
Electronic Structure, Spectroscopy, Cold Ion–Atom Elastic Collision Properties, and Photoassociation Formation Prediction of the (MgCs)+ Molecular Ion

Abstract
:1. Introduction
2. Theory and Computational Details

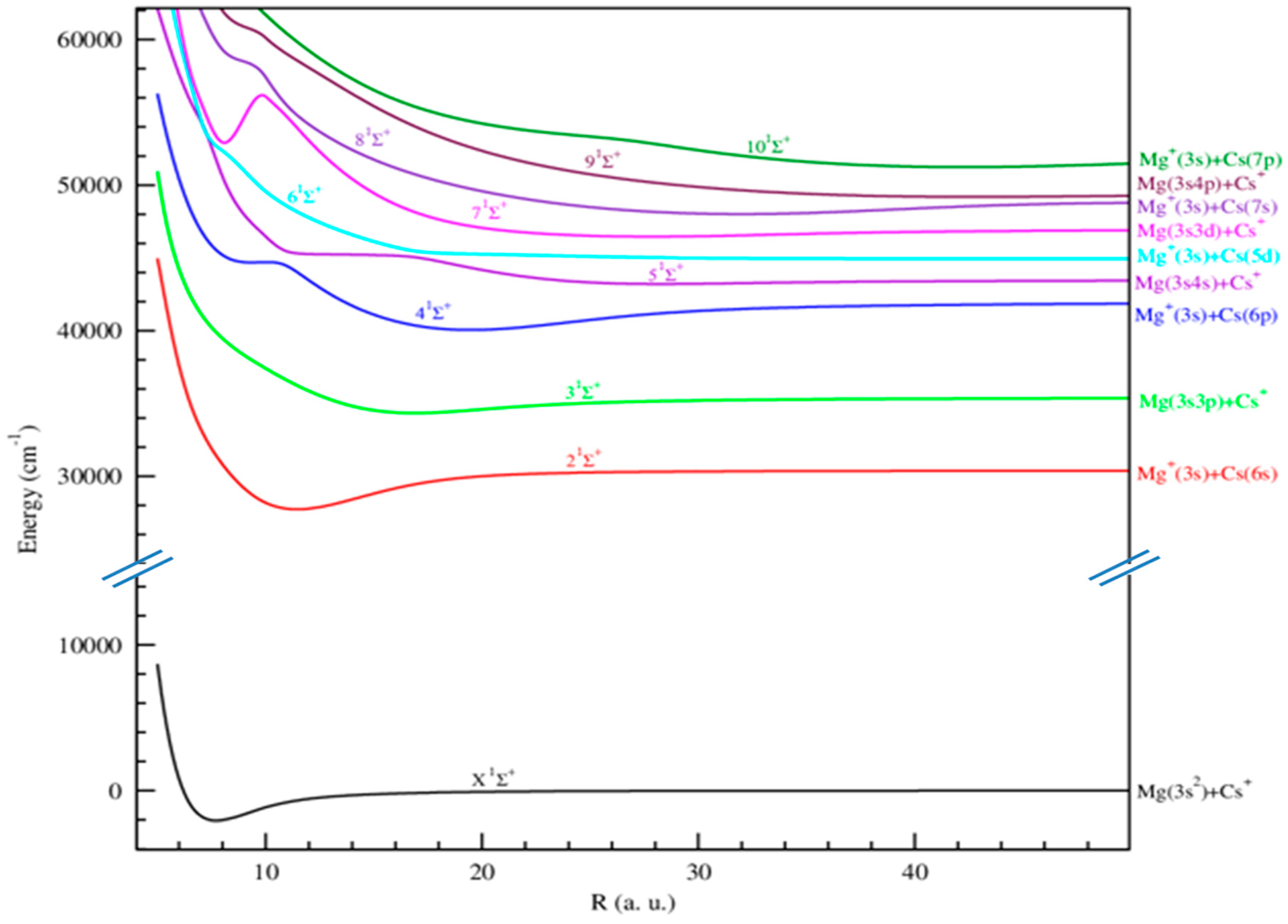{kind=link}
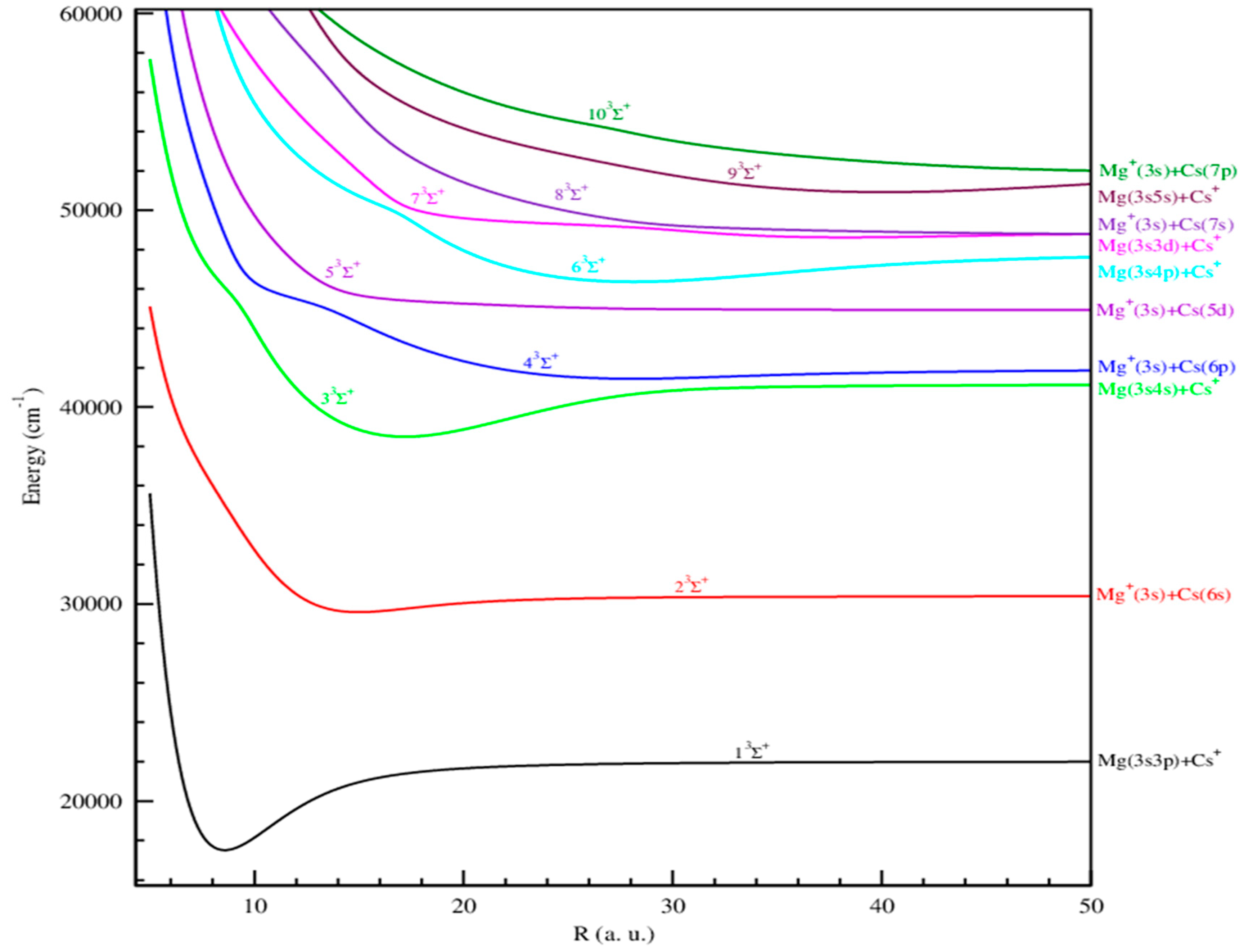{kind=link}
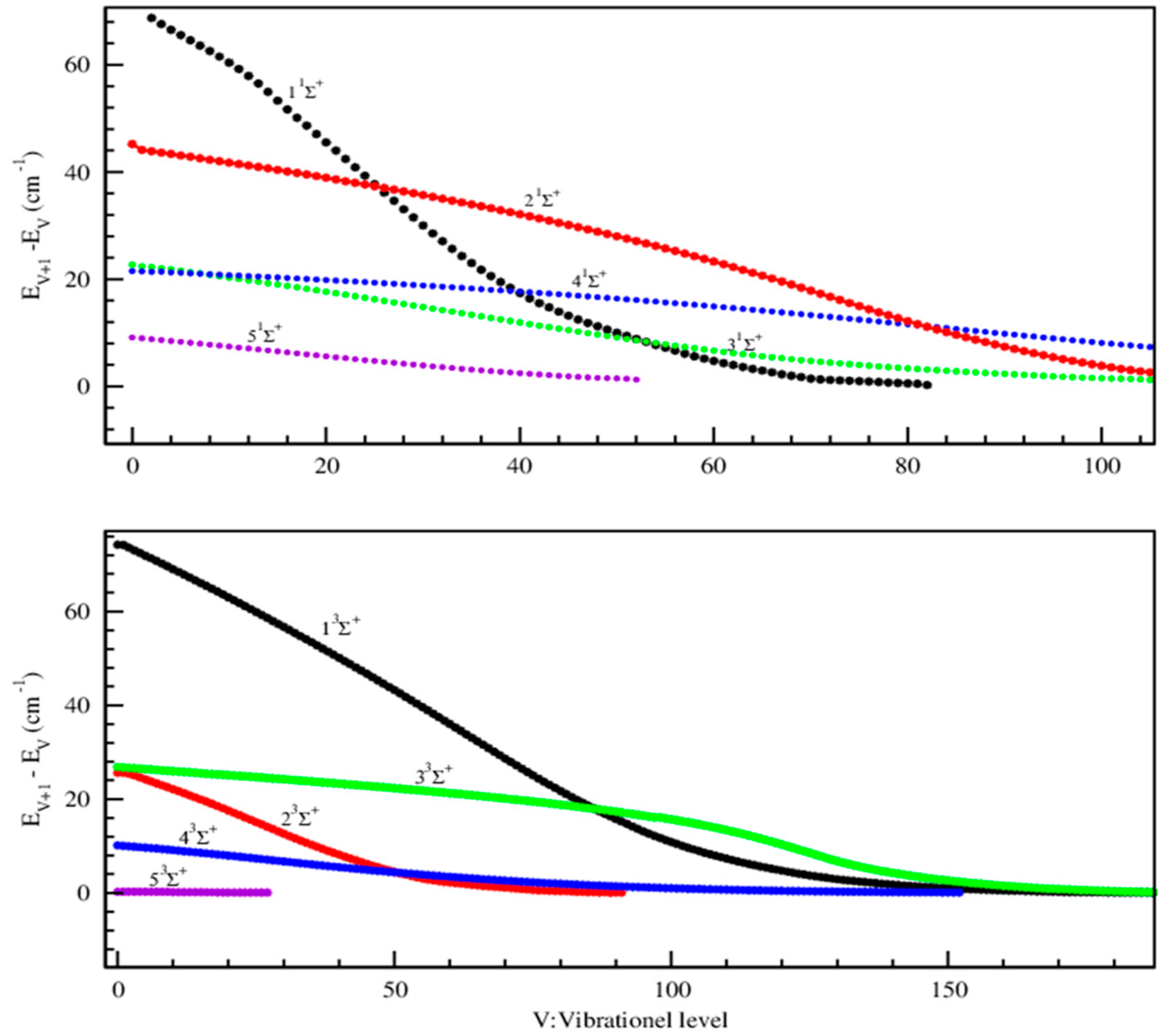{kind=link}
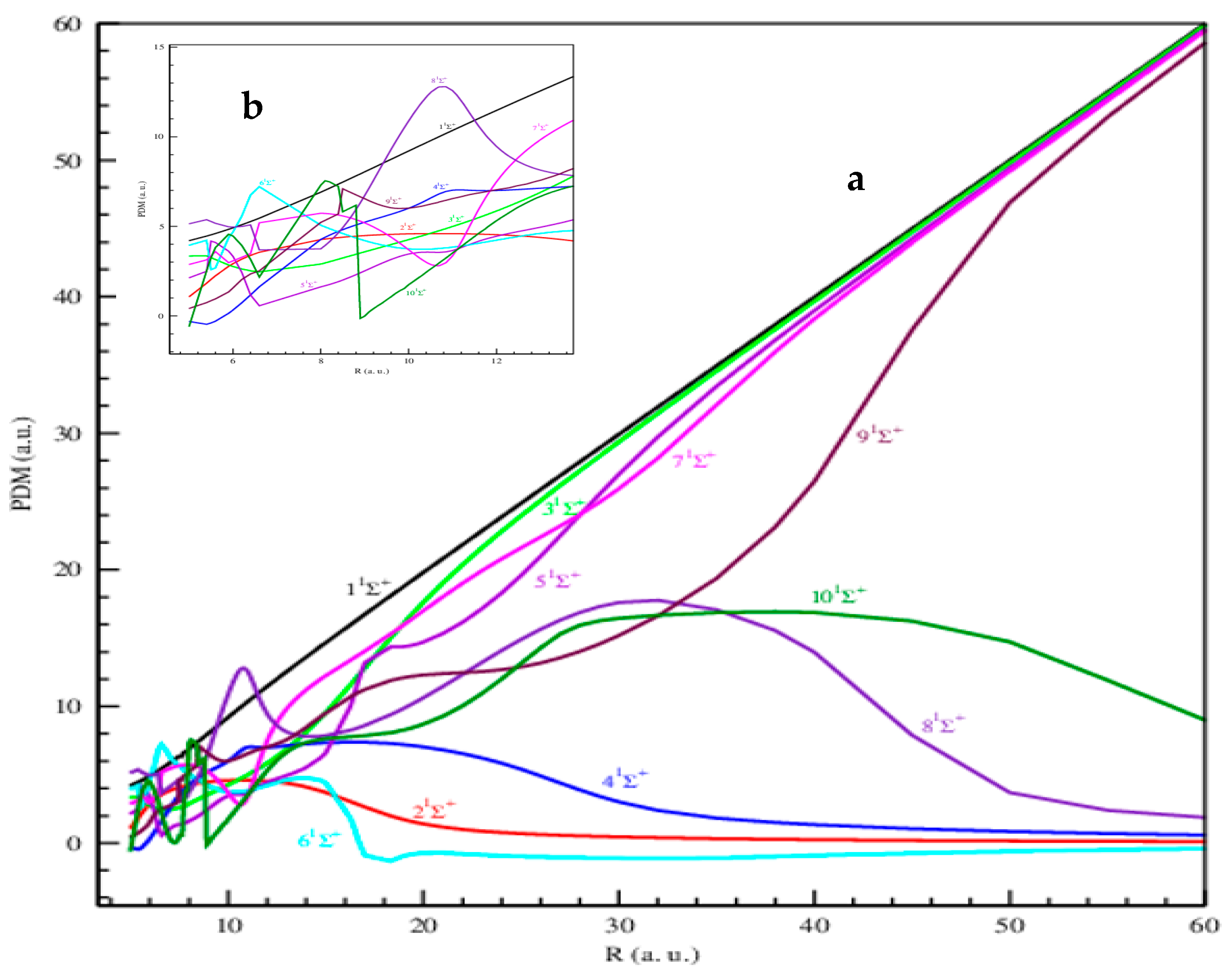{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Asymptotic Molecular State | This Work | Experiment [68,70] |
|---|---|---|---|
| X (1) 1Σ+ | Mg (3s2) + Cs+ | 0 | 0 |
| A (2) 1Σ+ | Mg+ (3s) + Cs (6s) | 30,263.62 | 30,398.60 |
| C (3) 1Σ+ | Mg (3s3p) + Cs+ | 35,050.59 | 35,319.67 |
| D (4) 1Σ+ | Mg+ (3s) + Cs (6p) | 41,811.30 | 41,946.72 |
| E (5) 1Σ+ | Mg (3s4s) + Cs+ | 43,502.57 | 43,472.29 |
| F (6) 1Σ+ | Mg+ (3s) + Cs (5d) | 44,821.40 | 44,956.60 |
| G (7) 1Σ+ | Mg (3s3d) + Cs+ | 46,402.50 | 47,967.79 |
| H (8) 1Σ+ | Mg+ (3s) + Cs (7s) | 48,802.02 | 48,934.58 |
| I (9) 1Σ+ | Mg (3s4p) + Cs+ | 49,346.10 | 49,487.00 |
| J (10) 1Σ+ | Mg+ (3s) + Cs (7p) | 52,143.74 | 52,284.65 |
| a (1) 3Σ+ | Mg (3s3p) + Cs+ | 21,876.60 | 21,994.46 |
| c (2) 3Σ+ | Mg+ (3s) + Cs (6s) | 30,263.62 | 30,398.60 |
| d (3) 3Σ+ | Mg (3s4s) + Cs+ | 41,196.77 | 41,154.85 |
| e (4) 3Σ+ | Mg+ (3s) + Cs (6p) | 41,811.30 | 41,946.72 |
| f (5) 3Σ+ | Mg+ (3s) + Cs (5d) | 44,821.40 | 44,956.60 |
| g (6) 3Σ+ | Mg (3s4p) + Cs+ | 47,857.84 | 48,197.80 |
| h (7) 3Σ+ | Mg (3s3d) + Cs+ | 47,956.38 | 46,647.87 |
| I (8) 3Σ+ | Mg+ (3s) + Cs (7s) | 48,802.02 | 48,934.58 |
| J (9) 3Σ+ | Mg (3s5s) + Cs+ | 51,871.81 | 51,976.72 |
| k (10) 3Σ+ | Mg+ (3s) + Cs (7p) | 52,143.74 | 52,284.65 |
2.1. Results and Discussions
2.1.1. Adiabatic Potential Energy and Spectroscopic Constants
2.1.2. Vibrational Levels
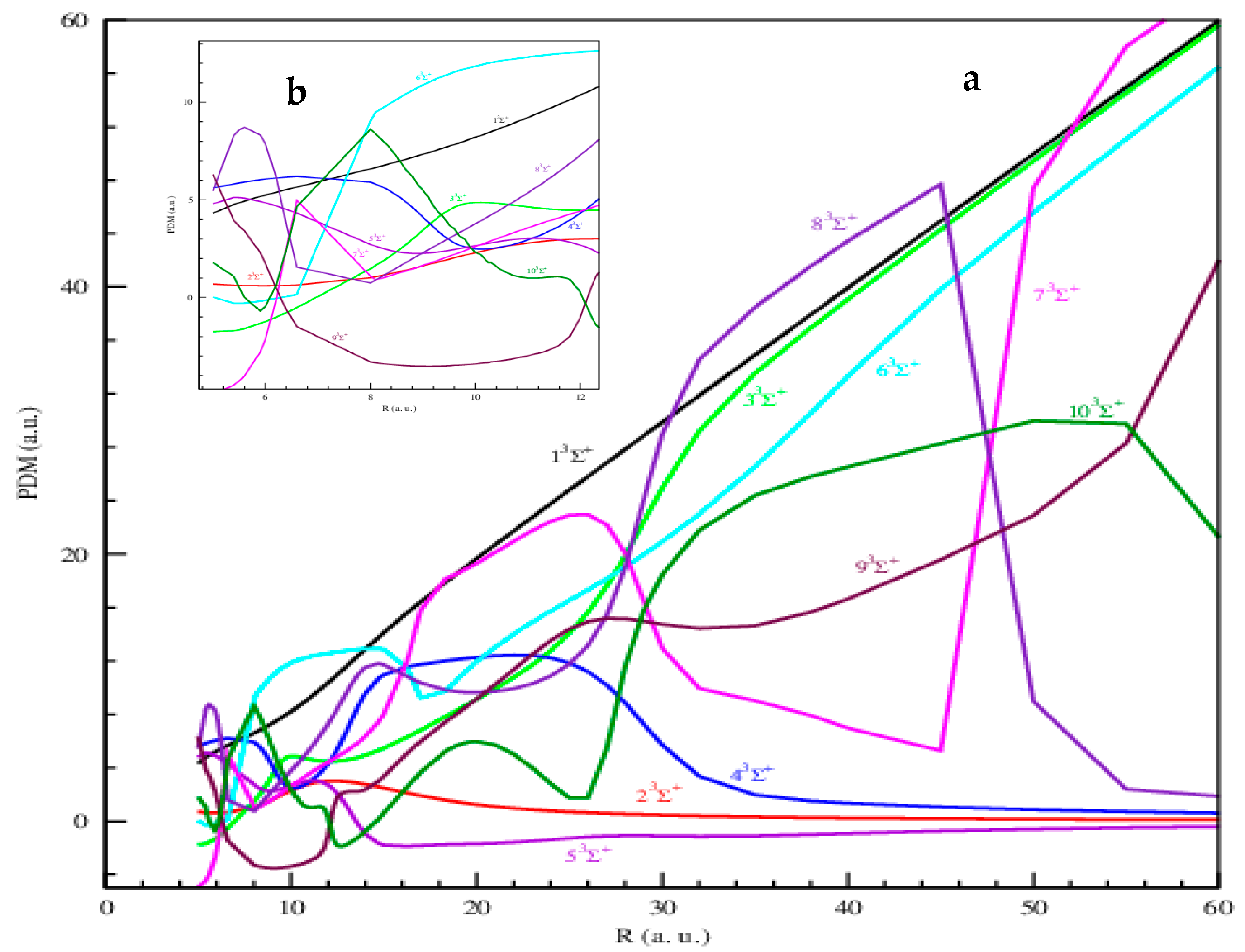
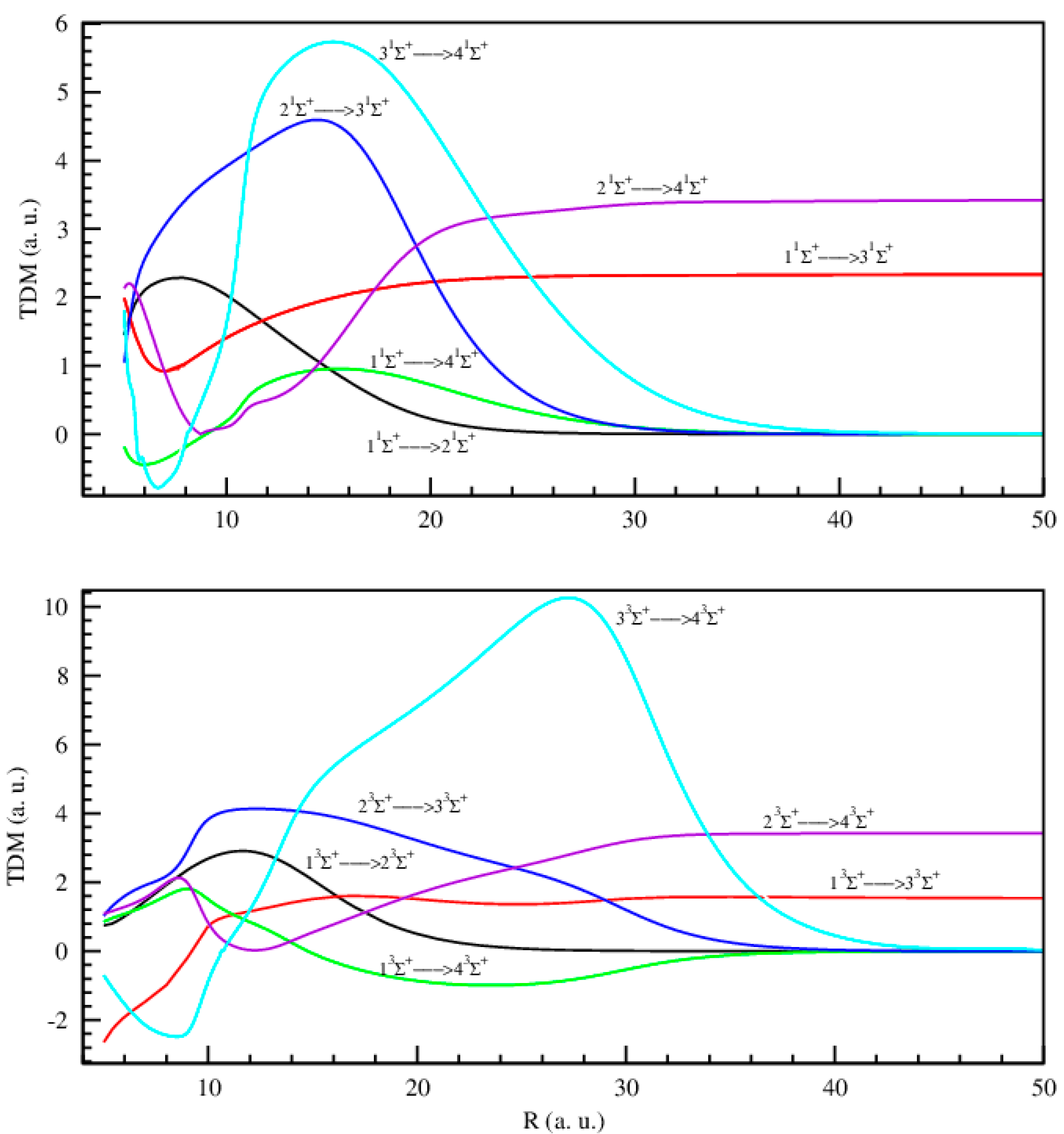
2.1.3. Permanent and Transition Dipole Moments
Permanent Dipole Moments
Transition Dipole Moments
2.1.4. Radiative Lifetime
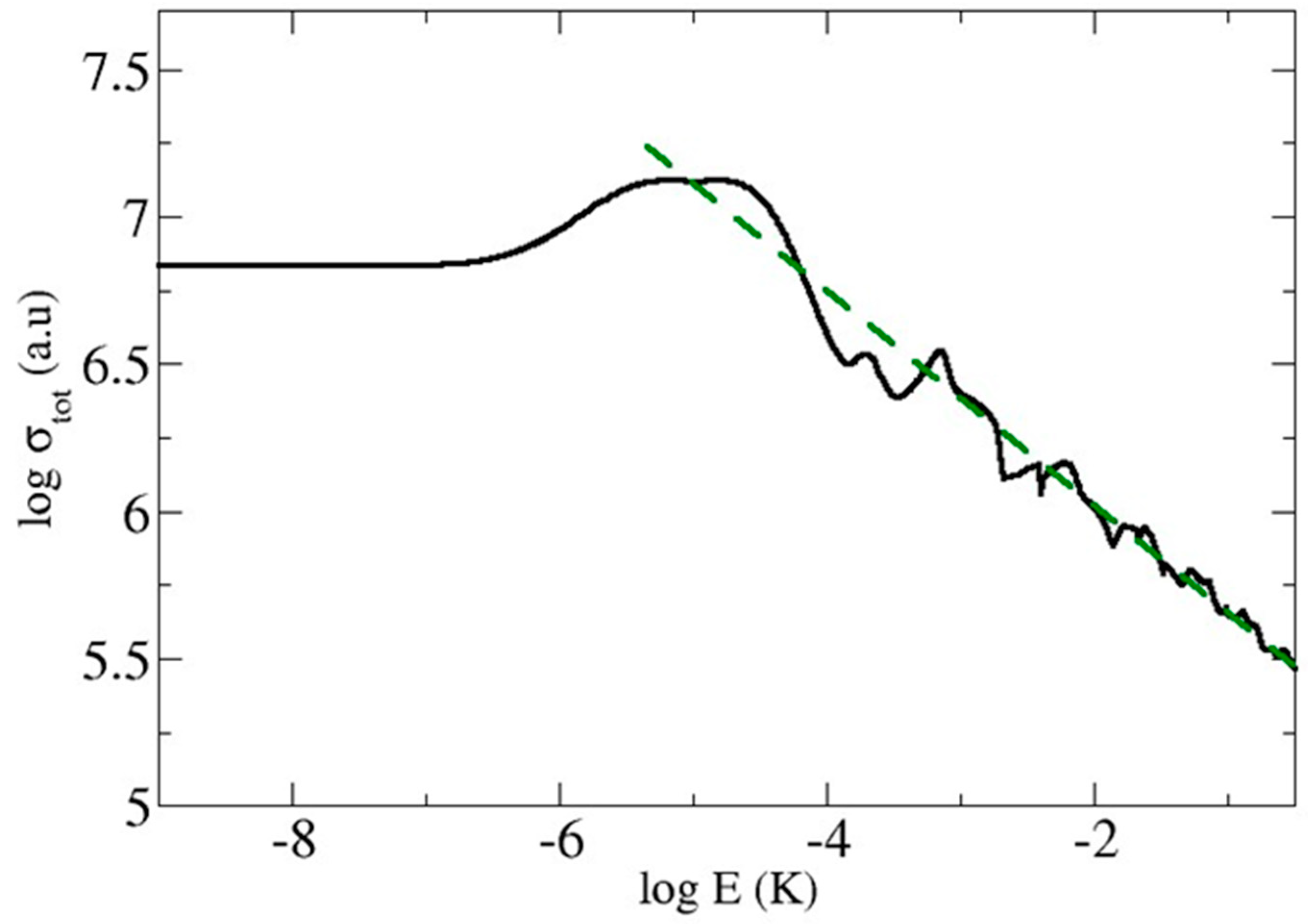
2.1.5. Ion–Atom Elastic Collisions
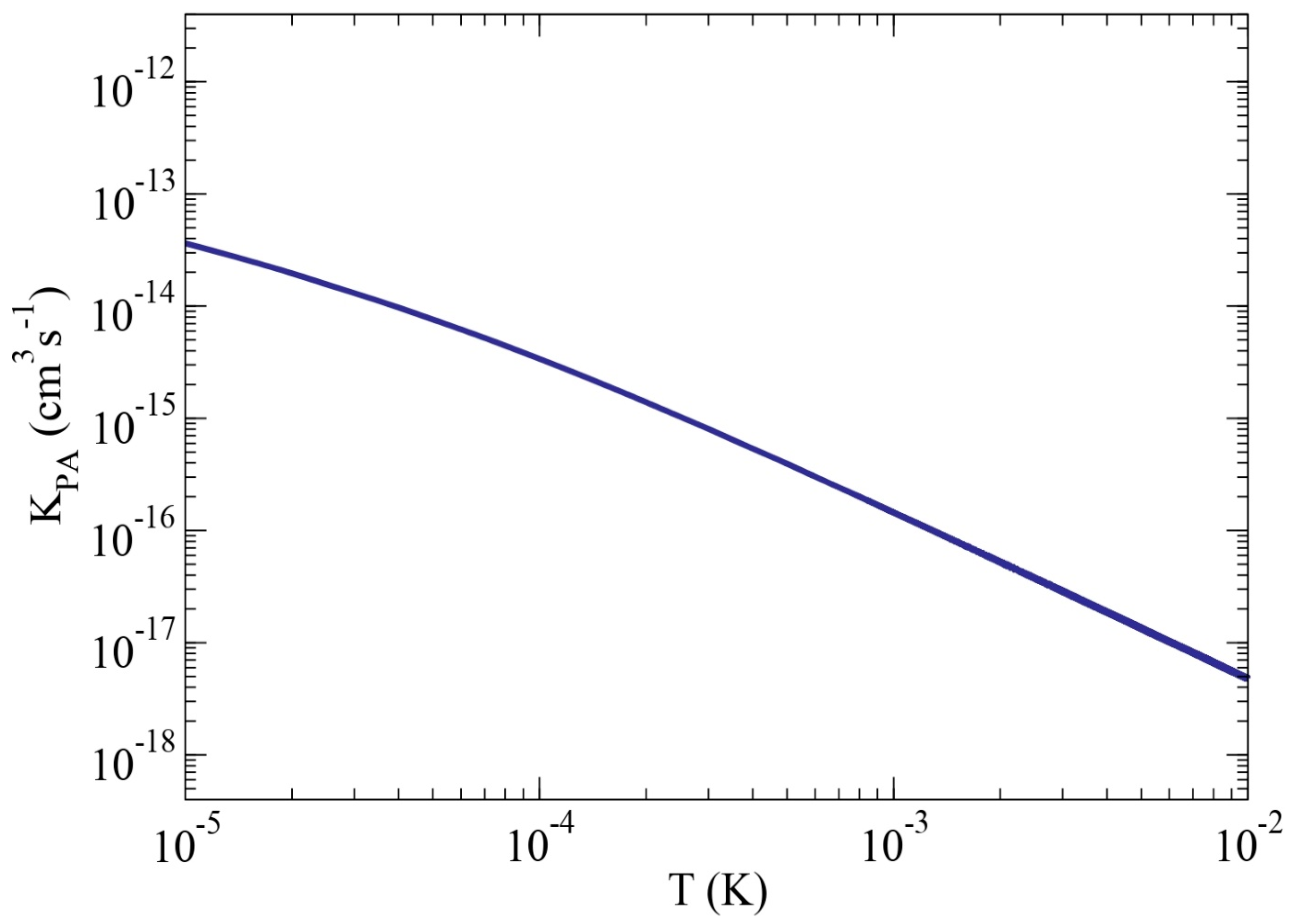
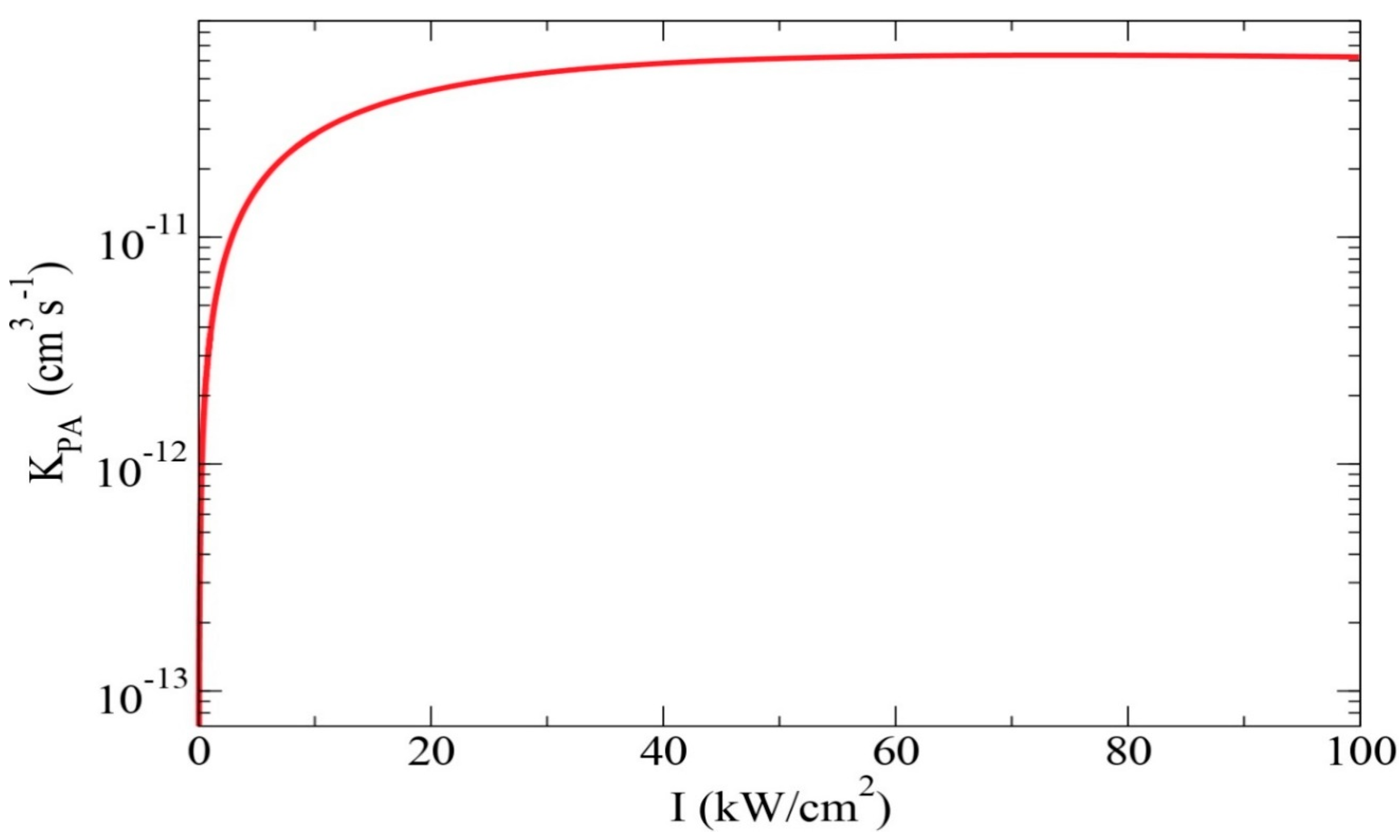
2.1.6. Two-Photon Photoassociation: Molecular Ion Formation
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carr, L.D.; DeMille, D.; Krems, R.V.; Ye, J. Cold and ultracold molecules: Science, technology and applications. New J. Phys. 2009, 11, 055049. [Google Scholar] [CrossRef]
- Hummon, M.T.; Yeo, M.; Stuhl, B.K.; Collopy, A.L.; Xia, Y.; Ye, J. 2D Magneto-Optical Trapping of Diatomic Molecules. Phys. Rev. Lett. 2013, 110, 143001. [Google Scholar] [CrossRef] [PubMed]
- Stwalleyand, W.C.; Wang, H. Photoassociation of Ultracold Atoms: A New Spectroscopic Technique. J. Mol. Spectrosc. 1999, 195, 194. [Google Scholar] [CrossRef] [PubMed]
- Côté, R.; Dalgarno, A. Ultracold atom-ion collisions. Phys. Rev. A 2000, 62, 012709. [Google Scholar] [CrossRef]
- Idziaszek, Z.; Calarco, T.; Julienne, P.S.; Simoni, A. Quantum theory of ultracold atom-ion collisions. Phys. Rev. A 2009, 79, 010702. [Google Scholar] [CrossRef]
- Krych, M.; Skomorowski, W.; Pawłowski, F.; Moszynski, R.; Idziaszek, Z. Sympathetic cooling of the Ba+ ion by collisions with ultracold Rb atoms: Theoretical prospects. Phys. Rev. A 2011, 83, 032723. [Google Scholar] [CrossRef]
- Ravi, K.; Lee, S.; Sharma, A.; Werth, G.; Rangwala, S. Cooling and stabilization by collisions in a mixed ion–atom system. Nat. Commun. 2012, 3, 1126. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, I.; Goodman, D.S.; Wells, J.E.; Narducci, F.A.; Smith, W.W. Evidence of sympathetic cooling of Na+ ions by a Na magneto-optical trap in a hybrid trap. Phys. Rev. A 2012, 86, 063419. [Google Scholar] [CrossRef]
- Boyarkin, O.V.; Mercier, S.R.; Kamariotis, A.; Rizzo, T.R. Electronic spectroscopy of cold, protonated tryptophan and tyrosine. J. Am. Chem. Soc. 2006, 128, 2816–2817. [Google Scholar] [CrossRef]
- Willitsch, S.; Bell, T.M.; Gingell, A.D.; Procter, S.R.; Softley, T.P. Cold Reactive Collisions between Laser-Cooled Ions and Velocity-Selected Neutral Molecules. Phys. Rev. Lett. 2008, 100, 043203. [Google Scholar] [CrossRef]
- Schmid, S.; Härter, A.; Denschlag, J.H. Dynamics of a Cold Trapped Ion in a Bose-Einstein Condensate. Phys. Rev. Lett. 2010, 105, 133202. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.M.; Tiesinga, E.; Lett, P.D.; Julienne, P.S. Ultracold photoassociation spectroscopy: Long-range molecules and atomic scattering, Rev. Mod. Phys. 2006, 78, 483. [Google Scholar] [CrossRef]
- Chin, C.; Grimm, R.; Julienne, P.; Tiesinga, E. Feshbach resonances in ultracold gases. Rev. Mod. Phys. 2010, 82, 1225–1286. [Google Scholar] [CrossRef]
- Da Silva, H., Jr.; Raoult, M.; Aymar, M.; Dulieu, O. Formation of molecular ions by radiative association of cold trapped atoms and ions. New J. Phys. 2015, 17, 045015. [Google Scholar] [CrossRef]
- Hall, F.H.J.; Aymar, M.; Bouloufa-Maafa, N.; Dulieu, O.; Willitsch, S. Light-Assisted Ion-Neutral Reactive Processes in the Cold Regime: Radiative Molecule Formation versus Charge Exchange. Phys. Rev. Lett. 2011, 107, 243202. [Google Scholar] [CrossRef] [PubMed]
- Hall, F.H.; Eberle, P.; Hegi, G.; Raoult, M.; Aymar, M.; Dulie, O.; Willitsch, S. Formation of molecular ions by radiative association of cold trapped atoms and ions. Mol. Phys. 2013, 111, 2020. [Google Scholar] [CrossRef]
- Mohammadi, A.; Krükow, A.; Mahdian, A.; Deiß, M.; Pérez-Ríos, J., Jr.; da Silva, H. Life and death of a cold BaRb+ molecule inside an ultracold cloud of Rb atoms. Phys. Rev. Res. 2021, 3, 013196. [Google Scholar] [CrossRef]
- Schmid, S.; Härter, A.; Frisch, A.; Hoinka, S.; Denschlag, J.H. An apparatus for immersing trapped ions into an ultracold gas of neutral atoms. Rev. Sci. Instrum. 2012, 83, 053108. [Google Scholar] [CrossRef]
- Baba, T.B.T.; Waki, I.W.I. Cooling and Mass-Analysis of Molecules Using Laser-Cooled Atoms. Jpn. J. Appl. Phys. 1996, 35, L1134. [Google Scholar] [CrossRef]
- Bertelsen, A.; Vogelius, I.S.; Jørgensen, S.; Kosloff, R.; Drewsen, M. Photo-dissociation of Cold MgH+ ions. Eur. Phys. J. D 2004, 31, 403. [Google Scholar] [CrossRef]
- Mølhave, K.; Drewsen, M. Formation of translationally cold MgH+ and MgD+ molecules in an ion trap. Phys. Rev. A. 2000, 62, 011401. [Google Scholar] [CrossRef]
- Aymar, M.; Guérout, R.; Sahlaoui, M.; Dulieu, O. Electronic structure of the magnesium hydride molecular ion. J. Phys. B: At. Mol. Opt. Phys. 2009, 42, 154025. [Google Scholar] [CrossRef]
- Khemiri, N.; Dardouri, R.; Oujia, B.; Gadéa, F.X. Ab Initio Investigation of Electronic Properties of the Magnesium Hydride Molecular Ion. J. Phys. Chem. A 2013, 117, 8915–8924. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.I.; Simons, J.; von, R. Schleyer, P. Ab initio study of the electronic structures of lithium containing diatomic molecules and ions. J. Chem. Phys. 1993, 99, 8793–8804. [Google Scholar] [CrossRef]
- Pekka Pyykk, Ö. Ab initio study of bonding trends among the 14-electron diatomic systems: From B24- to F24+. Mol. Phys. 1989, 67, 871. [Google Scholar] [CrossRef]
- Gao, Y.; Gao, T. Ab initio study of ground and low-lying excited states of MgLi and MgLi+ molecules with valence full configuration interaction and MRCI method. Mol. Phys. 2014, 112, 3015. [Google Scholar] [CrossRef]
- Fedorov, D.A.; Barnes, D.K.; Varganova, S.A. Ab initio calculations of spectroscopic constants and vibrational state lifetimes of diatomic alkali-alkaline-earth cations. J. Chem. Phys. 2017, 147, 124304. [Google Scholar] [CrossRef] [PubMed]
- Smialkowski, M.; Tomza, M. Interactions and chemical reactions in ionic alkali-metal and alkaline-earth-metal diatomic AB+ and triatomic A2B+ systems. Phys. Rev. A. 2020, 101, 012501. [Google Scholar] [CrossRef]
- ElOualhazi, R.; Berriche, H. Electronic Structure and Spectra of the MgLi+ Ionic Molecule. J. Phys. Chem. A 2016, 120, 452. [Google Scholar] [CrossRef]
- Bala, R.; Nataraj, H.S.; Abe, M.; Kajita, M. Calculations of electronic properties and vibrational parameters of alkaline-earth lithides: MgLi+ and CaLi+. Mol. Phys. 2018, 117, 712–725. [Google Scholar] [CrossRef]
- Grier, A.T.; Cetina, M.; Oručević, F.; Vuletić, V. Observation of Cold Collisions between Trapped Ions and Trapped Atoms. Phys. Rev. Lett. 2009, 102, 223201. [Google Scholar] [CrossRef]
- Sullivan, S.T.; Rellergert, W.G.; Kotochigova, S.; Hudson, E.R. Role of Electronic Excitations in Ground-State-Forbidden Inelastic Collisions Between Ultracold Atoms and Ions. Phys. Rev. Lett. 2012, 109, 223002. [Google Scholar] [CrossRef]
- Rellergert, W.G.; Sullivan, S.T.; Kotochigova, A.; Petrov, K.S.; Chen, S.; Schowalter, J.; Hudson, E.R.; Millikelvin, E. Reactive Collisions between Sympathetically Cooled Molecular Ions and Laser-Cooled Atoms in an Ion-Atom Hybrid Trap. Phys. Rev. Lett. 2011, 107, 24320. [Google Scholar]
- Zipkes, C.; Palzer, S.; Sias, C.; Köhl, M. A trapped single ion inside a Bose–Einstein condensate. Nature 2010, 464, 388–391. [Google Scholar] [CrossRef]
- Haze, S.; Hata, S.; Fujinaga, M.; Mukaiyama, T. Observation of elastic collisions between lithium atoms and calcium ions. Phys. Rev. A 2013, 87, 052715. [Google Scholar] [CrossRef]
- Smith, W.; Goodman, D.; Sivarajah, I.; Wells, J.; Banerjee, S.; Côté, R.; Michels, H.; Mongtomery, J.A.; Narducci, F. Experiments with an ion-neutral hybrid trap: Cold charge-exchange collisions. Appl. Phys. B 2014, 114, 75. [Google Scholar] [CrossRef]
- Meir, Z.; Sikorsky, T.; Ben-Shlomi, R.; Akerman, N.; Dallal, Y.; Ozeri, R. Dynamics of a Ground-State Cooled Ion Colliding with Ultracold Atoms. Phys. Rev. Lett. 2016, 117, 243401. [Google Scholar] [CrossRef] [PubMed]
- Fürst, H.; Feldker, T.; Ewald, N.V.; Joger, J.; Tomza, M.; Gerritsma, R. Dynamics of a single ion-spin impurity in a spin-polarized atomic bath. Phys. Rev. A 2018, 98, 012713. [Google Scholar] [CrossRef]
- yothi, S.; Egodapitiya, K.N.; Bondurant, B.; Jia, Z.; Pretzsch, E.; Chiappina, P.; Shu, G.; Brown, K.R. A hybrid ion-atom trap with integrated high resolution mass spectrometer. Rev. Sci. Instruments 2019, 90, 103201. [Google Scholar]
- Schmidt, J.; Weckesser, P.; Thielemann, F.; Schaetz, T.; Karpa, L. Optical Traps for Sympathetic Cooling of Ions with Ultracold Neutral Atoms. Phys. Rev. Lett. 2020, 124, 053402. [Google Scholar] [CrossRef]
- Härter, A.; Krükow, A.; Brunner, A.; Schnitzler, W.; Schmid, S.; Denschlag, J.H. Single Ion as a Three-Body Reaction Center in an Ultracold Atomic Gas. Phys. Rev. Lett. 2012, 109, 123201. [Google Scholar] [CrossRef]
- Jyothi, S.; Ray, T.; Dutta, S.; Allouche, A.R.; Vexiau, R.; Dulieu, O.; Rangwala, S.A. Photodissociation of Trapped Rb+2: Implications for Simultaneous Trapping of Atoms and Molecular Ions. Phys. Rev. Lett. 2016, 117, 213002. [Google Scholar] [CrossRef] [PubMed]
- da Silva, H., Jr.; Raoult, M.; Aymar, M.; Dulieu, O. Formation of molecular ions by radiative association of cold trapped atoms and ions. Mol. Phys. 2013, 111, 1683. [Google Scholar] [CrossRef]
- Alharzali, N.; Sardar, D.; Mlika, R.; Deb, B.; Berriche, H. Spectroscopic properties and cold elastic collisions of alkaline-earth Mg + Mg+ system. J. Phys. B At. Mol. Opt. Phys. 2018, 51, 195201. [Google Scholar] [CrossRef]
- Singer, K.; Poschinger, U.; Murphy, M.; Ivanov, P.; Ziesel, F.; Calarco, T.; Schmidt-Kaler, F. Trapped ions as quantum bits: Essential numerical tools. Rev. Mod. Phys. 2010, 82, 2609–2632. [Google Scholar] [CrossRef]
- Schneider, C.; Porras, D.; Schaetz, T. Experimental quantum simulations of many-body physics with trapped ions. Rep. Prog. Phys. 2012, 75, 024401. [Google Scholar] [CrossRef] [PubMed]
- Idziaszek, Z.; Simoni, A.; Calarco, T.; Julienne, P.S. Multichannel quantum-defect theory for ultracold atom–ion collisions. New J. Phys. 2011, 13, 083005. [Google Scholar] [CrossRef]
- Tomza, M.; Koch, C.P.; Moszynski, R. Quantum gas microscopy with spin, atom-number, and multilayer readout. Phys. Rev. A 2015, 91, 042706. [Google Scholar] [CrossRef]
- Farjallah, M.; Sardar, D.; El-Kork, N.; Deb, B.; Berriche, H. Electronic structure and photoassociation scheme of ultracold (MgK+) molecular ions. J. Phys. B At. Mol. Opt. Phys. 2018, 52, 045201. [Google Scholar] [CrossRef]
- Gacesa, M.; Montgomery, J.A.; Michels, H.H.; Côté, R. Controlling charge transfer in ultracold gases via Feshbach resonances. Phys. Rev. A 2016, 94, 013407. [Google Scholar] [CrossRef]
- Petrov, A.; Makrides, C.; Kotochigova, S.J. A new approach to molecular dynamics with non-adiabatic and spin-orbit effects with applications to QM/MM simulations of thiophene and selenophene. Chem. Phys. 2017, 146, 08430. [Google Scholar]
- Zrafi, W.; Ladjimi, H.; Said, H.; Berriche, H.; Tomza, M. Ab initio electronic structure and prospects for the formation of ultracold calcium–alkali-metal-atom molecular ions. New J. Phys. 2020, 22, 073015. [Google Scholar] [CrossRef]
- Ladjimi, H.; Farjallah, M.; Berriche, H. Spectroscopic, structural and lifetime calculations of the ground and low-lying excited states of the BeNa+, BeK+, and BeRb+ molecular ions. Phys. Scrip. 2020, 95, 055404. [Google Scholar] [CrossRef]
- Ladjimi, H.; Sardar, D.; Farjallah, M.; Alharzali, N.; Naskar, S.; Mlika, R.; Berriche, H.; Deb, B. Spectroscopic properties of the molecular ions BeX+ (X = Na, K, Rb): Forming cold molecular ions from an ion–atom mixture by stimulated Raman adiabatic process. Mol. Phys. 2018, 116, 1812–1826. [Google Scholar] [CrossRef]
- Ladjimi, H.; Zrafi, W.; Farjallah, M.; Bejaoui, M.; Berriche, H. Electronic structure, cold ion–atom elastic collision properties and possibility of laser cooling of BeCs+ molecular ion. Phys. Chem. Chem. Phys. 2022, 24, 18511–18522. [Google Scholar] [CrossRef] [PubMed]
- Ghanmi, C.; Farjallah, M.; Berriche, H. Theoretical study of the alkaline-earth (LiBe)+ ion: Structure, spectroscopy and dipole moments. J. Phys. B 2017, 50, 055101. [Google Scholar] [CrossRef]
- Farjallah, M.; Ghanmi, C.; Berriche, H. Structure and spectroscopic properties of the beryllium hydride ion BeH+: Potential energy curves, spectroscopic constants, vibrational levels and permanent dipole moments. Eur. Phys. J. D 2013, 67, 245. [Google Scholar] [CrossRef]
- Evangelisti, S.; Daudey, J.-P.; Malrieu, J.-P. Convergence of an improved CIPSI algorithm. Chem. Phys. 1983, 75, 91–102. [Google Scholar] [CrossRef]
- Durand, P.; Barthelat, J.C. A theoretical method to determine atomic pseudopotentials for electronic structure calculations of molecules and solids. Theor. Chim. Acta 1975, 38, 283–302. [Google Scholar] [CrossRef]
- Durand, P.; Barthelat, J.C. Pseudopotentials in atomic and molecular physics. Chem. Gazz. Chim. Ital. 1978, 108, 225–236. [Google Scholar]
- Barthelat, J.C.; Durand, P.; Serafini, A. Non-empirical pseudopotentials for molecular calculations. Mol. Phys. 1977, 33, 159–180. [Google Scholar] [CrossRef]
- Müller, W.; Flesch, J.; Meyer, W. Treatment of intershell correlation effects in ab initio calculations by use of core polarization potentials. Method and application to alkali and alkaline earth atoms. J. Chem. Phys. 1984, 80, 3297–3310. [Google Scholar] [CrossRef]
- Foucrault, M.; Millié, P.; Daudey, J.P. Non-perturbative method for core–valence correlation in pseudopotential calculations: Application to the Rb2 and Cs2 molecules. J. Chem. Phys. 1992, 96, 1257–1264. [Google Scholar] [CrossRef]
- Wilson, J.N.; Curtis, R.M. Dipole polarizabilities of ions in alkali halide crystals. J. Phys. Chem. 1970, 74, 187–196. [Google Scholar] [CrossRef]
- Pavolini, D.; Gustavsson, T.; Spiegelmann, F.; Daudey, J.P. Theoretical study of the excited states of the heavier alkali dimers. I. The RbCs molecule. J. Phys. B Mol. Opt. 1989, 77, 221721. [Google Scholar] [CrossRef]
- Ghanmi, C.; Berriche, H.; Ben Ouada, H. Evaluation of the Atomic Polarisabities of Li and K using the Long Range Lik+ Electronic States Behaviour. In Proceedings of the International Conference on Computational and Mathematical Methods in Science and Engineering, CMMSE, Alicante, Spain, 27–30 June 2005; pp. 166–174. [Google Scholar]
- Bouzouita, H.; Ghanmi, C.; Berriche, H. Ab initio study of the alkali-dimer cation Li2+. J. Mol. Struct. Theochem. 2006, 777, 75–80. [Google Scholar] [CrossRef]
- Moore, C.E.; Mack, J.E. Atomic energy levels. At. Energy Levels Deriv. Anal. Opt. Spectra. Phys. Today 1952, 5, 23. [Google Scholar] [CrossRef]
- Mitroy, J.; Zhang, J.Y. Long range interactions of the Mg+ and Ca+ ions. Eur. Phys. J. D 2007, 46, 415–424. [Google Scholar] [CrossRef]
- Moore, C.E. Atomic Energy Levels; NSRDS-NBS No. 467; US Government Printing Office: Washington, DC, USA, 1989. [Google Scholar]
- Zemke, W.T.; Crooks, J.B.; Stwalley, W.C. Radiative and nonradiative lifetimes for vibrational levels of the A 1Σ+ state of 7LiH. J. Chem. Phys. 1978, 68, 4628. [Google Scholar] [CrossRef]
- Partridge, H.; Langhoff, S.R. Theoretical treatment of the X 1Σ+, A 1Σ+, and B 1Π states of LiH. J. Chem. Phys. 1981, 74, 2361. [Google Scholar] [CrossRef]
- Berriche, H.; Gadéa, F.X. Transition dipole function and radiative lifetimes for the A and C 1Σ+ states of the LiH molecule. Eur. Phys. J. D 2016, 70, 1–9. [Google Scholar] [CrossRef]
- Berriche, H. Ab Initio Adiabatic and Diabatic Permanent Dipoles for the Low-Lying States of the LiH Molecule. Ph.D. Thesis, Paul Sabatier University, Toulouse, France, 1995. [Google Scholar]
- Stolyarov, A.V.; Pupyshev, V.I. Approximate sum rule for diatomic vibronic states. Phys. Rev. A 1994, 49, 1693. [Google Scholar] [CrossRef]
- Pazyuk, E.; Stolyarov, A.; Pupyshev, V. Approximate sum rule for diatomic vibronic states as a tool for the evaluation of molecular properties. Chem. Phys. Lett. 1994, 228, 219–224. [Google Scholar] [CrossRef]
- Johnson, R.B. New numerical methods applied to solving the one-dimensional eigenvalue problem. J. Chem. Phys. 1977, 67, 4086. [Google Scholar] [CrossRef]
- Napolitano, R.; Weiner, J.; Williams, C.J.; Julienne, P.S. Line Shapes of High Resolution Photoassociation Spectra of Optically Cooled Atoms. Phys. Rev. Lett. 1994, 73, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, A.; Deb, B. Formation of cold molecular ions by radiative processes in cold ion-atom collisions. Phys. Rev. A 2011, 83, 022703. [Google Scholar] [CrossRef]
- Sardar, D.; Naskar, S.; Pal, A.; Berriche, H.; Deb, B. Formation of a molecular ion by photoassociative Raman processes. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 245202. [Google Scholar] [CrossRef]
- Sardar, D.; Naskar, S. Cold collisions between alkali metals and alkaline-earth metals in the heteronuclear atom-ion system Li + Ba+. Phys. Rev. A 2023, 107, 043323. [Google Scholar] [CrossRef]









| Atom | α | ρs | ρp | ρd |
|---|---|---|---|---|
| Mg(Z = 12) | 0.46904 | 0.9 | 1.25499 | 1.500 |
| Cs(Z = 55) | 15.117 | 2.69 | 1.85 | 2.810 |
| State | Re (a.u.) | De (cm−1) | Te (cm−1) | ωe (cm−1) | ωexe (cm−1) | Be (cm−1) |
|---|---|---|---|---|---|---|
| X1Σ+ | 7.70 | 2047 | 0 | 73.2 | 0.63 | 0.049364 |
| 7.85 [28] | 1861 [28] | 73.2 [28] | 0.0481 [28] | |||
| 21Σ+ | 11.45 | 2662 | 29,782 | 44.64 | 0.25 | 0.022354 |
| 31Σ+ | 16.88 | 1054 | 36,391 | 21.92 | 0.16 | 0.010280 |
| 41Σ+ | 9.33 | −2750 | ||||
| 19.46 | 1891 | 42,101 | 21.73 | 0.06 | 0.007700 | |
| 51Σ+ | 28.24 | 263 | 45,256 | 9.12 | 0.07 | 0.003674 |
| 61Σ+ | 58.89 | 4 | 46,990 | −0.48 | 14.61 | 0.001160 |
| 71Σ+ | 8.68 | −3301 | ||||
| 27.83 | 509 | 48,507 | 8.92 | 26.51 | 0.038919 | |
| 81Σ+ | 8.68 | −8647 | ||||
| 31.99 | 923 | 50,059 | 10.13 | 29.95 | 0.038864 | |
| 91Σ+ | 8.69 | −10,460 | ||||
| 42.75 | 300 | 51,240 | 5.44 | 30.84 | 0.038839 | |
| 101Σ+ | 8.71 | −10,297 | ||||
| 42.27 | 1105 | 53,289 | 8.07 | 40.10 | 0.038614 | |
| 13Σ+ | 8.58 | 4504 | 19,556 | 74.78 | 0.52 | 0.039826 |
| 23Σ+ | 14.98 | 813 | 31,630 | 25.56 | 0.17 | 0.013050 |
| 33Σ+ | 17.20 | 2658 | 40,542 | 26.85 | 0.05 | 0.009905 |
| 43Σ+ | 28.15 | 506 | 43,486 | 10.12 | 0.04 | 0.003698 |
| 53Σ+ | 58.89 | 4 | 46,990 | −0.48 | 14.61 | 0.001160 |
| 63Σ+ | 28.13 | 1600 | 48,413 | 13.19 | 0.02 | 0.003704 |
| 73Σ+ | 38.20 | 31 | 50,667 | 6.81 | 0.03 | 0.002008 |
| 83Σ+ | 49.32 | 131 | 50,850 | 4.59 | 0.02 | 0.001205 |
| 93Σ+ | 40.09 | 1055 | 52,967 | 8.79 | 0.01 | 0.001823 |
| 103Σ+ | 56.76 | 395 | 53,999 | 3.95 | 0.01 | 0.000910 |
| Vibrational Level | Ev − Ev−1 (cm−1) | Franck–Condon (ns) | Sum Rule (ns) |
|---|---|---|---|
| 0 | 10.579 | 10.582 | |
| 1 | 44.110 | 10.658 | 10.661 |
| 2 | 43.878 | 10.739 | 10.742 |
| 3 | 43.599 | 10.825 | 10.828 |
| 4 | 43.367 | 10.912 | 10.915 |
| 5 | 43.080 | 11.003 | 11.006 |
| 6 | 42.848 | 11.096 | 11.098 |
| 7 | 42.550 | 11.191 | 11.194 |
| 8 | 42.278 | 11.290 | 11.294 |
| 9 | 42.016 | 11.394 | 11.399 |
| 10 | 41.744 | 11.502 | 11.511 |
| 11 | 41.477 | 11.634 | 11.666 |
| 12 | 41.213 | 11.770 | 11.824 |
| 13 | 40.941 | 12.055 | 12.253 |
| 14 | 40.666 | 12.294 | 12.589 |
| 15 | 40.389 | 12.989 | 13.787 |
| 16 | 40.109 | 13.549 | 14.649 |
| 17 | 39.832 | 14.486 | 16.450 |
| 18 | 39.534 | 15.301 | 17.708 |
| 19 | 39.237 | 15.689 | 18.283 |
| 20 | 38.926 | 15.967 | 18.465 |
| 21 | 38.604 | 16.259 | 18.689 |
| 22 | 38.293 | 17.042 | 19.857 |
| 23 | 37.992 | 17.532 | 20.454 |
| 24 | 37.668 | 18.321 | 21.644 |
| 25 | 37.352 | 18.660 | 21.795 |
| 26 | 37.041 | 19.142 | 22.251 |
| 27 | 36.707 | 19.728 | 22.916 |
| 28 | 36.381 | 20.504 | 23.969 |
| 29 | 36.044 | 21.158 | 24.555 |
| 30 | 35.699 | 21.574 | 24.792 |
| 31 | 35.361 | 22.446 | 25.859 |
| 32 | 35.012 | 23.075 | 26.408 |
| 33 | 34.666 | 23.816 | 27.190 |
| 34 | 34.312 | 24.277 | 27.467 |
| 35 | 33.959 | 25.217 | 28.468 |
| 36 | 33.599 | 26.400 | 29.687 |
| 37 | 33.235 | 26.976 | 30.140 |
| 38 | 32.868 | 27.668 | 30.647 |
| 39 | 32.493 | 28.386 | 31.252 |
| 40 | 32.114 | 29.689 | 32.700 |
| 41 | 31.726 | 30.554 | 33.375 |
| 42 | 31.333 | 31.446 | 34.124 |
| 43 | 30.932 | 32.939 | 35.617 |
| 44 | 30.526 | 33.864 | 36.390 |
| 45 | 30.118 | 34.807 | 37.156 |
| 46 | 29.705 | 35.796 | 37.999 |
| 47 | 29.287 | 37.175 | 39.363 |
| 48 | 28.863 | 38.664 | 40.724 |
| 49 | 28.435 | 39.788 | 41.692 |
| 50 | 28.000 | 41.625 | 43.490 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farjallah, M.; Sardar, D.; Deb, B.; Berriche, H. Electronic Structure, Spectroscopy, Cold Ion–Atom Elastic Collision Properties, and Photoassociation Formation Prediction of the (MgCs)+ Molecular Ion. Atoms 2023, 11, 121. https://doi.org/10.3390/atoms11090121
Farjallah M, Sardar D, Deb B, Berriche H. Electronic Structure, Spectroscopy, Cold Ion–Atom Elastic Collision Properties, and Photoassociation Formation Prediction of the (MgCs)+ Molecular Ion. Atoms. 2023; 11(9):121. https://doi.org/10.3390/atoms11090121
Chicago/Turabian StyleFarjallah, Mohamed, Dibyendu Sardar, Bimalendu Deb, and Hamid Berriche. 2023. "Electronic Structure, Spectroscopy, Cold Ion–Atom Elastic Collision Properties, and Photoassociation Formation Prediction of the (MgCs)+ Molecular Ion" Atoms 11, no. 9: 121. https://doi.org/10.3390/atoms11090121