The Status of p53 Oligomeric and Aggregation States in Cancer


 ,
,
Abstract
:
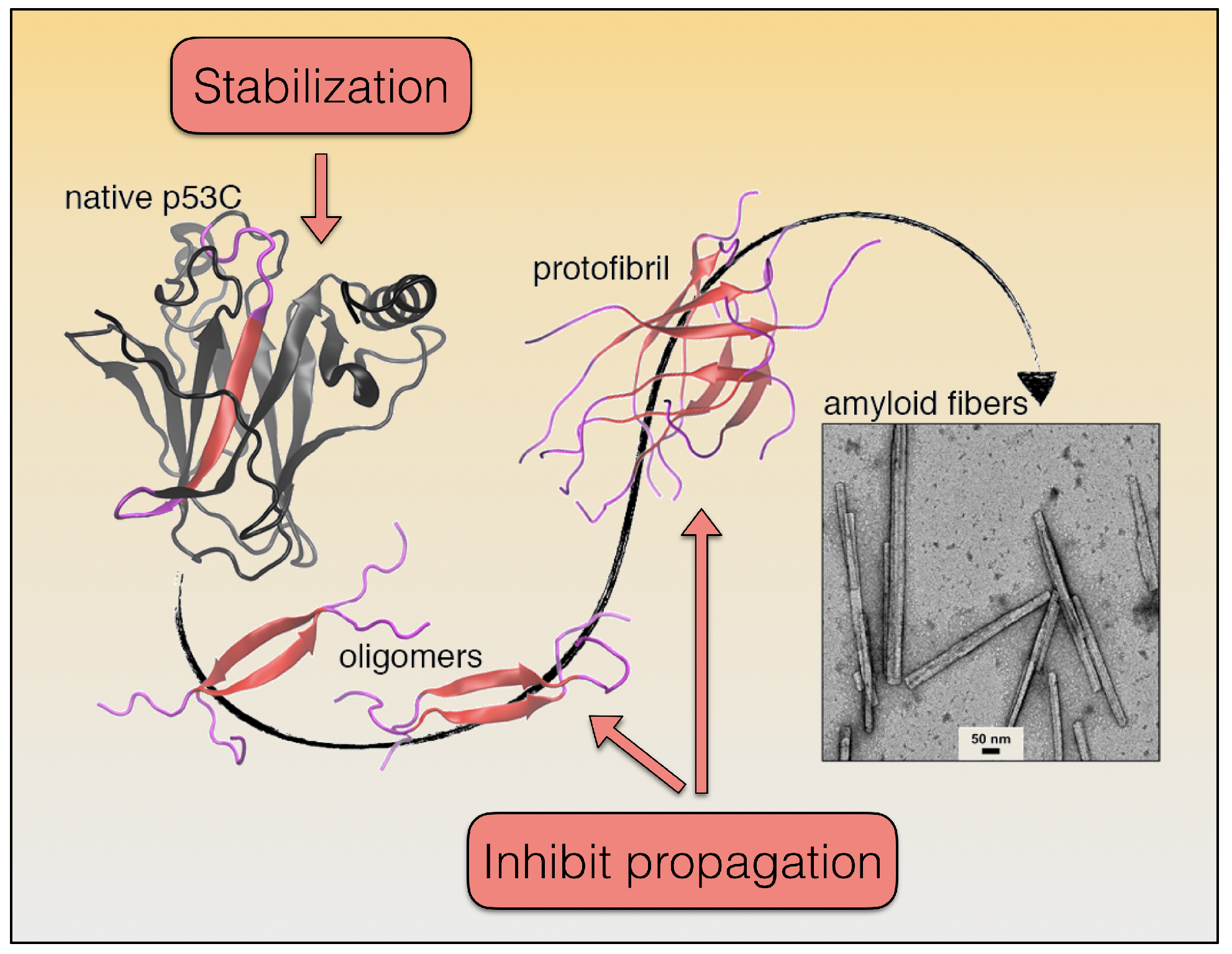{kind=link}
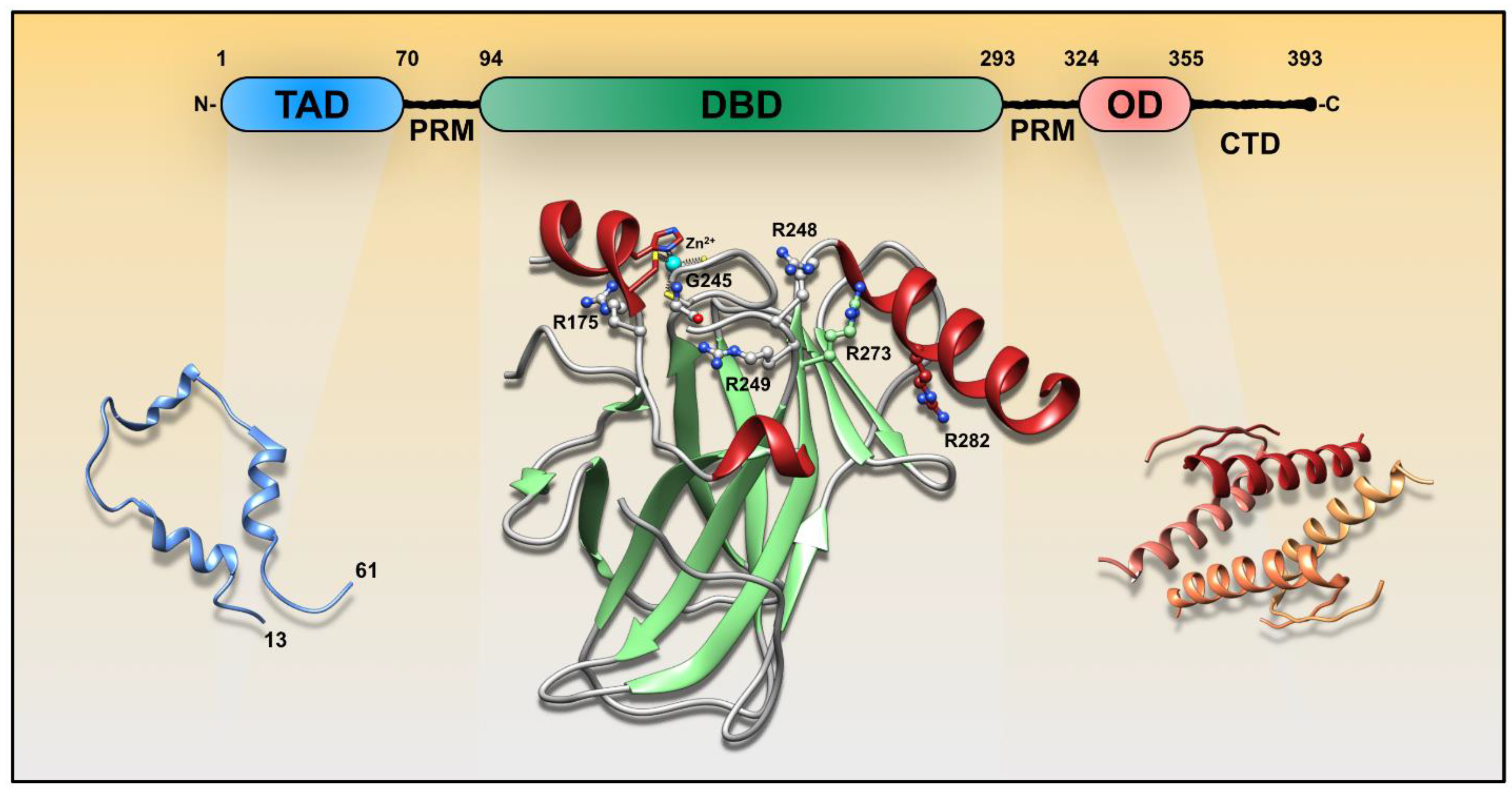{kind=link}
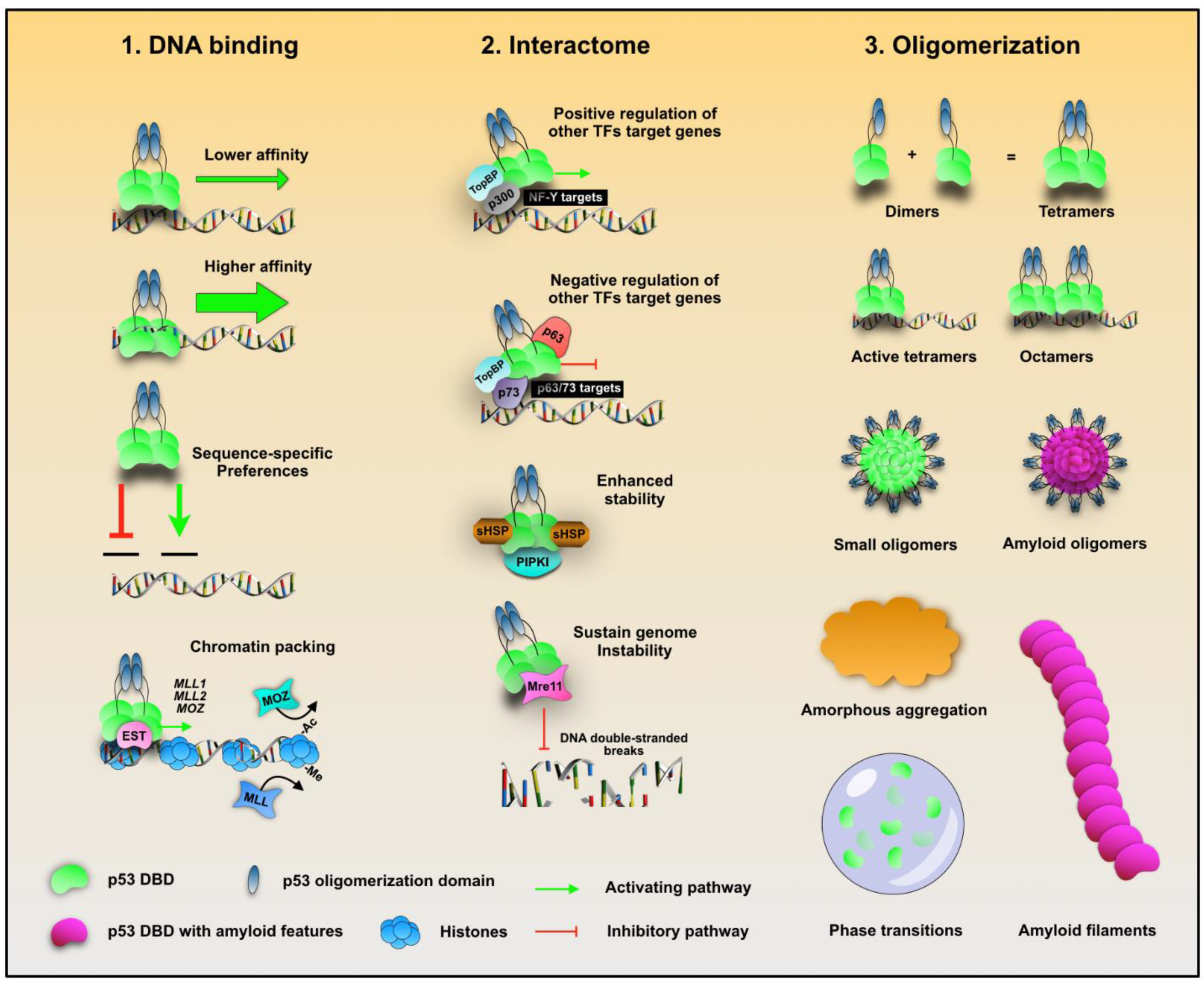{kind=link}
{kind=link}
{kind=link}
1. Overview of Mutated p53 Domains in Cancer
2. Renegade p53 Outcomes
3. p53 Oligomeric States
4. Other Factors Contributing to p53 Aggregation
5. p53 Aggregation Mechanism and Treatment Options
Author Contributions
Funding
Conflicts of Interest
References
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- de Oliveira, G.A.; Rangel, L.P.; Costa, D.C.; Silva, J.L. Misfolding, Aggregation, and Disordered Segments in c-Abl and p53 in Human Cancer. Front. Oncol. 2015, 5, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Costa, D.C.; de Oliveira, G.A.; Cino, E.A.; Soares, I.N.; Rangel, L.P.; Silva, J.L. Aggregation and Prion-Like Properties of Misfolded Tumor Suppressors: Is Cancer a Prion Disease? Cold Spring Harb. Perspect. Biol. 2016, 8, a023614. [Google Scholar] [CrossRef]
- Kastan, M.B.; Zhan, Q.; el-Deiry, W.S.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J., Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597. [Google Scholar] [CrossRef]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Kandil, A.N. Conformational shifts propagate from the oligomerization domain of p53 to its tetrameric DNA binding domain and restore DNA binding to select p53 mutants. EMBO J. 1993, 12, 5057–5064. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Saito, S.; Yamaguchi, H.; Higashimoto, Y.; Chao, C.; Xu, Y.; Fornace, A.J., Jr.; Appella, E.; Anderson, C.W. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 2003, 278, 37536–37544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 2001, 268, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamuro, D.; Sabbatini, P.; White, E.; Prendergast, G.C. The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene 1997, 15, 887–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, J.M.; Emerson, B.M. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell 2001, 8, 57–69. [Google Scholar] [CrossRef]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Taylor, J.A.; Li, Y.; He, M.; Mason, T.; Mettlin, C.; Vogler, W.J.; Maygarden, S.; Liu, E. p53 mutations in bladder tumors from arylamine-exposed workers. Cancer Res. 1996, 56, 294–298. [Google Scholar]
- Sun, X.F.; Johannsson, O.; Hakansson, S.; Sellberg, G.; Nordenskjold, B.; Olsson, H.; Borg, A. A novel p53 germline alteration identified in a late onset breast cancer kindred. Oncogene 1996, 13, 407–411. [Google Scholar]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; AP de Oliveira, G. Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer. Acc. Chem. Res. 2018, 51, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.; Medcalf, E.A. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991, 65, 765–774. [Google Scholar] [CrossRef]
- Vaughan, C.A.; Singh, S.; Windle, B.; Sankala, H.M.; Graves, P.R.; Andrew Yeudall, W.; Deb, S.P.; Deb, S. p53 mutants induce transcription of NF-kappaB2 in H1299 cells through CBP and STAT binding on the NF-kappaB2 promoter and gain of function activity. Arch. Biochem. Biophys. 2012, 518, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Will, K.; Warnecke, G.; Wiesmuller, L.; Deppert, W. Specific interaction of mutant p53 with regions of matrix attachment region DNA elements (MARs) with a high potential for base-unpairing. Proc. Natl. Acad. Sci. USA 1998, 95, 13681–13686. [Google Scholar] [CrossRef] [Green Version]
- Dell’Orso, S.; Fontemaggi, G.; Stambolsky, P.; Goeman, F.; Voellenkle, C.; Levrero, M.; Strano, S.; Rotter, V.; Oren, M.; Blandino, G. ChIP-on-chip analysis of in vivo mutant p53 binding to selected gene promoters. OMICS 2011, 15, 305–312. [Google Scholar] [CrossRef]
- Liu, K.; Ling, S.; Lin, W.C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [Green Version]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef]
- Martynova, E.; Pozzi, S.; Basile, V.; Dolfini, D.; Zambelli, F.; Imbriano, C.; Pavesi, G.; Mantovani, R. Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget 2012, 3, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Chen, M.; Cryns, V.L.; Anderson, R.A. A nuclear phosphoinositide kinase complex regulates p53. Nat. Cell Biol. 2019, 21, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Restle, A.; Farber, M.; Baumann, C.; Bohringer, M.; Scheidtmann, K.H.; Muller-Tidow, C.; Wiesmuller, L. Dissecting the role of p53 phosphorylation in homologous recombination provides new clues for gain-of-function mutants. Nucleic Acids Res. 2008, 36, 5362–5375. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Huang, F.; Fersht, A.R. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011, 39, 2294–2303. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef]
- Stenger, J.E.; Mayr, G.A.; Mann, K.; Tegtmeyer, P. Formation of stable p53 homotetramers and multiples of tetramers. Mol. Carcinog. 1992, 5, 102–106. [Google Scholar] [CrossRef]
- Wang, Y.; Schwedes, J.F.; Parks, D.; Mann, K.; Tegtmeyer, P. Interaction of p53 with its consensus DNA-binding site. Mol. Cell Biol. 1995, 15, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Stenger, J.E.; Tegtmeyer, P.; Mayr, G.A.; Reed, M.; Wang, Y.; Wang, P.; Hough, P.V.; Mastrangelo, I.A. p53 oligomerization and DNA looping are linked with transcriptional activation. EMBO J. 1994, 13, 6011–6020. [Google Scholar] [CrossRef]
- Okorokov, A.L.; Sherman, M.B.; Plisson, C.; Grinkevich, V.; Sigmundsson, K.; Selivanova, G.; Milner, J.; Orlova, E.V. The structure of p53 tumour suppressor protein reveals the basis for its functional plasticity. EMBO J. 2006, 25, 5191–5200. [Google Scholar] [CrossRef] [Green Version]
- Kearns, S.; Lurz, R.; Orlova, E.V.; Okorokov, A.L. Two p53 tetramers bind one consensus DNA response element. Nucleic Acids Res. 2016, 44, 6185–6199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, P.; Beno, I.; Xi, Z.; Stein, Y.; Golovenko, D.; Kessler, N.; Rotter, V.; Shakked, Z.; Haran, T.E. Diverse p53/DNA binding modes expand the repertoire of p53 response elements. Proc. Natl. Acad. Sci. USA 2017, 114, 10624–10629. [Google Scholar] [CrossRef] [Green Version]
- Pedrote, M.M.; Motta, M.F.; Ferretti, G.D.S.; Norberto, D.R.; Spohr, T.; Lima, F.R.S.; Gratton, E.; Silva, J.L.; de Oliveira, G.A.P. Oncogenic Gain of Function in Glioblastoma Is Linked to Mutant p53 Amyloid Oligomers. iScience 2020, 23, 100820. [Google Scholar] [CrossRef]
- Gaglia, G.; Guan, Y.; Shah, J.V.; Lahav, G. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. USA 2013, 110, 15497–15501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Katz, C.; Low-Calle, A.M.; Choe, J.H.; Laptenko, O.; Tong, D.; Joseph-Chowdhury, J.N.; Garofalo, F.; Zhu, Y.; Friedler, A.; Prives, C. Wild-type and cancer-related p53 proteins are preferentially degraded by MDM2 as dimers rather than tetramers. Genes Dev. 2018, 32, 430–447. [Google Scholar] [CrossRef] [Green Version]
- Marchenko, N.D.; Wolff, S.; Erster, S.; Becker, K.; Moll, U.M. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007, 26, 923–934. [Google Scholar] [CrossRef]
- Ano Bom, A.P.; Rangel, L.P.; Costa, D.C.; de Oliveira, G.A.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.; Cepeda, A.O.; Stumbo, A.C.; et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef] [Green Version]
- Lasagna-Reeves, C.A.; Clos, A.L.; Castillo-Carranza, D.; Sengupta, U.; Guerrero-Munoz, M.; Kelly, B.; Wagner, R.; Kayed, R. Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 2013, 430, 963–968. [Google Scholar] [CrossRef]
- Yang-Hartwich, Y.; Soteras, M.G.; Lin, Z.P.; Holmberg, J.; Sumi, N.; Craveiro, V.; Liang, M.; Romanoff, E.; Bingham, J.; Garofalo, F.; et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene 2015, 34, 3605–3616. [Google Scholar] [CrossRef]
- Melo Dos Santos, N.; de Oliveira, G.A.P.; Ramos Rocha, M.; Pedrote, M.M.; Diniz da Silva Ferretti, G.; Pereira Rangel, L.; Morgado-Diaz, J.A.; Silva, J.L.; Rodrigues Pereira Gimba, E. Loss of the p53 transactivation domain results in high amyloid aggregation of the Delta40p53 isoform in endometrial carcinoma cells. J. Biol. Chem. 2019, 294, 9430–9439. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, D.A.L.R.; Teixeira, L.S.P.; Quesado, P.A.; Maiolino, L.M.; Lopez, P.M.; Cordeiro, Y.; Costa, L.T.; Heckl, W.M.; Weissmüller, G.; Foguel, D.; et al. Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry 2003, 42, 9022–9027. [Google Scholar] [CrossRef]
- Ishimaru, D.; Maia, L.F.; Maiolino, L.M.; Quesado, P.A.; Lopez, P.C.M.; Almeida, F.C.L.; Valente, A.P.; Silva, J.L. Conversion of Wild-type p53 Core Domain into a Conformation that Mimics a Hot-spot Mutant. J. Mol. Biol. 2003, 333, 443–451. [Google Scholar] [CrossRef]
- Levy, C.B.; Stumbo, A.C.; Ano Bom, A.P.; Portari, E.A.; Cordeiro, Y.; Silva, J.L.; De Moura-Gallo, C.V. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011, 43, 60–64. [Google Scholar] [CrossRef]
- Ham, S.W.; Jeon, H.Y.; Jin, X.; Kim, E.J.; Kim, J.K.; Shin, Y.J.; Lee, Y.; Kim, S.H.; Lee, S.Y.; Seo, S.; et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019, 26, 409–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, J.X.; Liu, Y.H.; You, C.; Mao, Q. Mutant TP53 enhances the resistance of glioblastoma cells to temozolomide by up-regulating O(6)-methylguanine DNA-methyltransferase. Neurol. Sci. 2013, 34, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, S.A.; O’Hagan, H.M.; Ljungman, M. Accumulation of soluble and nucleolar-associated p53 proteins following cellular stress. J. Cell Sci. 2001, 114, 1867–1873. [Google Scholar] [PubMed]
- Latonen, L. Phase-to-Phase with Nucleoli—Stress Responses, Protein Aggregation and Novel Roles of RNA. Front. Cell Neurosci. 2019, 13, 151. [Google Scholar] [CrossRef]
- de Oliveira, G.A.P.; Cordeiro, Y.; Silva, J.L.; Vieira, T. Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases. Adv. Protein Chem. Struct. Biol. 2019, 118, 289–331. [Google Scholar] [CrossRef]
- Audas, T.E.; Audas, D.E.; Jacob, M.D.; Ho, J.J.; Khacho, M.; Wang, M.; Perera, J.K.; Gardiner, C.; Bennett, C.A.; Head, T.; et al. Adaptation to Stressors by Systemic Protein Amyloidogenesis. Dev. Cell 2016, 39, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.L.; Cordeiro, Y. The “Jekyll and Hyde” Actions of Nucleic Acids on the Prion-like Aggregation of Proteins. J. Biol. Chem. 2016, 291, 15482–15490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamagata, K.; Kanbayashi, S.; Honda, M.; Itoh, Y.; Takahashi, H.; Kameda, T.; Nagatsugi, F.; Takahashi, S. Liquid-like droplet formation by tumor suppressor p53 induced by multivalent electrostatic interactions between two disordered domains. Sci. Rep. 2020, 10, 580. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Vieira, T.; Kovachev, P.S.; Sanyal, S.; Silva, J.L. Modulation of p53 and prion protein aggregation by RNA. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 933–940. [Google Scholar] [CrossRef]
- Yang, D.S.S.A.; Davtyan, A.; Fathi, M.; Safari, M.S.; Klindziuk, A.; Barton, M.C.; Varadarajan, N.; Kolomeisky, A.B.; Vekilov, P.G. Mesoscopic Liquid Clusters Represent a Distinct Condensate of Mutant p53. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yoshida, Y.; Izumi, H.; Torigoe, T.; Ishiguchi, H.; Yoshida, T.; Itoh, H.; Kohno, K. Binding of RNA to p53 regulates its oligomerization and DNA-binding activity. Oncogene 2004, 23, 4371–4379. [Google Scholar] [CrossRef] [Green Version]
- Kovachev, P.S.; Banerjee, D.; Rangel, L.P.; Eriksson, J.; Pedrote, M.M.; Martins-Dinis, M.; Edwards, K.; Cordeiro, Y.; Silva, J.L.; Sanyal, S. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J. Biol. Chem. 2017, 292, 9345–9357. [Google Scholar] [CrossRef] [Green Version]
- Safari, M.S.; Wang, Z.; Tailor, K.; Kolomeisky, A.B.; Conrad, J.C.; Vekilov, P.G. Anomalous Dense Liquid Condensates Host the Nucleation of Tumor Suppressor p53 Fibrils. iScience 2019, 12, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Suwiwat, S.; Ricciardelli, C.; Tammi, R.; Tammi, M.; Auvinen, P.; Kosma, V.M.; LeBaron, R.G.; Raymond, W.A.; Tilley, W.D.; Horsfall, D.J. Expression of extracellular matrix components versican, chondroitin sulfate, tenascin, and hyaluronan, and their association with disease outcome in node-negative breast cancer. Clin. Cancer Res. 2004, 10, 2491–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerotziafas, G.T.; Papageorgiou, C.; Hatmi, M.; Samama, M.M.; Elalamy, I. Clinical studies with anticoagulants to improve survival in cancer patients. Pathophysiol. Haemost. Thromb. 2008, 36, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M. Heparin in tumor progression and metastatic dissemination. Semin. Thromb. Hemost. 2007, 33, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Alur, I.; Dodurga, Y.; Secme, M.; Elmas, L.; Bagci, G.; Goksin, I.; Avci, C.B. Anti-tumor effects of bemiparin in HepG2 and MIA PaCa-2 cells. Gene 2016, 585, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.K.; Hsu, K.H.; Cheng, Y.M.; Suen, H.Y.; Peng, S.F. Development and In Vitro Evaluation of Linear PEI-Shelled Heparin/Berberine Nanoparticles in Human Osteosarcoma U-2 OS Cells. Molecules 2018, 23, 3122. [Google Scholar] [CrossRef] [Green Version]
- Karbownik, M.S.; Nowak, J.Z. Hyaluronan: Towards novel anti-cancer therapeutics. Pharmacol. Rep. 2013, 65, 1056–1074. [Google Scholar] [CrossRef]
- Zhao, Y.; Qiao, S.; Hou, X.; Tian, H.; Deng, S.; Ye, K.; Nie, Y.; Chen, X.; Yan, H.; Tian, W. Bioengineered tumor microenvironments with naked mole rats high-molecular-weight hyaluronan induces apoptosis in breast cancer cells. Oncogene 2019, 38, 4297–4309. [Google Scholar] [CrossRef]
- Pedrote, M.M.; de Oliveira, G.A.P.; Felix, A.L.; Mota, M.F.; Marques, M.A.; Soares, I.N.; Iqbal, A.; Norberto, D.R.; Gomes, A.M.O.; Gratton, E.; et al. Aggregation-primed molten globule conformers of the p53 core domain provide potential tools for studying p53C aggregation in cancer. J. Biol. Chem. 2018, 293, 11374–11387. [Google Scholar] [CrossRef] [Green Version]
- Baud, M.G.J.; Bauer, M.R.; Verduci, L.; Dingler, F.A.; Patel, K.J.; Horil Roy, D.; Joerger, A.C.; Fersht, A.R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018, 152, 101–114. [Google Scholar] [CrossRef]
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef] [Green Version]
- Ferraz da Costa, D.C.; Campos, N.P.C.; Santos, R.A.; Guedes-da-Silva, F.H.; Martins-Dinis, M.; Zanphorlin, L.; Ramos, C.; Rangel, L.P.; Silva, J.L. Resveratrol prevents p53 aggregation in vitro and in breast cancer cells. Oncotarget 2018, 9, 29112–29122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cino, E.A.; Soares, I.N.; Pedrote, M.M.; de Oliveira, G.A.; Silva, J.L. Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds. Sci. Rep. 2016, 6, 32535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Chen, J.; Keshamouni, V.G.; Kanapathipillai, M. Polyarginine and its analogues inhibit p53 mutant aggregation and cancer cell proliferation in vitro. Biochem. Biophys. Res. Commun. 2017, 489, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.; Navalkar, A.; Maji, S.K.; Silva, J.L.; de Oliveira, G.A.P.; Cino, E.A. Biophysical characterization of p53 core domain aggregates. Biochem. J. 2020, 477, 111–120. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, G.A.P.; Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Vieira, T.C.R.G.; Cino, E.A.; Silva, J.L. The Status of p53 Oligomeric and Aggregation States in Cancer. Biomolecules 2020, 10, 548. https://doi.org/10.3390/biom10040548
de Oliveira GAP, Petronilho EC, Pedrote MM, Marques MA, Vieira TCRG, Cino EA, Silva JL. The Status of p53 Oligomeric and Aggregation States in Cancer. Biomolecules. 2020; 10(4):548. https://doi.org/10.3390/biom10040548
Chicago/Turabian Stylede Oliveira, Guilherme A. P., Elaine C. Petronilho, Murilo M. Pedrote, Mayra A. Marques, Tuane C. R. G. Vieira, Elio A. Cino, and Jerson L. Silva. 2020. "The Status of p53 Oligomeric and Aggregation States in Cancer" Biomolecules 10, no. 4: 548. https://doi.org/10.3390/biom10040548