GSK3α: An Important Paralog in Neurodegenerative Disorders and Cancer

Abstract
:1. Introduction
2. The Relevance of GSK3α
3. General Aspects of GSK3α in Neurodegenerative Disorders and Cancer
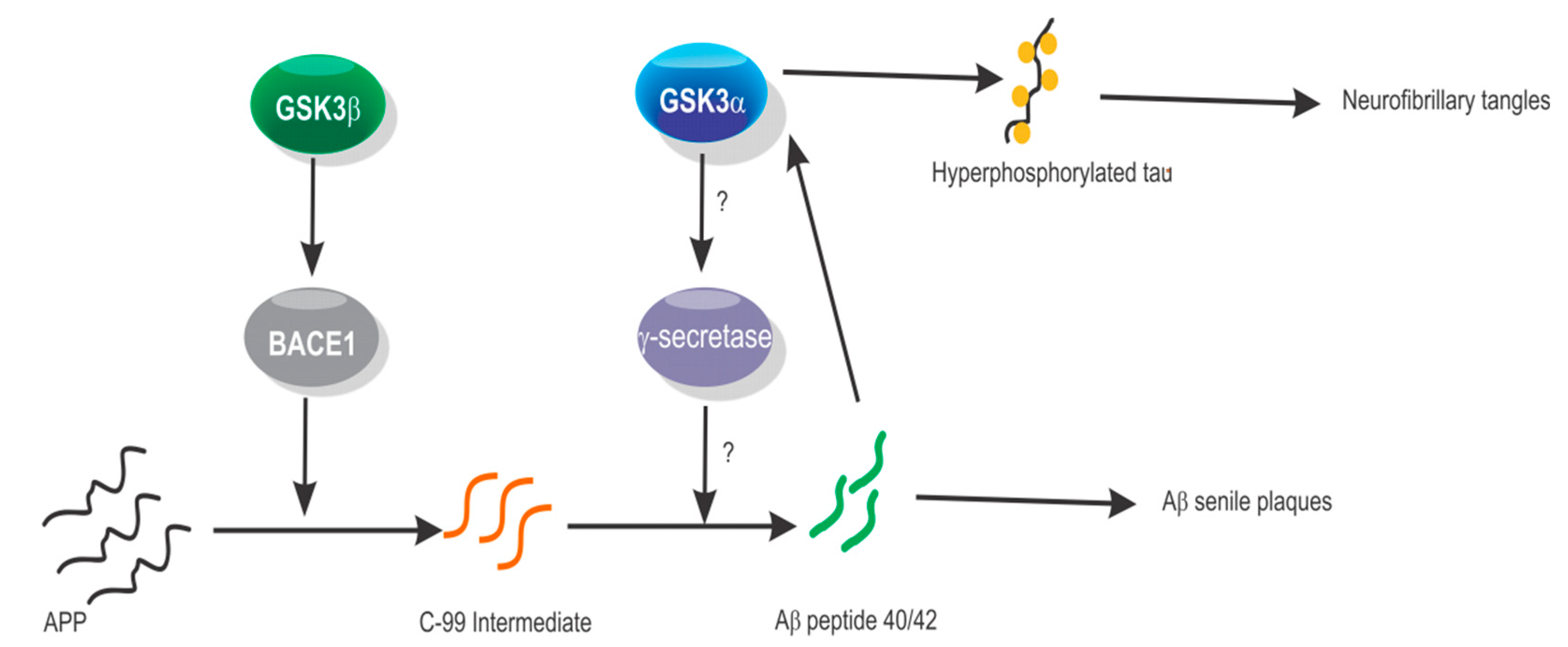
4. GSK3α in Alzheimer’s Disease and Neurodegenerative Disorders
5. GSK3 in Cancer
5.1. GSK3α in Brain Cancer
5.2. GSK3α in Pancreatic Ductal Adenocarcinoma
5.3. GSK3α in Prostate Cancer
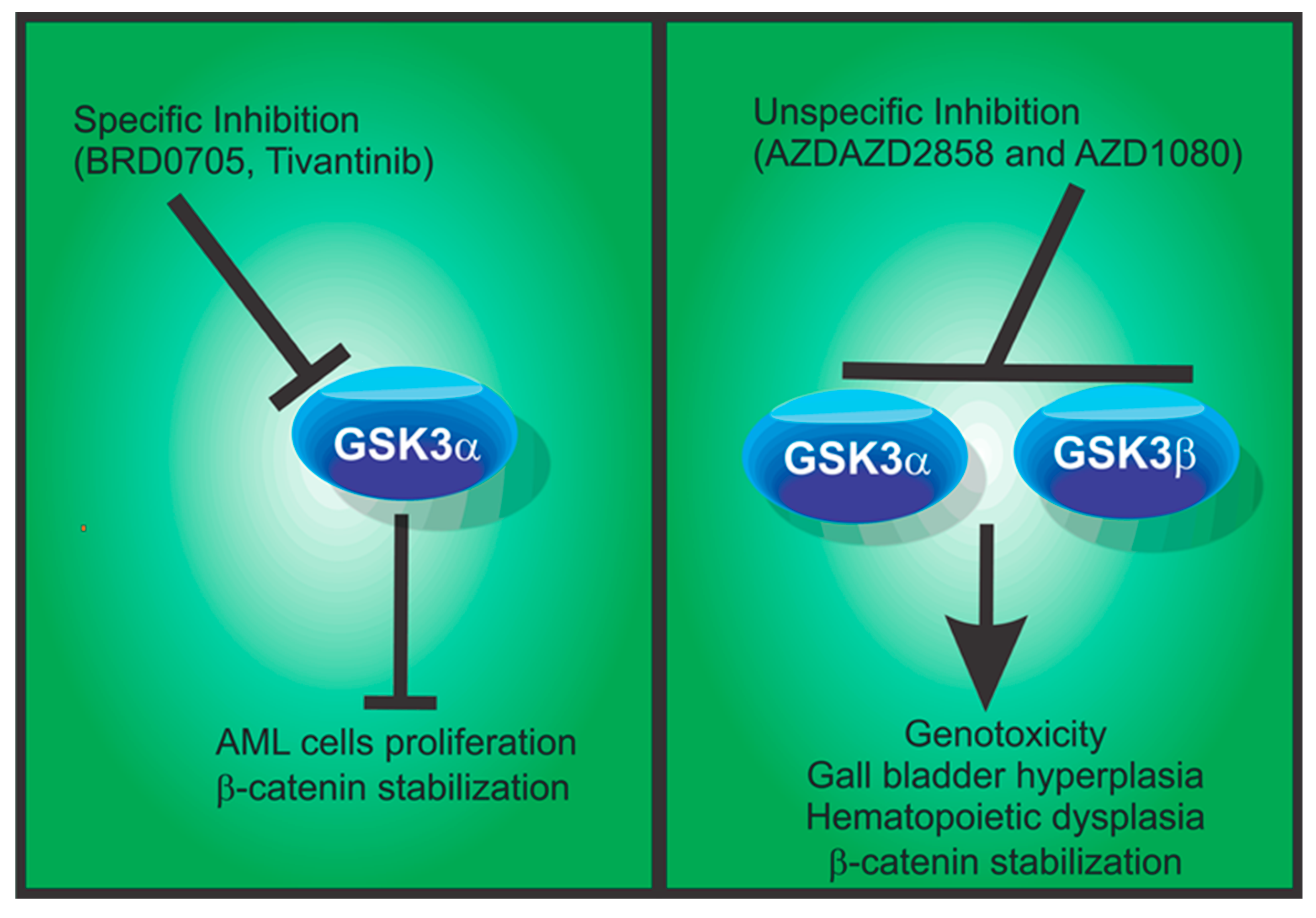
5.4. GSK3α in Acute Myeloid Leukemia
5.5. GSK3α in Multiple Myeloma
5.6. GSK3α in Lung Cancer

{kind=link}
{kind=link}
{kind=link}
| Cancer Type | GSK3α Biological Effect | References |
|---|---|---|
| Hepatocellular carcinoma | Overactive | [37] |
| Cervical carcinoma | Overactive | [39] |
| Thyroid tumor cells | Overactive | [40] |
| Prostate carcinoma | (1) Overactive (2) Associated with the androgen receptor transcriptional activity (3) KD represses proliferation and Ki67 expression | [41,82,83] |
| Acute myeloid leukemia | (1) Overactive (2) KD or chemical inhibition induces cell differentiation | [29,42] |
| Glioblastoma | Its activity represses cell proliferation | [73] |
| Neuroblastoma | Chemical inhibition with AR-A014418 downregulates p-Tyr-279 and induces apoptosis | [77] |
| Pancreatic ductal adenocarcinoma | Its inhibition represses NF-κB activity and induces apoptosis via Kras mutations | [78,79] |
| Multiple myeloma | (1) Levels of expression higher than GSK3β (2) KD sensitizes cells to cytotoxic effects triggered by Bortezomib | [92] |
| Lung cancer | (1) Its overexpression is a marker of a poor prognosis (2) Promotes the expression of cyclins A2, B1, D1, and E2 | [93] |
6. Targeting a Single GSK3 for Therapy
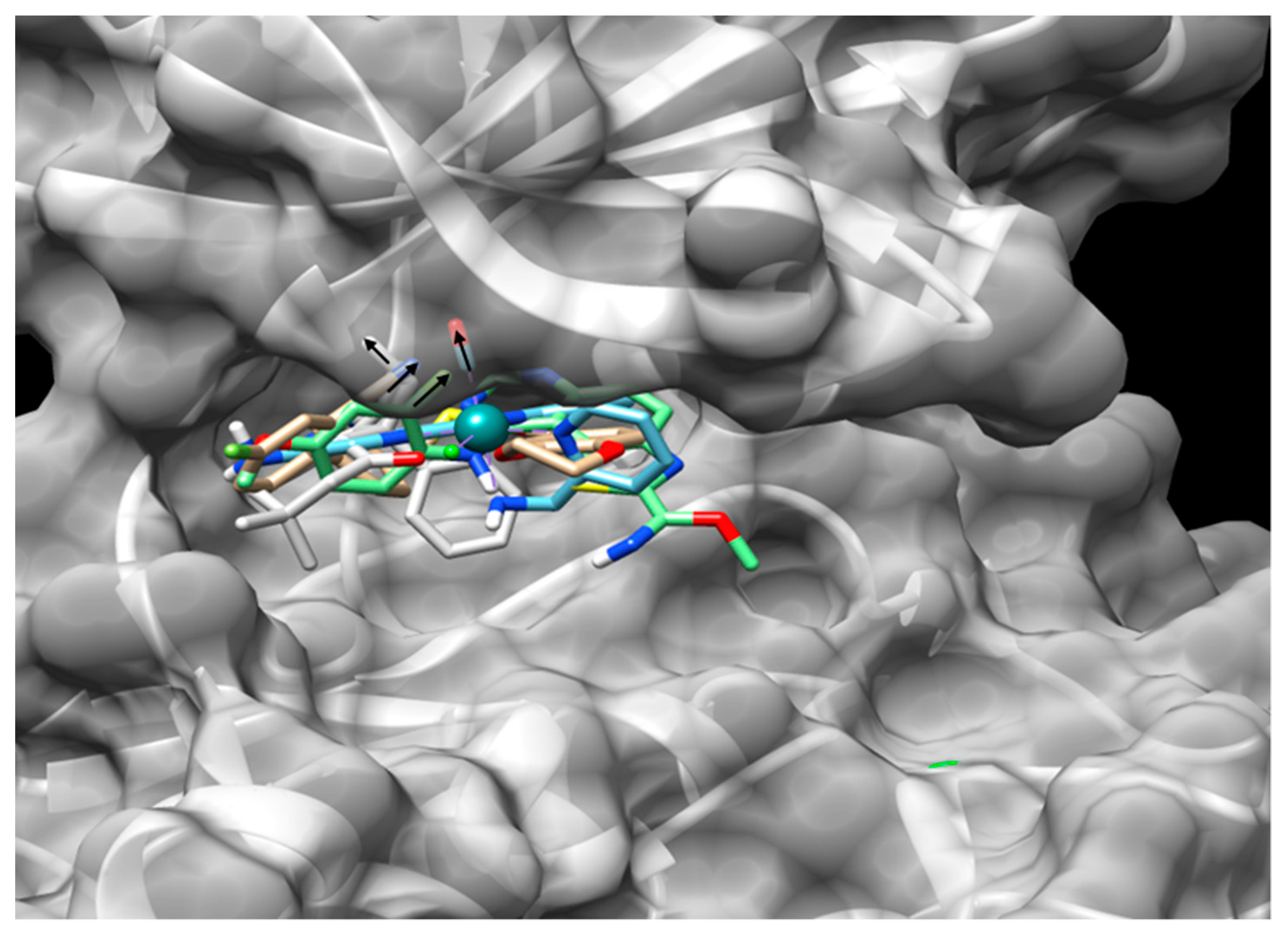
6.1. The Substrate Binding Domain
6.2. The Glycine-Rich Loop Domain
6.3. The Hinge Domain
6.4. Non-Conventional Pockets as Targets for Inhibitors
6.5. GSK3α Selective Inhibitors
| Molecule | GSK3α Inhibition, IC50 (nM) | GSK3β Inhibition, IC50 (nM) | Reported Biological Effect | References |
|---|---|---|---|---|
| BRD0705 | 66 | 515 | Induces cell differentiation of AML cell lines and impairs colony formation of AML patient cells | [29] |
| EHT5372 | 7.44 | 221 | Reduces phosphorylation of tau and Aβ production on recombinant expressing HEK293 cells | [97] |
| AZD2858 | 0.9 | 5 | Reduces tau hyperphosphorylation in rat brains, and induces differentiation of mesenchymal progenitors to osteoblasts | [112] [113] |
| EHT1610 | 9.11 | 143 | Reduces phosphorylation of tau and Aβ production on recombinant expressing HEK293 cells | [97] |
| 8g | 35 | 966 | Unknown | [111] |
| 8a | 4 | 90 | Unknown | [111] |
| 8b | 9 | 225 | Unknown | [111] |
| 27 | 42 | 140 | Induces differentiation and impairs colony formation of AML, HL-60, and NB4 cells | [89] |
| 26d | 2 | 17 | Unknown | [89] |
| 14a | 9 | 176 | Unknown | [104] |
| 15b | 2 | 185 | Unknown | [104] |
| 14d | 6 | 316 | Unknown | [104] |
| G28_14 | 33 | 218 | Induces differentiation and impairs colony formation of AML, HL-60, and NB4 cells | [114] |
| Λ-OS1 | 0.9 | 6 | Unknown | [96] |
| Tivantinib | 659 | 1865 | Apoptosis of AML cells | [91] |
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Hemmings, B.A.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Methods Enzymol. 1983, 99, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jørgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimer’s Dis. 2011, 2011, 505607. [Google Scholar] [CrossRef] [Green Version]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Yellowlees, D.; Kernohan, J.C.; Cohen, P. Purification of glycogen synthase kinase 3 from rabbit skeletal muscle. Copurification with the activating factor (FA) of the (Mg-ATP) dependent protein phosphatase. Eur. J. Biochem. 1981, 119, 443–451. [Google Scholar] [CrossRef]
- Guan, R.J.; Khatra, B.S.; Cohlberg, J.A. Phosphorylation of bovine neurofilament proteins by protein kinase FA (glycogen synthase kinase 3). J. Biol. Chem. 1991, 266, 8262–8267. [Google Scholar]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Ruel, L.; Bourouis, M.; Heitzler, P.; Pantesco, V.; Simpson, P. Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature 1993, 362, 557–560. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Force, T.; Woodgett, J.R. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J. Biol. Chem. 2009, 284, 9643–9647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Pandey, M.K.; DeGrado, T.R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Vieyra, R.; Silva-García, O.; Oviedo-Boyso, J.; Huante-Mendoza, A.; Bravo-Patiño, A.; Valdez-Alarcón, J.J.; Finlay, B.B.; Baizabal-Aguirre, V.M. The Glycogen Synthase Kinase 3α and β Isoforms Differentially Regulates Interleukin-12p40 Expression in Endothelial Cells Stimulated with Peptidoglycan from Staphylococcus aureus. PLoS ONE 2015, 10, e0132867. [Google Scholar] [CrossRef] [Green Version]
- Silva-García, O.; Rico-Mata, R.; Maldonado-Pichardo, M.C.; Bravo-Patiño, A.; Valdez-Alarcón, J.J.; Aguirre-González, J.; Baizabal-Aguirre, V.M. Glycogen Synthase Kinase 3α Is the Main Isoform That Regulates the Transcription Factors Nuclear Factor-Kappa B and cAMP Response Element Binding in Bovine Endothelial Cells Infected with Staphylococcus aureus. Front. Immunol. 2018, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Oviedo-Boyso, J.; Cortés-Vieyra, R.; Huante-Mendoza, A.; Yu, H.B.; Valdez-Alarcón, J.J.; Bravo-Patiño, A.; Cajero-Juárez, M.; Finlay, B.B.; Baizabal-Aguirre, V.M. The phosphoinositide-3-kinase-Akt signaling pathway is important for Staphylococcus aureus internalization by endothelial cells. Infect. Immun. 2011, 79, 4569–4577. [Google Scholar] [CrossRef] [Green Version]
- McAlpine, C.S.; Huang, A.; Emdin, A.; Banko, N.S.; Beriault, D.R.; Shi, Y.; Werstuck, G.H. Deletion of Myeloid GSK3α Attenuates Atherosclerosis and Promotes an M2 Macrophage Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Doble, B.W.; MacAulay, K.; Sinclair, E.M.; Drucker, D.J.; Woodgett, J.R. Tissue-specific role of glycogen synthase kinase 3 beta in glucose homeostasis and insulin action. Mol. Cell. Biol. 2008, 28, 6314–6328. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Freeman, T.A.; Ahmad, F.; Shang, X.; Mangano, E.; Gao, E.; Farber, J.; Wang, Y.; Ma, X.L.; Woodgett, J.; et al. GSK-3α is a central regulator of age-related pathologies in mice. J. Clin. Investig. 2013, 123, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Gao, S.; Holle, E.; Yu, X.; Yatani, A.; Wagner, T.; Sadoshima, J. Glycogen synthase kinase-3alpha reduces cardiac growth and pressure overload-induced cardiac hypertrophy by inhibition of extracellular signal-regulated kinases. J. Biol. Chem. 2007, 282, 33181–33191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maejima, Y.; Galeotti, J.; Molkentin, J.D.; Sadoshima, J.; Zhai, P. Constitutively active MEK1 rescues cardiac dysfunction caused by overexpressed GSK-3α during aging and hemodynamic pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H979–H988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, R.; Goswami, S.; Dudiki, T.; Popkie, A.P.; Phiel, C.J.; Kline, D.; Vijayaraghavan, S. Targeted disruption of glycogen synthase kinase 3A (GSK3A) in mice affects sperm motility resulting in male infertility. Biol. Reprod. 2015, 92, 65. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Xiong, Z.; Dai, H.; Zou, Z.; Jia, C.; Bai, X.; Chen, Z. mTORC1 Activation Promotes Spermatogonial Differentiation and Causes Subfertility in Mice. Biol. Reprod. 2016, 95, 97. [Google Scholar] [CrossRef]
- Hurcombe, J.A.; Lay, A.C.; Ni, L.; Barrington, A.F.; Woodgett, J.R.; Quaggin, S.E.; Welsh, G.I.; Coward, R.J. Podocyte GSK3α is important for autophagy and its loss detrimental for glomerular function. FASEB BioAdv. 2019, 1, 498–510. [Google Scholar] [CrossRef]
- Liang, M.H.; Chuang, D.M. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J. Biol. Chem. 2006, 281, 30479–30484. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, R.; Liu, X.; Wu, Y.; Zhou, T.; Yang, Y.; Perez, A.; Chen, Y.C.; Hu, L.; Chadarevian, J.P.; et al. A Chemical-Genetic Approach Reveals the Distinct Roles of GSK3α and GSK3β in Regulating Embryonic Stem Cell Fate. Dev. Cell 2017, 43, 563–576.e564. [Google Scholar] [CrossRef] [Green Version]
- Wagner, F.F.; Benajiba, L.; Campbell, A.J.; Weïwer, M.; Sacher, J.R.; Gale, J.P.; Ross, L.; Puissant, A.; Alexe, G.; Conway, A.; et al. Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci. Transl. Med. 2018, 10, eaam8460. [Google Scholar] [CrossRef] [Green Version]
- Giambelluca, M.S.; Bertheau-Mailhot, G.; Laflamme, C.; Rollet-Labelle, E.; Servant, M.J.; Pouliot, M. TNF-α expression in neutrophils and its regulation by glycogen synthase kinase-3: A potentiating role for lithium. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 3679–3690. [Google Scholar] [CrossRef]
- Gulen, M.F.; Bulek, K.; Xiao, H.; Yu, M.; Gao, J.; Sun, L.; Beurel, E.; Kaidanovich-Beilin, O.; Fox, P.L.; DiCorleto, P.E.; et al. Inactivation of the enzyme GSK3α by the kinase IKKi promotes AKT-mTOR signaling pathway that mediates interleukin-1-induced Th17 cell maintenance. Immunity 2012, 37, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Dunning, C.J.; McGauran, G.; Willén, K.; Gouras, G.K.; O’Connell, D.J.; Linse, S. Direct High Affinity Interaction between Aβ42 and GSK3α Stimulates Hyperphosphorylation of Tau. A New Molecular Link in Alzheimer’s Disease? ACS Chem. Neurosci. 2016, 7, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alon, L.T.; Pietrokovski, S.; Barkan, S.; Avrahami, L.; Kaidanovich-Beilin, O.; Woodgett, J.R.; Barnea, A.; Eldar-Finkelman, H. Selective loss of glycogen synthase kinase-3α in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 2011, 585, 1158–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, D.E.; Molina-Porcel, L.; Carroll, J.C.; Macdonald, C.; Aboagye, A.K.; Trojanowski, J.Q.; Lee, V.M. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 7392–7402. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.D.; Yu, J.S.; Yang, C.C.; Lee, S.C.; Lee, T.T.; Ni, M.H.; Kuan, C.Y.; Chen, H.C. Overexpression of protein kinase FA/GSK-3 alpha (a proline-directed protein kinase) correlates with human hepatoma dedifferentiation/progression. J. Cell. Biochem. 1996, 61, 238–245. [Google Scholar] [CrossRef]
- Huang, K.T.; Huang, Y.H.; Li, P.; He, B.; Chen, Z.K.; Yu, X.; Chen, J.O.; Zhang, Q.Y.; Shi, H.Q.; Shan, Y.F. Correlation between tuberous sclerosis complex 2 and glycogen synthase kinase 3 beta levels, and outcomes of patients with hepatocellular carcinoma treated by hepatectomy. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2014, 44, 1142–1150. [Google Scholar] [CrossRef]
- Yang, S.D.; Yu, J.S.; Lee, T.T.; Ni, M.H.; Yang, C.C.; Ho, Y.S.; Tsen, T.Z. Association of protein kinase FA/GSK-3alpha (a proline-directed kinase and a regulator of protooncogenes) with human cervical carcinoma dedifferentiation/progression. J. Cell. Biochem. 1995, 59, 143–150. [Google Scholar] [CrossRef]
- Lee, T.T.; Ho, Y.S.; Yu, J.S.; Yang, S.D. Overexpression of cellular activity and protein level of protein kinase FA/GSK-3 alpha correlates with human thyroid tumor cell dedifferentiation. J. Cell. Biochem. 1995, 58, 474–480. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsu, C.P.; Sheu, J.C.; Mai, X.Y.; Yang, S.D. Differential tyrosine phosphorylation/activation of oncogenic proline-directed protein kinase F(A)/GSK-3alpha in well and poorly differentiated human prostate carcinoma cells. J. Protein Chem. 1998, 17, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Hsueh, S.F.; Yang, C.C.; Yang, S.D. Suppression of proline-directed protein kinase F(A) expression inhibits the growth of human chronic myeloid leukaemia cells. Br. J. Cancer 2000, 82, 1480–1484. [Google Scholar] [CrossRef] [Green Version]
- Banerji, V.; Frumm, S.M.; Ross, K.N.; Li, L.S.; Schinzel, A.C.; Hahn, C.K.; Kakoza, R.M.; Chow, K.T.; Ross, L.; Alexe, G.; et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Investig. 2012, 122, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3α promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-κB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W., 3rd; Baldwin, A.S. Maintenance of Constitutive IκB Kinase Activity by Glycogen Synthase Kinase-3α/β in Pancreatic Cancer. Cancer Res. 2008, 68, 8156–8163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassilli, E.; Ianzano, L.; Bonomo, S.; Missaglia, C.; Cerrito, M.G.; Giovannoni, R.; Masiero, L.; Lavitrano, M. GSK3A is redundant with GSK3B in modulating drug resistance and chemotherapy-induced necroptosis. PLoS ONE 2014, 9, e100947. [Google Scholar] [CrossRef]
- Behrens, M.I.; Lendon, C.; Roe, C.M. A common biological mechanism in cancer and Alzheimer’s disease? Curr. Alzheimer Res. 2009, 2009 6, 196–204. [Google Scholar] [CrossRef]
- Papageorgakopoulos, T.N.; Moraitou, D.; Papanikolaou, M.; Tsolaki, M. The association between Alzheimer’s disease and cancer: Systematic review-Meta-analysis. Hell. J. Nucl. Med. 2017, 20, 45–57. [Google Scholar]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Takashima, A.; Noguchi, K.; Sato, K.; Hoshino, T.; Imahori, K. Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 1993, 90, 7789–7793. [Google Scholar] [CrossRef] [Green Version]
- Feyt, C.; Kienlen-Campard, P.; Leroy, K.; N’Kuli, F.; Courtoy, P.J.; Brion, J.P.; Octave, J.N. Lithium chloride increases the production of amyloid-beta peptide independently from its inhibition of glycogen synthase kinase 3. J. Biol. Chem. 2005, 280, 33220–33227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Barrachina, M.; Puig, B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002, 104, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, T.; Dewachter, I.; Lechat, B.; Gees, M.; Kremer, A.; Demedts, D.; Borghgraef, P.; Devijver, H.; Kügler, S.; Patel, S.; et al. GSK-3α/β kinases and amyloid production in vivo. Nature 2011, 480, E4–E5. [Google Scholar] [CrossRef]
- Rao, C.V.; Farooqui, M.; Madhavaram, A.; Zhang, Y.; Asch, A.S.; Yamada, H.Y. GSK3-ARC/Arg3. 1 and GSK3-Wnt signaling axes trigger amyloid-β accumulation and neuroinflammation in middle-aged Shugoshin 1 mice. Aging Cell 2020, 19, e13221. [Google Scholar] [CrossRef]
- Lal, H.; Zhou, J.; Ahmad, F.; Zaka, R.; Vagnozzi, R.J.; DeCaul, M.; Woodgett, J.; Gao, E.; Force, T. Glycogen Synthase Kinase-3α Limits Ischemic Injury, Cardiac Rupture, Post–Myocardial Infarction Remodeling and Death. Circulation 2012, 125, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Lal, H.; Zhou, J.; Vagnozzi, R.J.; Yu, J.E.; Shang, X.; Woodgett, J.R.; Gao, E.; Force, T. Cardiomyocyte-Specific Deletion of Gsk3α Mitigates Post–Myocardial Infarction Remodeling, Contractile Dysfunction, and Heart Failure. J. Am. Collge Cardiol. 2014, 64, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Maurin, H.; Lechat, B.; Dewachter, I.; Ris, L.; Louis, J.V.; Borghgraef, P.; Devijver, H.; Jaworski, T.; Van Leuven, F. Neurological characterization of mice deficient in GSK3α highlight pleiotropic physiological functions in cognition and pathological activity as Tau kinase. Mol. Brain 2013, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Lynn, C.W.; Bassett, D.S. The physics of brain network structure, function and control. Nat. Rev. Phys. 2019, 1, 318–332. [Google Scholar] [CrossRef] [Green Version]
- Garrido, J.J.; Simón, D.; Varea, O.; Wandosell, F. GSK3 alpha and GSK3 beta are necessary for axon formation. FEBS Lett. 2007, 581, 1579–1586. [Google Scholar] [CrossRef]
- Peineau, S.; Nicolas, C.S.; Bortolotto, Z.A.; Bhat, R.V.; Ryves, W.J.; Harwood, A.J.; Dournaud, P.; Fitzjohn, S.M.; Collingridge, G.L. A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: Evidence for a role of GSK-3 but not other serine/threonine kinases. Mol. Brain 2009, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shahab, L.; Plattner, F.; Irvine, E.E.; Cummings, D.M.; Edwards, F.A. Dynamic range of GSK3α not GSK3β is essential for bidirectional synaptic plasticity at hippocampal CA3-CA1 synapses. Hippocampus 2014, 24, 1413–1416. [Google Scholar] [CrossRef] [Green Version]
- Cymerman, I.A.; Gozdz, A.; Urbanska, M.; Milek, J.; Dziembowska, M.; Jaworski, J. Structural plasticity of dendritic spines requires GSK3α and GSK3β. PLoS ONE 2015, 10, e0134018. [Google Scholar] [CrossRef]
- Draffin, J.E.; Sánchez-Castillo, C.; Fernández-Rodrigo, A.; Sánchez-Sáez, X.; Ávila, J.; Wagner, F.F.; Esteban, J.A. GSK3α, not GSK3β, drives hippocampal NMDAR-dependent LTD via tau-mediated spine anchoring. EMBO J. 2020, e105513. [Google Scholar] [CrossRef]
- Maqbool, M.; Hoda, N. GSK3 Inhibitors in the Therapeutic Development of Diabetes, Cancer and Neurodegeneration: Past, Present and Future. Curr. Pharm. Des. 2017, 23, 4332–4350. [Google Scholar] [CrossRef]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Acikgoz, E.; Güler, G.; Camlar, M.; Oktem, G.; Aktug, H. Glycogen synthase kinase-3 inhibition in glioblastoma multiforme cells induces apoptosis, cell cycle arrest and changing biomolecular structure. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 209, 150–164. [Google Scholar] [CrossRef]
- Vashishta, V.; Jinghan, N.; Yadav, A.K. Antagonistic role of GSK3 isoforms in glioma survival. J. Cancer 2018, 9, 1846–1855. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Alfaguter, I.; Yaffe, Y.; Licht-Murava, A.; Urbanska, M.; Jaworski, J.; Pietrokovski, S.; Hirschberg, K.; Eldar-Finkelman, H. Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: Functional role in calcium/calpain signaling. J. Biol. Chem. 2011, 286, 13470–13480. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Li, W.; Zhang, M. The function of the RNA-binding protein hnRNP in cancer metastasis. J. Cancer Res. Ther. 2013, 9, S129–S134. [Google Scholar] [CrossRef]
- Zhou, J.; Lal, H.; Chen, X.; Shang, X.; Song, J.; Li, Y.; Kerkela, R.; Doble, B.W.; MacAulay, K.; DeCaul, M.; et al. GSK-3alpha directly regulates beta-adrenergic signaling and the response of the heart to hemodynamic stress in mice. J. Clin. Investig. 2010, 120, 2280–2291. [Google Scholar] [CrossRef] [Green Version]
- Carter, Y.M.; Kunnimalaiyaan, S.; Chen, H.; Gamblin, T.C.; Kunnimalaiyaan, M. Specific glycogen synthase kinase-3 inhibition reduces neuroendocrine markers and suppresses neuroblastoma cell growth. Cancer Biol. Ther. 2014, 15, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.S.; Herreros-Villanueva, M.; Koeing, A.; Deng, Z.; Narvajas, A.A.; Gomez, T.S.; Meng, X.; Bujanda, L.; Ellenrieder, V.; Li, X.K.; et al. Differential activity of GSK-3 isoforms regulates NF-κB and TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell Death Dis. 2014, 5, e1142. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Darrington, R.S.; Campa, V.M.; Walker, M.M.; Bengoa-Vergniory, N.; Gorrono-Etxebarria, I.; Uysal-Onganer, P.; Kawano, Y.; Waxman, J.; Kypta, R.M. Distinct expression and activity of GSK-3α and GSK-3β in prostate cancer. Int. J. Cancer 2012, 131, E872–E883. [Google Scholar] [CrossRef]
- Li, B.; Thrasher, J.B.; Terranova, P. Glycogen synthase kinase-3: A potential preventive target for prostate cancer management. Urol. Oncol. 2015, 33, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Al-Azayzih, A.; Somanath, P.R. Discrete functions of GSK3α and GSK3β isoforms in prostate tumor growth and micrometastasis. Oncotarget 2015, 6, 5947–5962. [Google Scholar] [CrossRef] [PubMed]
- Campa, V.M.; Baltziskueta, E.; Bengoa-Vergniory, N.; Gorroño-Etxebarria, I.; Wesołowski, R.; Waxman, J.; Kypta, R.M. A screen for transcription factor targets of glycogen synthase kinase-3 highlights an inverse correlation of NFκB and androgen receptor signaling in prostate cancer. Oncotarget 2014, 5, 8173–8187. [Google Scholar] [CrossRef] [PubMed]
- George, J.N. Interferon-induced thrombotic microangiopathy. Blood 2016, 128, 2753–2754. [Google Scholar] [CrossRef] [Green Version]
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell 2016, 29, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, R.; Ramakrishnan, P.; Moreton, S.A.; Xia, Z.; Hou, Y.; Lee, D.A.; Gupta, K.; deLima, M.; Beck, R.C.; Wald, D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016, 7, 11154. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; Maestro, R.; et al. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2014, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Ueda, M.; Stetson, L.; Ignatz-Hoover, J.; Moreton, S.; Chakrabarti, A.; Xia, Z.; Karan, G.; Lima, M.; Agrawal, M.K.; et al. A novel glycogen synthase kinase-3 inhibitor optimized for acute myeloid leukemia differentiation activity. Mol. Cancer Ther. 2016, 7, 1485–1494. [Google Scholar] [CrossRef] [Green Version]
- Neumann, T.; Benajiba, L.; Göring, S.; Stegmaier, K.; Schmidt, B. Evaluation of Improved Glycogen Synthase Kinase-3α Inhibitors in Models of Acute Myeloid Leukemia. J. Med. Chem. 2015, 58, 8907–8919. [Google Scholar] [CrossRef] [Green Version]
- Bhat, R.V.; Andersson, U.; Andersson, S.; Knerr, L.; Bauer, U.; Sundgren-Andersson, A.K. The Conundrum of GSK3 Inhibitors: Is it the Dawn of a New Beginning? J. Alzheimer’s Dis. 2018, 64, S547–S554. [Google Scholar] [CrossRef]
- Kuenzi, B.M.; Rix, L.L.R.; Kinose, F.; Kroeger, J.L.; Lancet, J.E.; Padron, E.; Rix, U. Off-target based drug repurposing opportunities for tivantinib in acute myeloid leukemia. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Piazza, F.; Manni, S.; Tubi, L.Q.; Montini, B.; Pavan, L.; Colpo, A.; Gnoato, M.; Cabrelle, A.; Adami, F.; Zambello, R.; et al. Glycogen Synthase Kinase-3 regulates multiple myeloma cell growth and bortezomib-induced cell death. BMC Cancer 2010, 10, 526. [Google Scholar] [CrossRef]
- Park, S.A.; Lee, J.W.; Herbst, R.S.; Koo, J.S. GSK-3α Is a Novel Target of CREB and CREB-GSK-3α Signaling Participates in Cell Viability in Lung Cancer. PLoS ONE 2016, 11, e0153075. [Google Scholar] [CrossRef] [Green Version]
- Thanacoody, H.K.R. Chronic valproic acid intoxication: Reversal by naloxone. Bmj Case Rep. 2007, 24, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.; Brooks, T.W.A.; Yam, P.; Kelley, K. Management and treatment of lithium-induced nephrogenic diabetes insipidus. Therapy 2005, 2, 669–675. [Google Scholar] [CrossRef]
- Feng, L.; Geisselbrecht, Y.; Blanck, S.; Wilbuer, A.; Atilla-Gokcumen, G.E.; Filippakopoulos, P.; Kräling, K.; Celik, M.A.; Harms, K.; Maksimoska, J.; et al. Structurally sophisticated octahedral metal complexes as highly selective protein kinase inhibitors. J. Am. Chem. Soc. 2011, 133, 5976–5986. [Google Scholar] [CrossRef] [Green Version]
- Chaikuad, A.; Diharce, J.; Schröder, M.; Foucourt, A.; Leblond, B.; Casagrande, A.S.; Désiré, L.; Bonnet, P.; Knapp, S.; Besson, T. An Unusual Binding Model of the Methyl 9-Anilinothiazolo[5,4-f] quinazoline-2-carbimidates (EHT 1610 and EHT 5372) Confers High Selectivity for Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases. J. Med. Chem. 2016, 59, 10315–10321. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T.; Di Daniel, E.; Culbert, A.A.; Reith, A.D. Expression and characterization of GSK-3 mutants and their effect on beta-catenin phosphorylation in intact cells. J. Biol. Chem. 2002, 277, 23330–23335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frame, S.; Cohen, P.; Biondi, R.M. A Common Phosphate Binding Site Explains the Unique Substrate Specificity of GSK3 and Its Inactivation by Phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Buescher, J.L.; Phiel, C.J. A noncatalytic domain of glycogen synthase kinase-3 (GSK-3) is essential for activity. J. Biol. Chem. 2010, 285, 7957–7963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamann, M.; Alonso, D.; Martín-Aparicio, E.; Fuertes, A.; Pérez-Puerto, M.J.; Castro, A.; Morales, S.; Navarro, M.L.; del Monte-Millán, M.; Medina, M.; et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure-activity relationship (SAR) studies of the manzamine alkaloids. Potential for Alzheimer’s disease. J. Nat. Prod. 2007, 70, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Bossemeyer, D. The glycine-rich sequence of protein kinases: A multifunctional element. Trends Biochem. Sci. 1994, 19, 201–205. [Google Scholar] [CrossRef]
- Atilla-Gokcumen, G.E.; Pagano, N.; Streu, C.; Maksimoska, J.; Filippakopoulos, P.; Knapp, S.; Meggers, E. Extremely tight binding of a ruthenium complex to glycogen synthase kinase 3. ChemBioChem A Eur. J. Chem. Biol. 2008, 9, 2933–2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Monte, F.; Kramer, T.; Gu, J.; Anumala, U.R.; Marinelli, L.; La Pietra, V.; Novellino, E.; Franco, B.; Demedts, D.; Van Leuven, F.; et al. Identification of glycogen synthase kinase-3 inhibitors with a selective sting for glycogen synthase kinase-3α. J. Med. Chem. 2012, 55, 4407–4424. [Google Scholar] [CrossRef] [PubMed]
- Fraser, E.; Young, N.; Dajani, R.; Franca-Koh, J.; Ryves, J.; Williams, R.S.; Yeo, M.; Webster, M.T.; Richardson, C.; Smalley, M.J.; et al. Identification of the Axin and Frat binding region of glycogen synthase kinase-3. J. Biol. Chem. 2002, 277, 2176–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.Q.; Korhonen, P.K.; Cai, H.; Young, N.D.; Nejsum, P.; von Samson-Himmelstjerna, G.; Boag, P.R.; Tan, P.; Li, Q.; Min, J.; et al. Genetic blueprint of the zoonotic pathogen Toxocara canis. Nat. Commun. 2015, 6, 6145. [Google Scholar] [CrossRef] [Green Version]
- Harwood, A.J.; Plyte, S.E.; Woodgett, J.; Strutt, H.; Kay, R.R. Glycogen synthase kinase 3 regulates cell fate in Dictyostelium. Cell 1995, 80, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Zapata, L.; Ding, J.; Willing, E.M.; Hartwig, B.; Bezdan, D.; Jiao, W.B.; Patel, V.; Velikkakam James, G.; Koornneef, M.; Ossowski, S.; et al. Chromosome-level assembly of Arabidopsis thaliana Ler reveals the extent of translocation and inversion polymorphisms. Proc. Natl. Acad. Sci. USA 2016, 113, E4052–E4060. [Google Scholar] [CrossRef] [Green Version]
- Palomo, V.; Soteras, I.; Perez, D.I.; Perez, C.; Gil, C.; Campillo, N.E.; Martinez, A. Exploring the binding sites of glycogen synthase kinase 3. Identification and characterization of allosteric modulation cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef] [Green Version]
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D.I.; Martínez, A.; Luque, F.J.; Medina, M. Evidence for a new binding mode to GSK-3: Allosteric regulation by the marine compound palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef]
- Lo Monte, F.; Kramer, T.; Gu, J.; Brodrecht, M.; Pilakowski, J.; Fuertes, A.; Dominguez, J.M.; Plotkin, B.; Eldar-Finkelman, H.; Schmidt, B. Structure-based optimization of oxadiazole-based GSK-3 inhibitors. Eur. J. Med. Chem. 2013, 61, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, A.; Nagaraju, C.K.; O’Shea, P.J.; Mohanty, S.T.; Kottam, L.; Pilling, J.; Sullivan, M.; Djerbi, M.; Koopmann, W.; Croucher, P.I.; et al. Glycogen synthase kinase-3α/β inhibition promotes in vivo amplification of endogenous mesenchymal progenitors with osteogenic and adipogenic potential and their differentiation to the osteogenic lineage. J. Bone Min. Res. 2011, 26, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.; Bergh, M.; Hellberg, S.; Högdin, K.; Lo-Alfredsson, Y.; Söderman, P.; Berg, S.V.; Weigelt, T.; Ormo, M.; Xue, Y.; et al. Discovery of novel potent and highly selective glycogen synthase kinase-3β (GSK3β) inhibitors for Alzheimer’s disease: Design, synthesis, and characterization of pyrazines. J. Med. Chem. 2012, 55, 9107–9119. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dou, X.; Jiang, L.; Jin, H.; Zhang, L.; Zhang, L.; Liu, Z. Discovery of novel glycogen synthase kinase-3α inhibitors: Structure-based virtual screening, preliminary SAR and biological evaluation for treatment of acute myeloid leukemia. Eur. J. Med. Chem. 2019, 171, 221–234. [Google Scholar] [CrossRef]
- Zhou, J.; Ahmad, F.; Parikh, S.; Hoffman, N.E.; Rajan, S.; Verma, V.K.; Song, J.; Yuan, A.; Shanmughapriya, S.; Guo, Y.; et al. Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy. Circ. Res. 2016, 118, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Huisamen, B.; Hafver, T.L.; Lumkwana, D.; Lochner, A. The Impact of Chronic Glycogen Synthase Kinase-3 Inhibition on Remodeling of Normal and Pre-Diabetic Rat Hearts. Cardiovasc. Drugs Ther. 2016, 30, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Flepisi, T.B.; Lochner, A.; Huisamen, B. The consequences of long-term glycogen synthase kinase-3 inhibition on normal and insulin resistant rat hearts. Cardiovasc. Drugs Ther. 2013, 27, 381–392. [Google Scholar] [CrossRef]
- Kühn, R.; Schwenk, F. Conditional Knockout Mice. In Transgenic Mouse: Methods and Protocols; Hofker, M.H., van Deursen, J., Eds.; Humana Press: Totowa, NJ, USA, 2002; pp. 159–185. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-García, O.; Cortés-Vieyra, R.; Mendoza-Ambrosio, F.N.; Ramírez-Galicia, G.; Baizabal-Aguirre, V.M. GSK3α: An Important Paralog in Neurodegenerative Disorders and Cancer. Biomolecules 2020, 10, 1683. https://doi.org/10.3390/biom10121683
Silva-García O, Cortés-Vieyra R, Mendoza-Ambrosio FN, Ramírez-Galicia G, Baizabal-Aguirre VM. GSK3α: An Important Paralog in Neurodegenerative Disorders and Cancer. Biomolecules. 2020; 10(12):1683. https://doi.org/10.3390/biom10121683
Chicago/Turabian StyleSilva-García, Octavio, Ricarda Cortés-Vieyra, Francisco N. Mendoza-Ambrosio, Guillermo Ramírez-Galicia, and Víctor M. Baizabal-Aguirre. 2020. "GSK3α: An Important Paralog in Neurodegenerative Disorders and Cancer" Biomolecules 10, no. 12: 1683. https://doi.org/10.3390/biom10121683