [CrossRef] [Green Version]
- Du, H.N.; Tang, L.; Luo, X.Y.; Li, H.T.; Hu, J.; Zhou, J.W.; Hu, H.Y. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry 2003, 42, 8870–8878. [Google Scholar] [CrossRef]
- Li, H.T.; Du, H.N.; Tang, L.; Hu, J.; Hu, H.Y. Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: Non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers 2002, 64, 221–226. [Google Scholar] [CrossRef]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of alpha-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.W.; Giasson, B.I.; Lewis, K.A.; Lee, V.M.; Demartino, G.N.; Thomas, P.J. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: Implications for pathogenesis of Parkinson disease. J. Biol. Chem. 2005, 280, 22670–22678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowther, R.A.; Jakes, R.; Spillantini, M.G.; Goedert, M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998, 436, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109-140 on α-synuclein aggregation in vitro. Biochemistry 2004, 43, 16233–16242. [Google Scholar] [CrossRef]
- Zibaee, S.; Jakes, R.; Fraser, G.; Serpell, L.C.; Crowther, R.A.; Goedert, M. Sequence Determinants for Amyloid Fibrillogenesis of Human alpha-Synuclein. J. Mol. Biol. 2007, 374, 454–464. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Irvine, G.B. Aggregation and neurotoxicity of α-synuclein and related peptides. Biochem. Soc. Trans. 2002, 30, 559–565. [Google Scholar] [CrossRef]
- Doherty, C.P.A.; Ulamec, S.M.; Maya-Martinez, R.; Good, S.C.; Makepeace, J.; Khan, G.N.; van Oosten-Hawle, P.; Radford, S.E.; Brockwell, D.J. A short motif in the N-terminal region of α-synuclein is critical for both aggregation and function. Nat. Struct. Mol. Biol. 2020, 27, 249–259. [Google Scholar] [CrossRef]
- Koehl, P.; Levitt, M. Structure-based conformational preferences of amino acids. Proc. Natl. Acad. Sci. USA 1999, 96, 12524–12529. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [Green Version]
- Biere, A.L.; Wood, S.J.; Wypych, J.; Steavenson, S.; Jiang, Y.; Anafi, D.; Jacobsen, F.W.; Jarosinski, M.A.; Wu, G.M.; Louis, J.C.; et al. Parkinson’s disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J. Biol. Chem. 2000, 275, 34574–34579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Gallardo, G.; Fernández-Chacón, R.; Schlüter, O.M.; Südhof, T.C. α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Agerschou, E.D.; Flagmeier, P.; Saridaki, T.; Galvagnion, C.; Komnig, D.; Heid, L.; Prasad, V.; Shaykhalishahi, H.; Willbold, D.; Dobson, C.M.; et al. An engineered monomer binding-protein for α-synuclein efficiently inhibits the proliferation of amyloid fibrils. eLife 2019, 8, e46112. [Google Scholar] [CrossRef]
- Mirecka, E.A.; Feuerstein, S.; Gremer, L.; Schröder, G.F.; Stoldt, M.; Willbold, D.; Hoyer, W. β-Hairpin of Islet Amyloid Polypeptide Bound to an Aggregation Inhibitor. Sci. Rep. 2016, 6, 33474. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 2019, 8, e43584. [Google Scholar] [CrossRef]
- Nespovitaya, N.; Mahou, P.; Laine, R.F.; Schierle, G.S.K.; Kaminski, C.F. Heparin acts as a structural component of β-endorphin amyloid fibrils rather than a simple aggregation promoter. Chem. Commun. 2017, 53, 1273–1276. [Google Scholar] [CrossRef] [Green Version]
- Maïza, A.; Chantepie, S.; Vera, C.; Fifre, A.; Huynh, M.B.; Stettler, O.; Ouidja, M.O.; Papy-Garcia, D. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 2018, 592, 3806–3818. [Google Scholar] [CrossRef] [Green Version]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Boström, F.; Hansson, O.; Gerhardsson, L.; Lundh, T.; Minthon, L.; Stomrud, E.; Zetterberg, H.; Londos, E. CSF Mg and Ca as diagnostic markers for dementia with Lewy bodies. Neurobiol. Aging 2009, 30, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Kragelund, B.B.; Schenstrøm, S.M.; Rebula, C.A.; Panse, V.G.; Hartmann-Petersen, R. DSS1/Sem1, a Multifunctional and Intrinsically Disordered Protein. Trends Biochem. Sci. 2016, 41, 446–459. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radii (Å) | ||||
|---|---|---|---|---|
| aSN | aSN1–61 | Reference | ||
| Rg | Folded | 14.4 | 10.5 | [57] |
| Unfolded | 36.8 | 22.4 | [58] | |
| SAXS | 36.5 | 24.0 ± 0.3 | [59], This work | |
| Rh | Folded | 20.1 | 15.8 | [45] |
| Unfolded | 36.8 | 22.9 | [45] | |
| IDP | 31.1 | 20.3 | [60] | |
| NMR * | 29.1 ± 0.1 | 20.1 ± 0.1 | This work | |
| Rg/Rh | Folded | 0.71 | 0.66 | [57] |
| Unfolded | 1 | 0.97 | [58] | |
| Nygaard | 0.98 | 1.05 | [56] | |
| SAXS/NMR | 1.08 | 1.2 | This work | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skaanning, L.K.; Santoro, A.; Skamris, T.; Martinsen, J.H.; D’Ursi, A.M.; Bucciarelli, S.; Vestergaard, B.; Bugge, K.; Langkilde, A.E.; Kragelund, B.B. The Non-Fibrillating N-Terminal of α-Synuclein Binds and Co-Fibrillates with Heparin. Biomolecules 2020, 10, 1192. https://doi.org/10.3390/biom10081192
Skaanning LK, Santoro A, Skamris T, Martinsen JH, D’Ursi AM, Bucciarelli S, Vestergaard B, Bugge K, Langkilde AE, Kragelund BB. The Non-Fibrillating N-Terminal of α-Synuclein Binds and Co-Fibrillates with Heparin. Biomolecules. 2020; 10(8):1192. https://doi.org/10.3390/biom10081192
Chicago/Turabian StyleSkaanning, Line K., Angelo Santoro, Thomas Skamris, Jacob Hertz Martinsen, Anna Maria D’Ursi, Saskia Bucciarelli, Bente Vestergaard, Katrine Bugge, Annette Eva Langkilde, and Birthe B. Kragelund. 2020. "The Non-Fibrillating N-Terminal of α-Synuclein Binds and Co-Fibrillates with Heparin" Biomolecules 10, no. 8: 1192. https://doi.org/10.3390/biom10081192