1. Introduction
Lung cancer is one of the leading causes of death worldwide, mostly due to a late diagnosis. In fact, an estimated nearly 136,000 patients are expected to die from lung cancer in 2020 in the United States [
1]. Even though it only contains 9% of the world’s population, Europe accounts for 25% of the global cancer burden, with an estimated 3.9 million new cancer cases and 1.9 million expected cancer deaths in 2018 [
2]. Within these cases, the most common cause of cancer death was lung cancer, and 280,000 are expected to die from lung cancer in 2019 [
3]. In Asia, and especially in Japan, the number of new cases of lung cancer in 2018 was 118,971 (13.5%), which is the worst number of cases among all cancers. The same was true for the risk of death; 81,820 (20.0%), as indicated by the statistics summarized by the World Health Organization (WHO) [
4].
Lung cancer can be classified into two major types: small-cell lung cancer (SCLC), which accounts for approximately 15% of cases and non-small-cell lung cancer (NSCLC), which accounts for approximately 85%. Therefore, NSCLC involves the majority of the lung cancer population, and adenocarcinoma is the most common type of NSCLC. Multiple mutations have been reported to occur in NSCLC, but needless to say, the spectrum of mutations is different between different subtypes [
5,
6]. Thus, knowing the clinical, pathological, and molecular biological outcomes in diverse aspects is quite important to achieve an improvement in the quality of life of cancer patients.
Recently, in the medical field, deep-learning-driven classification of cancer showed a great success [
7]. After that, many images-based machine-learning and deep-learning studies demonstrated their use for cancer prediction, prognosis, or even to assess treatment response in lung cancer [
8,
9,
10]. However, single-level omics data have limitations, particularly because cancer is a heterogeneous disease, so relying on results obtained from single-level omics data may be risky and misleading; thus, it could affect the understanding of cancer as a whole and possibly negatively affect patients.
One of the proposed approaches to overcome this problem is a multi-omics analysis, an approach that has rapidly emerged in disease-related biology. A new cancer subtyping method, with the integration of multi-omics data, has already been used to reveal molecular subtypes of cancer with TCGA dataset. Multi-omics analysis, using integrated TCGA data of RNA expression, DNA methylation, point mutations, and copy number variation, demonstrated a prediction capability for poor patient outcomes [
11]. Multi-omics analysis with a TCGA hepatocellular carcinoma (LIHC) dataset was also performed, using a deep-learning-based and machine-learning-based pipeline to predict patient survival, using RNA expression, DNA methylation, and miRNA expression [
12]. The authors implemented an autoencoder to reduce the dimension of multi-omics features as an unsupervised approach, and then, the reduced features were further analyzed via the Cox proportional hazards (Cox-PH) model, to select survival-associated features. A similar approach was applied by using gene expression and copy number variants to classify poor or good subtypes in neuroblastoma [
13].
Here, to develop a classifier for the prediction of lung-cancer-patient prognosis and to investigate a patient risk-dependent analysis, we applied a deep-learning- and machine-learning-based pipeline for multi-omics analysis of lung cancer data. We chose data of RNA expression and miRNA expression as input data, so that the result we received could be interpretable, since RNA expression is regulated by miRNA by functional duplexes. Firstly, we developed an SVM that was able to distinguish prognosis-related subtypes from the TCGA LUAD. Secondly, we performed a risk-dependent pathway analysis that can give us relevant information and knowledge about potential mechanisms related to the different subtypes. Lastly, using differentially expressing RNAs in the subtypes, we found novel genes that are associated with patient survival, and we demonstrated that newly identified genes were associated with prognosis.
4. Discussion
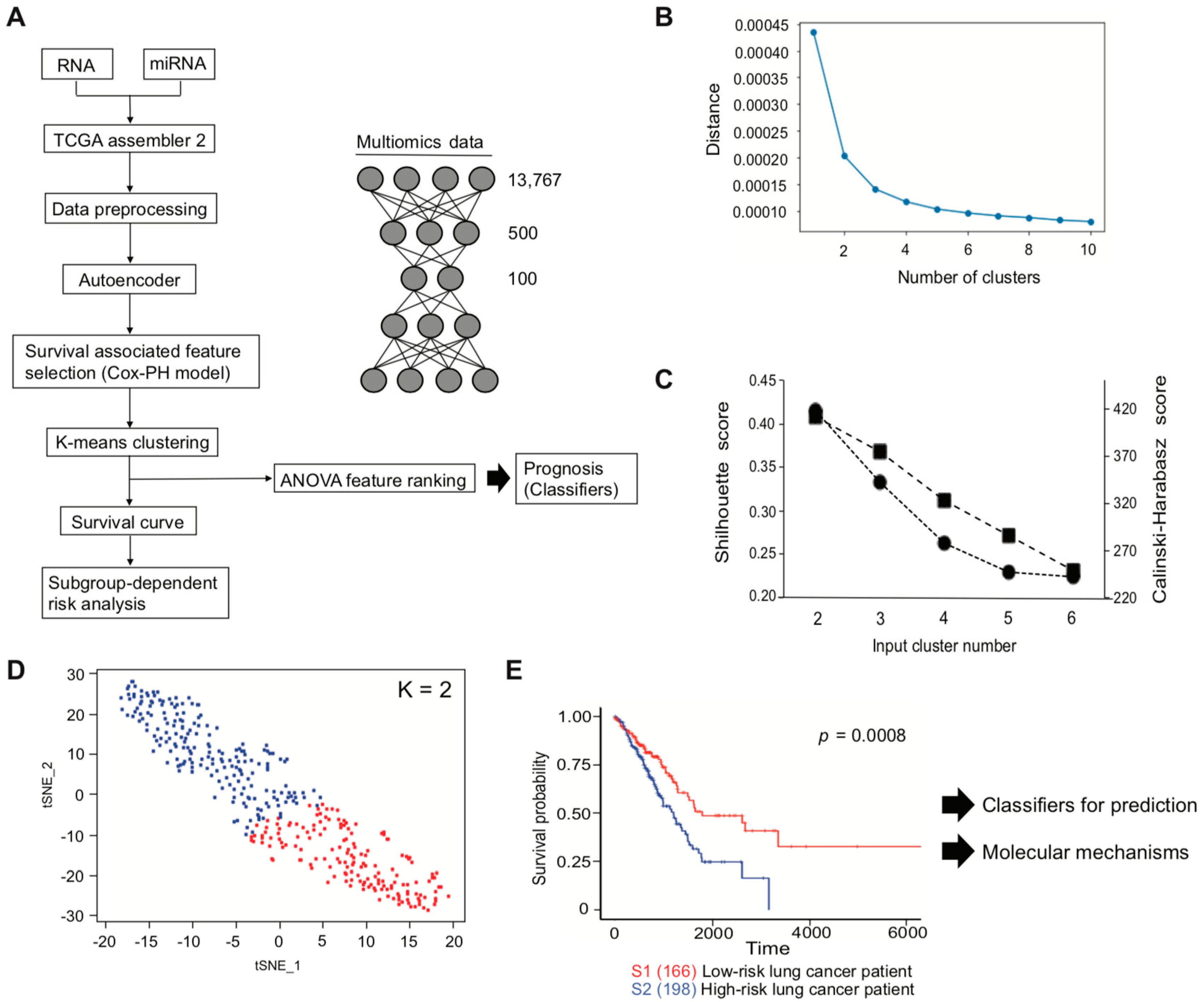
Here, we developed a pipeline, using a TCGA LUAD dataset, with the aim of efficiently identifying genes of interest that are associated with the lung cancer patients survival. Pipeline development started with multi-omics data to implement an autoencoder, followed by clinical associated feature selection by Cox-PH. Selected features were then labeled depending on the result of K-means clustering, which is later demonstrated to be associated with patient survival. The inferred labels, or two subtypes classed by K-means clustering, were applied to plot a Kaplan–Meier survival estimation, to visualize whether the labels were associated with a poor or a good patient survival subtype and used to develop an SVM that can successfully predict patient prognosis.
During autoencoder optimization, batch size, epochs, and activation function varied. Based on our results, a batch size of 1 and epochs of 150, or even between 100 and 150, gave reasonable results, while avoiding overfitting by early stopping [
47] and/or Rectified Linear Unit (ReLU) function replacing tanh function at the last layer [
48] did not work well in our autoencoder. Clustering analyses applied with clinically associated features demonstrated that K = 2 was the optimal number, and this is concordant with the previous report performing with the 10 TCGA cancer dataset [
49].
The multi-omics analysis with TCGA LIHC showed more
TP53 gene mutations in the high-risk subtype (Fisher test
p = 0.042), but unfortunately, other genes such as
EGFR were not investigated [
12]. In our case,
TP53 was slightly more mutated in the high-risk subtype (0.42%), compared with the low-risk subtype (0.39%), but not significance (Fisher test
p = 0.633). Whole-exome sequencing data of LUAD were analyzed independently in the oncogene-positive subset (
KRAS,
EGFR,
ERBB2,
BRAF,
MET,
ALK,
RET,
ROS,
HRAS,
NRAS, and
MAP2K1 driver mutations) and the oncogene-negative subset [
24]. The authors found that
TP53 and
NF1 co-mutations were enriched in the oncogene-negative subset. Additionally, RNA profiling provided new subtypes that the proximal-inflammatory subtype (formerly squamoid) was co-mutated with
TP53 and
NF1 [
6,
24]. In our analysis, we found
NF1 mutations were more enriched in the high-risk subtype, suggesting that the high-risk subtype we identified might correspond to the subset that has
TP53 and
NF1 co-mutations in [
24].
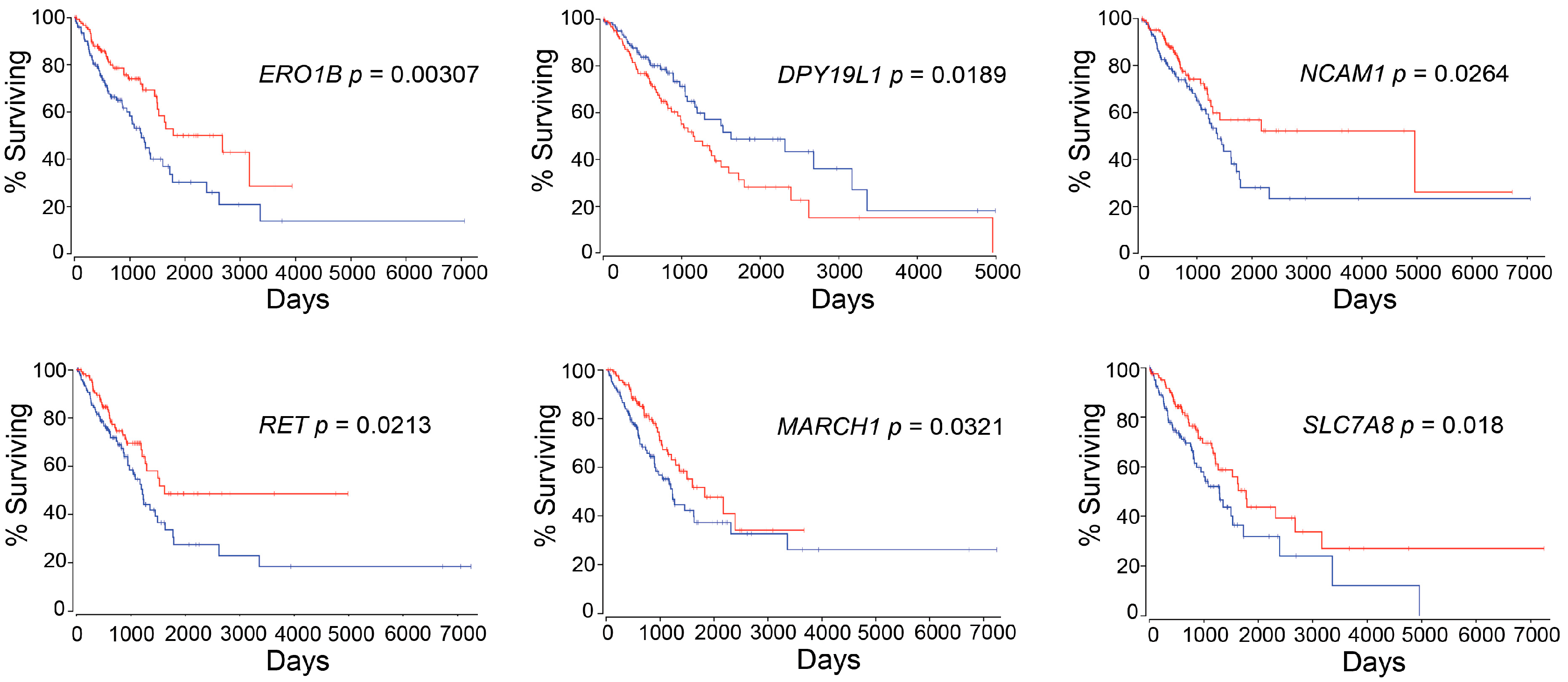
ERO1B was first reported as an endoplasmic reticulum disulfide oxidase [
50]. Later, additional biological functions, such as insulin biogenesis and glucose homeostasis, were demonstrated [
51]. In relation to lung cancer,
ERO1B has been recently identified as a gene that, together with an additional three genes identified using TCGA LUAD dataset, is able to predict patient prognosis [
52] and has been suggested to be a biomarker for pancreatic cancer [
53,
54].
DPY19L1 was firstly identified as an unclassified gene from human brain cDNA libraries in 1998 [
55]. Still, its function remains unknown, and no evidence has been reported so far on the link between
DPY19L1 and cancer prognosis. Therefore, to the best of our knowledge, this is the first report to reveal the association between
DPY19L1 expression and the prognosis in lung cancer patients.
NCAM1 or
CD56 is a member of the immunoglobulin superfamily involved in cell–cell interaction and cell–matrix interactions during the development. Additionally, it plays a fundamental role in processes such as cell migration and cell survival, in specific phenotypes of neural cells [
56].
NCAM1 may play an important role in EMT not only in intrahepatic cholangiocarcinoma but also in lung cancer via miR-200 (
Table 6 and [
57]). Recently, antibody-based anticancer treatment was analyzed with the expression levels of
NCAM1. The phase 1/2 study is ongoing, since
NCAM1 is expressed on several malignancies, including SCLC [
58,
59,
60], or could be available to predict prognosis in adult acute lymphoblastic leukemia patient [
61].
RET was identified in 1985.
RET is a receptor-type tyrosine kinase with multiple domains.
RET was first discovered in papillary thyroid carcinoma, and later in sporadic tumors, neurodegenerative diseases, and Hirschsprung’s disease [
62].
RET can be found in the rearrangement of genes generating RET fusion proteins in many cancers, including lung cancer, and thus an inhibitor was recently approved by the FDA for cancer therapy [
63]. It is important to note that, not only genetic factors, but also epigenetic factors, affect
RET expression that influences the probability of patient survival [
64]. It suggests that multi-omics analysis, including epigenetic data, could improve availability of output, in terms of precision medicine or personalized medicine, as we recently reported [
65]. The E3 ubiquitin ligase
MARCH1 plays an important role in immunology [
66], although only a few publications have focused on
MARCH1 in the context of cancer [
67,
68]. Therefore, further studies in this area are required and could have the potential to contribute to the field of cancer research, and more particularly lung cancer.
SLC7A8 or
LAT-2 is an L-type amino acid transporter-2 protein that binds and regulates mechanistic target of rapamycin kinase (mTOR) activation in pancreatic cancer [
69]. L-type amino acid transporters are known to be novel targets for cancer therapy [
70,
71]. However, as is the case for
DPY19L1 and
MARCH1, no publications have demonstrated the link between lung cancers.
We identified six genes with expression levels that were associated with patient survival, using the autoencoder, followed by bioinformatics analysis. The practice guidelines in oncology illustrate a strategy of patient treatment based on the result of gene mutations, such as
EGFR,
ALK,
ROS1, and
PD-L1 [
72], but not considering RNA or miRNA expression levels. It might be of great help to estimate survival outcome and to make treatment strategy for patients if several RNA-expression levels, such as
ERO1B,
DPY19L1,
NCAM1,
RET,
MARCH1, and/or
SLC7A8, are also examined at the time when patients are diagnosed.
To elucidate whether six genes were only associated with LUAD patient prognosis or whether these genes were key regulators of other types of NSCLC prognosis, survival analysis against TCGA lung squamous cell carcinoma (LUSC) was performed. The p-values for high expression and low expression of genes of interest were from 0.106 to 0.674, suggesting that the genes we identified were LUAD-specific survival-related genes. This result gave us confidence that the multi-omics analysis we developed truly identified input-data-specific survival-associated features. In other words, if we would like to identified genes of interest that are associated with LUSC patient survival, we need to use a LUSC dataset as an input.
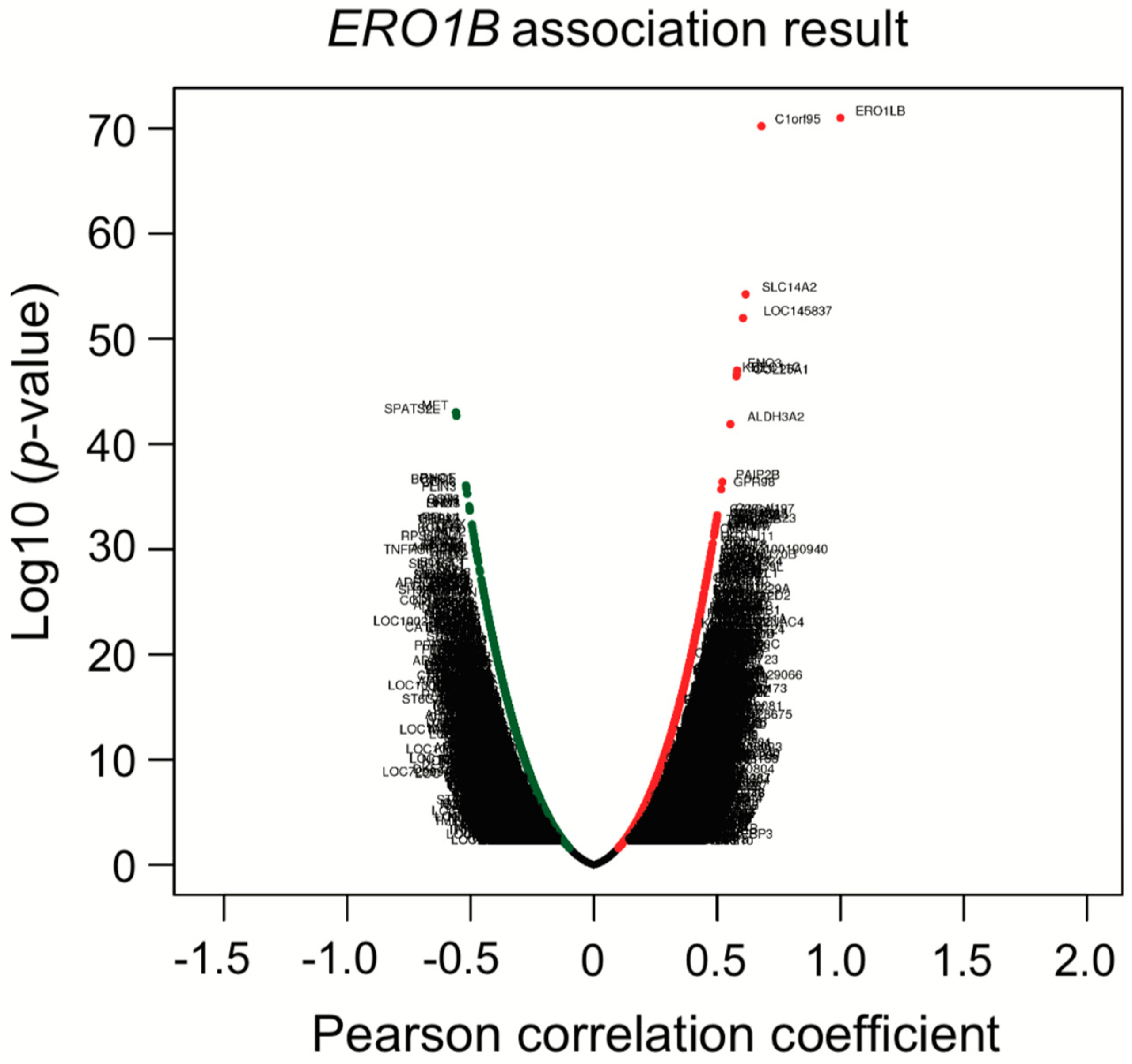
Co-expression analysis showed that
ERO1B,
ENO3,
RET, and
KCNE4 were co-upregulated. Later, we showed that
ENO3 and
KCNE4 have a target sequence for miR-6838. The functional role of miR-6838 has been recently investigated, showing that miR-6838 regulates EMT in triple-negative breast cancer by inhibiting the Wnt pathway [
73]. KEGG miRNA target analysis in
Table 6 indicated that miR-26 families were enriched in the high-risk subtype. Based on the TargetScanHuman analysis, miR-26 is one of four miRNAs that was predicted to bind to the
ERO1B transcript and suppress gene expression. As we mentioned in
Section 3.4, KEGG miRNA analysis revealed that miR-501, miR-26, miR-507, miR-33, and miR-200/miR-429 were involved in lung-cancer subtypes. The miRNAs we identified have been previously reported as regulating Wnt/β-catenin signaling and/or contributing EMT signaling. Taken together, not only KEGG analysis, but also co-expression analysis, gave us insight into the molecular mechanisms that underlie patient prognosis.
A limitation of this study is the difficulty with preparing the validation dataset. The SVM model we developed uses 20 miRNA and 25 RNA expressions. Thus, we need a validation dataset that includes miRNA expression, RNA expression, and clinical information. There are datasets available that include miRNA expression (GSE63805) and RNA expression (GSE63459), together with clinical information. However, some of the miRNA expression and RNA expression for the top 20 miRNA and top 25 RNAs used to develop the SVM model were missing, and therefore we were not able to evaluate the SVM with the abovementioned publicly available dataset. This constitutes a technical limitation of the study, since it makes it difficult to assess the robustness of the developed classifier. Therefore, we decided to use the TCGA dataset again, for the validation. All data (364 patients) were randomly split into 75% and 25%, and the 25% of patient data were used for validation. Result of the accuracy score of the developed SVM model was 0.92.
The second limitation of this study is the fact the frequency of certain gene mutations can vary depending on the patients’ race. For example, EGFR mutation is more often found in Asian American patients than Caucasian or African American patients [
74]. Therefore, the SVM model we developed may not be able to distinguish a high-risk subtype from a low-risk subtype if the model is applied to a different distributed dataset such as on containing an Asian population. In that case, the SVM model will need to be redeveloped.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
